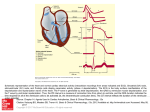
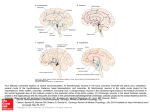
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Membrane potential wikipedia , lookup
Environmental enrichment wikipedia , lookup
Neural oscillation wikipedia , lookup
Endocannabinoid system wikipedia , lookup
Synaptogenesis wikipedia , lookup
Limbic system wikipedia , lookup
Functional magnetic resonance imaging wikipedia , lookup
Neuroinformatics wikipedia , lookup
Subventricular zone wikipedia , lookup
Neurophilosophy wikipedia , lookup
Neurolinguistics wikipedia , lookup
Artificial general intelligence wikipedia , lookup
Neuroeconomics wikipedia , lookup
Selfish brain theory wikipedia , lookup
Human brain wikipedia , lookup
Cognitive neuroscience wikipedia , lookup
Development of the nervous system wikipedia , lookup
Activity-dependent plasticity wikipedia , lookup
Brain Rules wikipedia , lookup
Nonsynaptic plasticity wikipedia , lookup
End-plate potential wikipedia , lookup
Neuropsychology wikipedia , lookup
Premovement neuronal activity wikipedia , lookup
Biochemistry of Alzheimer's disease wikipedia , lookup
Brain morphometry wikipedia , lookup
Holonomic brain theory wikipedia , lookup
Aging brain wikipedia , lookup
Neural correlates of consciousness wikipedia , lookup
Neuroplasticity wikipedia , lookup
Electrophysiology wikipedia , lookup
Spike-and-wave wikipedia , lookup
Feature detection (nervous system) wikipedia , lookup
Haemodynamic response wikipedia , lookup
Multielectrode array wikipedia , lookup
Single-unit recording wikipedia , lookup
Pre-Bötzinger complex wikipedia , lookup
Synaptic gating wikipedia , lookup
Optogenetics wikipedia , lookup
Nervous system network models wikipedia , lookup
Circumventricular organs wikipedia , lookup
Stimulus (physiology) wikipedia , lookup
History of neuroimaging wikipedia , lookup
Metastability in the brain wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Neuroanatomy wikipedia , lookup
Molecular neuroscience wikipedia , lookup
DOES ISCHEMIA CAUSE ACUTE NEURONAL DAMAGE BY CONVERTING THE NA+/K+ PUMP INTO A CHANNEL? by Danielle Kim A thesis submitted to the Department of Neuroscience In conformity with the requirements for the degree of Master of Science Queen’s University Kingston, Ontario, Canada (September, 2014) Copyright ©Danielle Kim, 2014 Abstract The gray matter of the higher brain undergoes spreading depolarization in response to ischemia, which increases metabolic demand and so promotes acute neuronal injury. The molecular mechanism linking ischemic failure of the Na+/K+ pump to the subsequent onset of a large inward current in neurons has remained a mystery because blockade of any conventional voltage- or ligand- gated channel does not prevent ischemic or “anoxic” depolarization (AD) propagating across grey matter. Recently our laboratory became aware of a marine poison whose molecular action is well characterized but has escaped scrutiny by neuroscientists. We hypothesized that this toxin could provide insight as to how ischemia acutely damages neurons at the molecular level. Palytoxin (PTX) specifically binds the Na+/K+ ATPase molecule at nanomolar concentrations, converting it from an ATP-requiring transporter to an open cationic channel. The result is sudden neuronal Na+ influx and K+ efflux. The double jeopardy of pump failure with the induction of a strong inward current should induce dramatic AD-like activity. Using light transmittance (LT) imaging, we show that bath application of 10 nM PTX to live coronal brain slices induces a propagating depolarization at 246±14 s in the neocortex, similar to AD induced by oxygen/glucose deprivation (OGD) at 289±10 s. In the neocortex, a distinct negative DC shift was recorded as the elevated LT front induced by OGD (4.2±1.0 mV) or PTX (4.1±0.8 mV) passed by an extracellular recording pipette, indicating strong depolarization of cells at the electrode. Both treatments induced strong spreading depolarization in the same higher brain regions and weaker events in lower brain gray matter. We also tested the drugs dibucaine and carbetapentane, which potently delay AD onset induced by OGD. One µM dibucaine pretreatment increased depolarization latency induced by OGD by 44±11 % and PTX by 61±20 % while 30 µM carbetapentane pretreatment increased depolarization latency induced by OGD by 51±16 % and PTX by 129±29 %. All of the above findings support our proposal that, like most biological poisons, palytoxin mimics (and takes advantage of) a natural process. In this case the process is ischemia itself where low ATP conditions might open the Na+/K+ pump to evoke AD and the brain damage that ensues. ii Co-Authorship Danielle Kim was responsible for brain slice preparation, obtaining all light transmittance imaging data and field recordings, and analyses of these data. Robin Britton assisted with the initial static bath PTX experiments and Kaitlyn Tresidder assisted with the collection and analysis of the light transmittance imaging data involving OGD and the brainstem. Dr. David Andrew collected the 2-photon microscopy data from slices prepared by DK. The first draft of this thesis was written by Danielle Kim, with all subsequent drafts written in collaboration with DA. iii Acknowledgements I would like to first and foremost thank Dr. David Andrew, whose unending support, advice and contributions were invaluable in the production of this thesis. Without your expertise and your guidance, this thesis would not have been possible. I would also like to thank Robin Brisson and Kaitlyn Tressider for all their help. iv Table of Contents Abstract ......................................................................................................................................................... ii Co-Authorship.............................................................................................................................................. iii Acknowledgements ...................................................................................................................................... iv List of Figures ............................................................................................................................................. vii List of Tables ............................................................................................................................................. viii List of Abbreviations ................................................................................................................................... ix Chapter 1 Introduction .................................................................................................................................. 1 1.1 Brain Ischemia and Stroke .................................................................................................................. 1 1.2 Spreading Depolarizations .................................................................................................................. 2 1.2.1 Spreading Depression .................................................................................................................. 3 1.2.2 Anoxic (Ischemic) Depolarization ............................................................................................... 3 1.3 Anoxic Depolarization in the Brainstem ............................................................................................. 5 1.4 The Search for the Mechanism Generating Spreading Depolarization ............................................... 5 1.5 Drugs that Inhibit AD Onset ............................................................................................................... 7 1.5.1 Dibucaine ..................................................................................................................................... 8 1.5.2 Carbetapentane (CP) .................................................................................................................... 9 1.5.3 Comparing Dibucaine and CP.................................................................................................... 10 1.6 The Na+/K+ Pump ............................................................................................................................. 11 1.6.1 Structure and Function of Na+/K+ ATPase ................................................................................. 11 1.6.2 The Na+/K+ Pump and Ouabain ................................................................................................. 12 1.6.3 Palytoxin (PTX) ......................................................................................................................... 13 1.7 Background of Experimental Techniques Utilized ........................................................................... 14 1.7.1 Brain Slice Technique ................................................................................................................ 14 1.7.2 Light Transmittance Imaging ..................................................................................................... 15 1.7.3 Recording the Negative Shift ..................................................................................................... 15 1.7.4 Fluorescence Microscopy and 2PLSM ...................................................................................... 16 1.8 Objectives ......................................................................................................................................... 18 Figures .................................................................................................................................................... 20 Chapter 2 Methods ...................................................................................................................................... 22 2.1 Live Brain Slice Preparation ............................................................................................................. 22 2.2 Experimental Solutions ..................................................................................................................... 23 2.3 Imaging Changes in Light Transmittance (∆LT) .............................................................................. 23 v 2.4 Recording the Negative Shift ............................................................................................................ 24 2.5 Two-Photon Laser Scanning Microscopy (2PLSM) ......................................................................... 25 2.6 Pretreatment of Slices with Dibucaine and Carbetapentane ............................................................. 25 2.7 Statistical Analysis ............................................................................................................................ 26 Figures .................................................................................................................................................... 27 Chapter 3 Results ........................................................................................................................................ 29 3.1 Light Transmittance (LT) Imaging of Anoxic and Palytoxin-induced Depolarization..................... 29 3.2 LT Imaging of OGD- and Palytoxin-Induced Depolarization in the Brainstem ............................... 31 3.3 Extracellular Recording of Anoxic- and Palytoxin-Induced Depolarization .................................... 32 3.4 Two-Photon Laser Scanning Microscopy (2PLSM) ......................................................................... 32 3.5 LT Imaging of AD and PD in Slices Pretreated with Dibucaine and Carbetapentane ...................... 33 Tables ...................................................................................................................................................... 34 Figures .................................................................................................................................................... 39 Chapter 4 Discussion .................................................................................................................................. 53 4.1 Anoxic Depolarization in Cortical Slices.......................................................................................... 53 4.2 Palytoxin Causes Spreading Depolarization in the Neocortex ......................................................... 55 4.3 The Negative Shift Confirms a Population Depolarization .............................................................. 57 4.4 2PLSM Confirms Similar Neuronal Injury Induced by OGD or Palytoxin ...................................... 57 4.5 Brainstem LT Imaging Reveals PTX Affects the Same Structures as OGD .................................... 58 4.6 2 Photon Laser Scanning Microscopy of Mesencephalic Neurons After Palytoxin Exposure ......... 60 4.7 Palytoxin Delay Using Dibucaine and Carbetapentane .................................................................... 62 4.8 Potential Significance of Our Findings ............................................................................................. 63 4.9 Future Directions .............................................................................................................................. 63 Figures .................................................................................................................................................... 66 References ................................................................................................................................................... 68 vi List of Figures Figure 1. Alternative-access model of the Na+/K+ pump. ........................................................................... 20 Figure 2. The subunits of the Na+/K+ pump. ............................................................................................... 21 Figure 3. A schematic of the equipment used to image changes in light transmittance. ............................ 27 Figure 4. Light transmittance (LT) imaging reveals damage in higher gray matter following AD induced by 10 minutes of oxygen-glucose deprivation (OGD). ............................................................................... 39 Figure 5. Palytoxin at 10 nM induces an OGD-like sequence of LT change. ............................................ 40 Figure 6. Palytoxin at 1 nM superfusion induces an OGD-like sequence of LT change. ........................... 41 Figure 7. Distribution of palytoxin-induced depolarization (PD) latency measured by LT imaging. ........ 42 Figure 8. Maximal and minimal LT changes in the neocortex induced by OGD or by PTX. .................... 43 Figure 9. Negative shift detected when elevated LT front passes recording electrode. .............................. 44 Figure 10. Amplitudes of the negative-shift in extracellular voltage during depolarization induced by OGD or by palytoxin. ................................................................................................................................. 45 Figure 11. Two-photon laser scanning microscopy (2PLSM) images of pyramidal neurons before and after OGD-aCSF. ........................................................................................................................................ 46 Figure 12. 2PLSM images of pyramidal neurons before and after 10 nM palytoxin aCSF exposure. ....... 47 Figure 13. Distribution of pyramidal neuron soma swelling induced by OGD or PTX. ............................ 48 Figure 14. 2PLSM images of midbrain mesencephalic neurons before and after exposure to 10 nM PTX. .................................................................................................................................................................... 49 Figure 15. 2PLSM images of midbrain mesencephalic neurons before and after 10 nM PTX. ................. 50 Figure 16. Distribution of swelling by mesencephalic neuronal cell bodies induced by OGD or by 10 nM PTX. ............................................................................................................................................................ 51 Figure 17. A comparison of the spreading depolarization latency in slices pretreated with dibucaine or carbetapentane to untreated slices. .............................................................................................................. 52 Figure 18. PTX transforms the Na+/K+ pump into a channel, one ATPase at a time. ................................. 66 Figure 19. Proportions of the different Na+/K+ pump isoforms in the cortex and brainstem. ..................... 67 vii List of Tables Table 1. Latency of spreading depolarization induced in slices using a static bath and measured by LT imaging. ...................................................................................................................................................... 34 Table 2. Regional frequency of spreading depolarization induced in slices using a static bath. ................ 35 Table 3. Regional generation of OGD- or palytoxin-induced spreading depolarization in higher brain gray matter using LT imaging of superfused slices. ........................................................................................... 36 Table 4. Regional generation of 10 nM palytoxin-induced spreading depolarization in brainstem gray matter. ......................................................................................................................................................... 37 Table 5. Spreading depolarization latency in slices pretreated with dibucaine or carbetapentane. ............ 38 viii List of Abbreviations aCSF Artificial cerebrospinal fluid AD Anoxic depolarization AP Action potential ATP Adenosine triphosphate CA1 Hippocampus CA1 CBF Cerebral blood flow CBV Cerebral blood volume CCD Charge-coupled device CNS Central nervous system CP Carbetapentane DIB Dibucaine EEG Electroencephalography GFP Green fluorescent protein Hipp Hippocampus LT Light transmittance Mes Mesencephalic trigeminal nucleus mPTP Mitochondrial permeability transition pore Na+/K+ ATPase Sodium-potassium activated adenosine triphosphatase OGD Oxygen-glucose deprivation PAG Periaqueductal gray PD Palytoxin-induced depolarization PID Peri-infarct depolarization PNS Peripheral nervous system Pretx Pretreatment PTX Palytoxin sAHP Slow afterhyperpolarization SD Spreading depression Str Striatum TBI Traumatic brain injury XS Cross-sectional ix YFP Yellow Fluorescent Protein 2PLSM Two-photon laser scanning microscopy σR σ receptor x Chapter 1 Introduction 1.1 Brain Ischemia and Stroke Ischemia refers to a decrease in blood flow to tissue which results in altered levels of cellular function (Woodruff et al., 2011). Global ischemia is a serious reduction in blood flow to the entire brain, most often caused by sudden cardiac arrest. Stroke is the diagnostic term for the brain dysfunction caused by ischemic neuronal injury and death (Bretón and Rodríguez, 2012). Stroke is the world’s second leading cause of mortality, with an estimated lifetime risk of 8-10% (Seshadri et al., 2006). It has roughly a 30% mortality rate (Dirnagl et al., 1999) resulting in 10% of deaths worldwide (Murray and Lopez, 1997). Stroke encompasses a heterogeneous group of conditions including large artery ischemia (from vessel occlusion), intracerebral and subarachnoid hemorrhage, cerebral venous sinus thrombosis and global brain ischemia from cardiac arrest (Bretón and Rodríguez, 2012; Moustafa and Baron, 2008; Brown et al., 2006). Ischemic stroke accounts for 85% of all strokes (Woodruff et al., 2011), over half of which are attributed to large-artery ischemia (Moustafa and Baron, 2008). The remaining 15% are hemorrhagic (Bretón and Rodríguez, 2012). Focal ischemic stroke results in a gradient of hypo-perfusion surrounding the areas supplied by the dysfunctional vessel(s). The ischemic “core” describes the region with severely low cerebral blood flow (CBF) (20% of normal levels or lower) (Hossmann, 1994) and low cerebral blood volume (CBV) with resulting low rates of oxygen and glucose turnover (Marchal et al., 1999). In the ischemic core, affected tissue undergoes irreversible damage in the absence of prompt treatment. Without adequate reperfusion, acute neuronal injury follows within minutes (Singhal et al., 2011). 1 Surrounding the ischemic core is the penumbra, a rim of moderately ischemic tissue with 20-40% CBF (Ginsberg, 1997) supported by collateral circulation. In this region cell damage progresses slowly relative to the ischemic core and cell death may take hours to days following stroke onset, depending on the severity and duration of ischemia (Dirnagl et al., 1999; Jones et al., 1981; Woodruff et al., 2011). The penumbra is the prime target of stroke therapeutics because it is potentially salvageable after the onset of stroke (Dirnagl et al., 1999). This may be achieved following timely reperfusion within 6-8 hours (Kaufmann et al., 1999). The brain injury and neurological deficit associated with stroke are the result of a complex sequence of pathophysiological events upon the onset of ischemia, called the ischemic cascade. Within this cascade, two of the major initial pathogenic events were thought to be excitotoxicity and peri-infarct depolarizations (PID) (Dirnagl et al., 1999). Excitotoxicity, defined as the cell injury and death arising from prolonged intense exposure to accumulating extracellular glutamate and its associated cellular ionic imbalance (Bretón and Rodríguez, 2012), was thought to be responsible for the initiation of ischemic damage (Jarvis et al., 2001). However, studies have found that acute neocortical damage occurs independent of glutamate (Obrenovitch, 2000; Obrenovitch and Urenjak, 1997; Jarvis et al., 2001; Obeidat et al., 2000; Murphy et al., 2008). PIDs are recurrent spreading depolarizations in the penumbra that expand the core (Hartings et al., 2003; Singhal et al., 2011). Inhibition of PIDs could be an effective and valuable neuroprotective strategy, given the window of opportunity of up to 48 hours to suppress them post-stroke (Hartings et al., 2003). 1.2 Spreading Depolarizations Spreading depolarization is a general term that includes spreading depression (SD), PIDs and anoxic depolarization (AD). It is observed in vivo in gray matter under metabolic stress, including various types of stroke, global ischemia (Brisson et al., 2014) and traumatic brain 2 injury (Dreier, 2011). It can also be evoked by hypo-osmolality, hyperthermia, sodium pump inhibiters, hypoglycemia, hypoxia or ischemia (Hossmann, 1994; Leao, 1947; Somjen, 2004). 1.2.1 Spreading Depression Spreading depression (SD) or cortical spreading depression (CSD) was originally described by Leao in 1944 as a marching wave of electrical silence across rabbit neocortex evoked by local application of KCl. The depressed electroencephalography (EEG) activity was observed to recover within 5 minutes (Leao, 1944). It was later shown that SD is a moving wavefront of sustained depolarization of neurons and glial cells, characterized by ion gradient breakdown, severe decrease of membrane resistance and a rapid negative shift in extracellular potential resulting from the sudden cellular depolarization (Marshall, 1959; Somjen et al., 1992) caused by temporary failure of the Na+/K+ pump (Figures 1, 2). In the presence of normal metabolic substrates, the tissue repolarizes in 1-2 minutes without permanent effects on the tissue (Marshall, 1959; Somjen et al., 1992). The sustained depolarization of SD inactivates Na+ channels within seconds (Urenjak and Obrenovitch, 1996), which stops action potential firing thereby silencing brain electrical activity. SD typically propagates at 2-6 mm/min (Lauritzen, 1994) as a concentric wave from its initiation site(s) through grey matter, halting at the border of white matter (Somjen et al., 1992). Experimentally SD can be provoked by focal mechanical, electrical or elevated [K+]o stimuli. SD is generally accepted as the pathophysiological process accounting for the marching sensory symptoms of migraine aura prior to pain onset in patients (Hadjikhani et al., 2001). Thus stopping SD is of therapeutic interest in that the pain may arise from the SD. 1.2.2 Anoxic (Ischemic) Depolarization Anoxic depolarization (AD) is evoked by severe hypoxia or ischemia in the CNS gray matter, initiating within a minute or so of blood flow loss (Dreier, 2011). It was first described by Leao (1947) as an ischemic version of SD. Characterized as a moving wavefront of electrical 3 depolarization, loss of membrane ion resistance, electrical silence and brain swelling, AD resembles SD in many ways because it also results from Na+/K+ pump failure (Figures 1, 2). Without prompt reversal to normoxic conditions, the depolarized neurons poorly recover and can be permanently damaged. AD and SD possess similar mechanisms of initiation, both caused by a local ionic imbalance across the membrane coupled with a metabolic overload (Somjen et al., 1992; Dreier, 2011). The deleterious effects of AD vs. the lack thereof following SD are determined by the levels of glucose and oxygen that enable the cell to repolarize. One aspect where AD and SD differ is the role of glutamate in SD and AD generation. Glutamate receptor antagonists are effective in inhibiting SD but not AD (Pietrobon and Moskowitz, 2014; Anderson et al., 2005). This difference is likely attributable to synaptic failure that occurs prior to AD onset induced by oxygen-glucose deprivation. Likely due to synaptic failure preceding AD, glutamate accumulation does not occur preceding AD and rather has been found to accumulate after AD (Obrenovitch and Urenjak, 1997; Obrenovitch, 1999; Martin et al., 1994). During SD and AD, the neuronal membrane potential undergoes a “slow depolarization” before a “fast depolarization” within 0.5 to 4 seconds, with K+, Na+ and Ca2+ ions rapidly flowing down their concentration gradients (Tanaka et al., 1997; Somjen et al., 1992). Within several minutes of AD, pyramidal cell dendrites lose their spines and form distinct dilations or “beads” (Davies et al., 2007; Jarvis et al., 2001). This dendritic beading is minimal following SD and is a key indicator of permanent damage in live brain slices (Jarvis et al., 2001). Although AD is difficult to treat because the onset of ischemia cannot be predicted ahead of time, understanding AD is still important as it may provide insight on how to inhibit peri-infarct depolarizations (PIDs). As noted earlier, PID is an AD-like event in the penumbra, but unlike AD, neurons may recover because there is a supply of glucose and oxygen from collateral blood vessels. PIDs often recur over many hours which expand the ischemic core (Hartings et al., 2003; Dreier, 2011). 4 1.3 Anoxic Depolarization in the Brainstem The brainstem’s response to ischemia is more resilient than the “higher”, more rostral structures of neocortex, thalamus, striatum and hippocampus. The most obvious clinical evidence comes from the thousands of patients whose brains are globally deprived of blood flow and enter a persistent vegetative state where only their hypothalamus and brainstem are adequately functioning. The hypothalamus and brainstem demonstrate a striking degree of resilience to ischemia while other higher brain structures show signs of injury in several brain imaging studies of patients following global ischemia (Luigetti et al., 2012; Falini et al., 1998; Wytrzes et al., 1989). Also in animal studies there is a general pattern of increased resistance to ischemic stress as one descends along the neuraxis (Bures and Buresova, 1981; Branston et al., 1984). In live brain slice studies, neurons in “lower” gray matter of the hypothalamus and brainstem demonstrate only a weak AD. That is, there is a slow depolarization in response to 10 minutes of OGD, with repolarization upon return of normoxic aCSF (Brisson et al., 2014; Brisson et al., 2013; Brisson and Andrew, 2012). For example, neurons in the locus ceruleus and mesencephalic neurons of the brainstem recover between 80-100% of their membrane potential, input resistance and action potential amplitude (Brisson et al., 2014). They proposed that this striking difference in response to ischemia may be tied to regionally different Na+/K+ pump isoforms. The more ischemia-resilient 1α2 and 1α3 isoforms are found in higher proportion in the hypothalamus and brainstem, while the 1α1 isoform is proportionally more abundant in rostral structures (Dobretsov and Stimers, 2005; Blanco, 2005). 1.4 The Search for the Mechanism Generating Spreading Depolarization In an effort to understand how AD arises, ion channels, altered ion concentrations and neurotransmitters have been studied. No individual neurotransmitter agonist or antagonist (including those involving glutamate receptors) blocks or delays AD onset in most brain regions (Anderson et al., 2005; Murphy et al., 2008) with the exception of delaying AD onset in the 5 cerebellum (Brisson et al., 2014). In fact synapses fail before AD onset and glutamate receptor inactivation does not delay AD (Anderson et al., 2005). Although a popular theory for over 20 years, excessive glutamate release (excitotoxicity) as the cause of ischemic damage has not been supported in either rodent brain slices or in vivo (Anderson et al., 2005; Jarvis et al., 2001; Obeidat et al., 2000; Lipton 1999; Murphy et al., 2008; Muller and Somjen, 2000). Studies examining K+, Na+ and Ca2+ ion concentrations (Tanaka et al., 1997; Rossi et al., 2000; Jarvis et al., 2001; Obeidat et al., 2000; Obrenovitch et al., 2000) and various types of Na+, K+ and Ca+ channel blockers (Xie et al., 1994; Xie et al., 1995; Yamamoto et al., 1997) collectively revealed that no one channel type drives AD in brain slices (Anderson et al., 2005). Studies have also found evidence that blockers of Na+, K+ and Ca2+ channels individually only delay AD (Xie et al., 1995; Somjen et al., 1992), suggesting that none of these channels are essential for the movement of these ions down their concentration gradients during depolarization. More recent research has therefore shifted to search for a non-specific channel that might open to conduct more than one ion under ischemic conditions. For instance, the mitochondrial permeability transition pore (mPTP) is a megachannel that opens in stressful conditions to mediate apoptosis and is activated during AD (Liu and Murphy, 2009). Pannexin is a hemichannel gap junction present in neurons that is normally closed, but opens during AD (Thompson et al., 2006). Pannexin in its open state allows ions and small molecules <2000 daltons to passively diffuse through it. Although it does not appear responsible for AD initiation (Madry et al., 2010), the concept makes the possibility of another channel opening to generate AD in response to ischemia more plausible. Two studies have reported that a cocktail of channel blockers and transmitter antagonists block AD in immature rodent hippocampal slices (Rossi et al., 2000; Madry et al., 2010). However, such treatment renders neurons functionally unresponsive so a comparable clinical application would be lethal (Anderson et al., 2005). Another study used a cocktail of Na+ and Ca2+ current inhibitors containing DNQX (an AMPA and kainate receptor antagonist), CPP (an 6 NMDA antagonist), TTX (Na+ channel blocker) and Ni2+ (a Ca2+ channel inhibitor) to prevent hypoxia-induced SD in rat hippocampal slices (Muller and Somjen, 1998). Simply decreasing oxygen to simulate depolarization does not adequately simulate an ischemic environment. Yet even in the metabolically less stressful state of hypoxia, any three of the four drugs combination was unable to block hypoxia-induced depolarization (Muller and Somjen, 2000; Jing et al., 1994). The conclusion is that AD is extremely difficult to stop by simply blocking channels that drive normal neuronal function. 1.5 Drugs that Inhibit AD Onset The time from ischemic onset to AD generation (AD latency) can be used as an indicator of tissue resistance to acute ischemia (Bures and Buresova, 1957). Some drugs found to be beneficial in the treatment of cerebral ischemia in rodents are able to delay AD onset (Balestrino and Somjen, 1986; Deshpande and Wieloch, 1986; Holler et al., 1986; Xie et al., 1995) but none have yet proven to be clinically neuroprotective after stroke. Two types of drugs that effectively delay AD are the “caine” family and certain σ-receptor ligands. Na+ channel blockers are not able to block AD generation outright but some do delay AD onset (Weber and Taylor, 1994; Taylor and Meldrum, 1995; Xie et al., 1995) and the length of delay is correlated with the Na+ channel binding strength (Douglas et al., 2011). The caine family of Na+ channel blockers in particular significantly delay AD onset (White et al., 2012; Douglas et al., 2011; Weber and Taylor, 1994). Like the caine family of drugs, certain σ receptor ligands delay AD onset in rodent cortical slices (Anderson et al., 2005). Although initially thought to be a type of opioid receptor, σ-receptors have non-opiate and non-phencyclidine (PCP) binding sites that are distinct from other neurotransmitter, neuropeptide and steroid receptors (Quirion et al., 1992; Leonard, 2004). There are two subclasses of receptors, σ 1 and σ 2, which are distinct in their drug selectivity 7 (Guitart et al., 2004). σ1 receptors are found in the plasma membrane and subcellular membranes such as the endoplasmic reticulum and translocate intracellularly to act as “receptor chaperones” that modulate receptors (Cobos et al., 2008; Ishikawa and Hashimoto, 2010). Although the specific biological function of sigma receptors and the identification of their endogenous ligands is still unclear, the most prominent action of σ1 receptors is the regulation of various voltagegated and ligand-gated ion channels including Ca2+, K+, Na+, Cl- and NMDA receptors (Maurice and Su, 2009) through protein-protein interactions not always involving G proteins (Maurice and Su, 2009; Fontanilla et al., 2009). Of the ion channels affected by σ 1 receptors, σ 1R agonists inhibit the voltage-gated channels and potentiate the ligand-gated channels (Maurice and Su, 2009). Present throughout the brain, σ receptors exhibit a wide range of actions within the CNS (Kume et al., 2002) and are implicated in a variety of neurological functions and disorders, including learning and memory (Senda et al., 1996), drug addiction (Matsumoto et al., 2002), schizophrenia, movement disorders (Guitart et al., 2004), Alzheimer`s and stroke (Maurice and Su, 2009). However, the actual biological function of the receptor is still unclear. 1.5.1 Dibucaine Dibucaine is a local anesthetic and Na+ channel blocker of the caine family. It is one of the most potent in its ability to block Na+ channels and delay AD onset induced by simulated ischemia in rodent brain slices (Douglas et al., 2011; Yamada et al., 2004) and human cortical slices (Risher et al., 2011). Bath-applied dibucaine acts like the other caines (Creveling et al., 1983) creating a use-dependent block by entering neurons through briefly opened Na+ channels and then binding to the channel on the intracellular side (White et al., 2012). But if AD initiates, dibucaine has no effect on AD propagation or the damage following AD (Douglas et al., 2011). Both ouabain- and OGD-induced AD are blocked or delayed by dibucaine in rodent brain slices. Our laboratory has proposed that dibucaine protects neurons in both cases by inhibiting AD-associated currents activated by Na+/K+ pump failure (Anderson et al., 2005; White et al., 8 2012). Although to date the specific channel(s) involved in AD remain unidentified, a component of the depolarization during AD involves Na+ channel opening. Correspondingly, one of the better understood effects of dibucaine is its ability to block voltage-gated Na+ channels (Kuroda et al., 2000; Ragsdale et al., 1994), although the specific Na+ channel sub-types inhibited by dibucaine have not yet been identified. Intracellular recordings of CA1 hippocampal neurons after 30 minutes of 10 µM dibucaine pretreatment show that both orthodromic and antidromic action potentials are blocked and the evoked EPSP amplitude is significantly decreased (White et al., 2012). Dibucaine likely inhibits AD onset at least partly through the same mechanism that suppresses AP firing, by blocking both sustained and slow Na+ currents (INaS) (White et al., 2012). Furthermore, of the caine family drugs that have been studied, the order of potency for blocking Na+ channels was similar to the order of potency of AD delay (Douglas et al., 2011; Yamada et al., 2004). Therefore, the neuroprotective effects of dibucaine through AD delay likely involves an interaction with Na+ channels. Lidocaine is another member of the caine family that effectively delays AD at a low concentration of 10 µM (Yamada et al., 2004). Lidocaine antagonizes the persistent sodium current, but based on the IC50 values for presynaptic volley inhibition, this concentration should not adequately suppress Na+ channels to provide protection against AD (Stafstrom et al., 1985; Yamada et al., 2004). Yamada et al. (2004) proposed that the caines may act neuroprotectively through another unknown mechanism. A study that looked at five caine family drugs that were reasonably potent AD inhibitors (including dibucaine) found that lidocaine had the highest order of affinity for σ1R. Therefore, it is possible that lidocaine acts on both Na+ channels and σ1Rs to delay AD. 1.5.2 Carbetapentane (CP) Of the few σR ligands that affect AD onset, the antitussive carbetapentane (CP) is the most effective. It is a σ1R ligand that delays AD onset in both cortical and hippocampal CA1 9 neurons (Anderson et al., 2005; White et al., 2012) but its mechanism of action during ischemia is still unknown (Katnik et al., 2006). The σ1R is found throughout the CNS, particularly on neuronal cell bodies and dendrites, but has not been detected on axons and axon terminals (Alonso et al., 2000). Based on these findings, CP and other σR ligands likely affect the somadendritic region to cause AP delay. Previous studies examining neurons exposed to 100 µM of σR ligands dextromethorphan (Wong et al., 1988) or OPC-24439 (Ishihara et al., 1999) found no change in CA1 membrane potential, cell input resistance or AP threshold during 50 min of drug exposure. Similarly CP decreases CA1 neuron excitability but rather than increase the AP threshold, it increases the slow afterhyperpolarization (sAHP) following an AP train, which likely decreases the firing rate (White et al., 2012). Carbetapentane showed no effect on antidromically stimulated APs, which corresponds with the lack σ1 receptors at the level of the axon (White et al., 2012). In addition, the evoked responsiveness of CA1 and neocortical layers were not altered by 30 minute exposure to 10-30 µM CP (Anderson et al., 2005), although these concentrations were effective in blocking or delaying AD. The neuron’s ability to function while AD is blocked may mean that the currents driving AD act beyond the standard channels and neurotransmitter receptors required for normal neuronal physiological functioning. Rather, an abnormal channel conductance could drive AD. How σ receptors delay AD has not yet been elucidated. It is likely through a σR ligandrelated action, because several σR agonists block or delay ouabain-induced AD. σR ligands have been found to inhibit voltage activated K+ currents (Wilke et al., 1999; Zhang and Cuevas, 2005) and attenuate intracellular Ca+ increases during ischemia (Hayashi et al., 2000; Katnik et al., 2006), which may contribute to AD inhibition. 1.5.3 Comparing Dibucaine and CP 10 Current literature point to different mechanisms of action in AD delay between the caines and σ1R drugs. Although there is a correlation between the strength of Na+ channel binding by caines and AD onset delay, there is no correlation between caine binding strength to σ1Rs and the ability to delay AD onset (Douglas et al., 2011). Furthermore, although both drug classes delay or block AD without affecting the baseline properties of neurons, dibucaine increases AP threshold while CP increases the sAHP (White et al., 2012). A common mechanism behind these two drugs delaying AD could be the inhibition of a persistent Na+ current, which would reduce excitability (Cheng et al., 2008). Similarly, both drugs might affect the neuron through other mechanisms to inhibit AD (Yamada et al., 2004; Taylor and Meldrum, 1995, Douglas et al., 2011). For instance, both drugs have subtle effects on calcium and potassium movements (Hirota et al., 1997; Oda et al., 1992; White et al., 2012), although how these specific interactions affect AD onset in neurons is not well studied. A possible mechanism could be that both drug types up-regulate the Na+/K+ pump, thereby delaying AD (Brisson et al., 2014), although this is speculation. 1.6 The Na+/K+ Pump 1.6.1 Structure and Function of Na+/K+ ATPase The sodium-potassium-activated adenosine triphosphatase (Na+/K+ ATPase or Na+/K+ pump) is a protein enzyme complex that spans the plasma membrane (Figure 2). It consists of a catalytic alpha (α) subunit (1000 residues) and beta (β) (300 residues) subunit (Rose and Valdes, 1994; Toyoshima et al., 2011). The beta subunit acts as a molecular chaperone and stabilizes the alpha subunit conformation as well as modulating pump function (such as cation binding affinity) (Toyoshima et al., 2011). There is also a tissue-specific auxiliary regulatory subunit known as the FXYD protein (70-180 residues), which modulates the pump’s kinetic properties by adjusting the cation affinities based on the specific demands of the tissue (Toyoshima et al., 2011). In each cycle, the enzyme transports sodium and potassium ions across the membrane against their 11 respective concentration gradients using the energy released from the hydrolysis of adenosine triphosphate (Figure 1). The enzyme functions by cycling between open conformations and an “occluded” state where both gates are closed which momentarily traps the ions being transported. The occluded state ensures that a channel-like short circuit where both gates are open (which has less than a 10-5 probability) cannot occur (Gadsby et al., 2009). With one complete conformational cycle required to transport 2 K+ in and 3 Na+ ions out of the cell (Figure 1), the pump is limited to the transporting approximately 100 ions/s (Gadsby et al., 2009). 1.6.2 The Na+/K+ Pump and Ouabain The human brain expends roughly 50% of its available energy maintaining the Na+/K+ ATPase pump (Gottron and Lo, 2009). Within 3 minutes of cardiac arrest, the ATP concentration in brain tissue decreases to 10% of normoxic values (Martin et al., 1994). Without oxygen and glucose, the pump is unable to maintain the voltage and chemical gradients between neurons and extracellular space (Balestrino, 1995; Martin et al., 1994). Ouabain, a cardiac glycoside that binds and inhibits the Na+/K+ ATPase, provides a valuable clue to AD generation. When applied to brain slices, ouabain causes a propagating depolarization with ensuing damage similar to AD as measured by extracellular voltage recordings (Balestrino, 1995) and light transmittance (LT) imaging (Jarvis et al., 2001). The extracellular K+ changes during ouabain-induced depolarization are also similar to AD (Balestrino, 1995). But how AD arises as the pump fails is unknown. So while failure of the Na+/K+ pump leads to the large inward current comprising AD (i.e. rapid Na+ influx and K+ efflux) (Brisson and Andrew, 2012; Martin et al., 1994; Tanaka et al., 1997), it is not known how the two events are linked nor the channel(s) conducting the formidable inward current. We argue below that AD generation must involve the opening of a previously unidentified channel or the formation of pores or membrane perforations. 12 1.6.3 Palytoxin (PTX) Originally isolated in 1961 from palythoa toxica corals found in Hawaii (Moore and Scheuer, 1971), PTX is one of the world’s most potent toxins (Deeds et al., 2011). It has been found in several species of palythoa zoanthids (corals) and up the food chain in certain anemones, fishes, crabs, sea urchins. It may originate further down the food chain in dinoflagellates which are plankton (Riobó and Franco, 2011; Tosteson, 2000). PTX is a large nonpeptide with both lipophilic and hydrophilic areas. An extremely low lethal dose ranges from 0.03 to 0.45 µg/kg in mice, rats, rabbits, dogs, and monkeys (Riobó and Franco, 2011). In early experiments of toxicity, PTX induced neuromuscular symptoms leading to follow-up studies on PTX’s ability to cause membrane depolarization (Rossini and Bigiani, 2011) which was minimally affected by voltage-gated sodium channel blockers (i.e., tetrodotoxin and saxitotoxin). The involvement of other monovalent cations in the depolarization made it unlikely that sodium channels were directly involved in PTX’s action (see Rossini and Bigiani, 2011). Subsequent investigations using erythrocytes showed that ouabain inhibited the effects induced by PTX and vice versa, which suggested competitive inhibition for the same receptor (Habermann and Chhatwal, 1982). These workers proposed that palytoxin converts the Na+/ K+ pump into a pore that allows the passive diffusion of cations (Chhatwal et al., 1983). A series of studies, first using yeast cells which naturally lack Na+/K+ ATPase (Scheiner-Bobis et al., 1994) and later using cell-free conditions (Hirsh and Wu, 1997) provided compelling evidence that PTX specifically and reversibly induces the formation of single channels converted from the Na+/K+ ATPase transporter (Rossini and Bigiani, 2011). PTX acts by binding tightly to each Na+/K+ ATPase transporter, transforming it into a passive, nonselective cation channel (Artigas and Gadsby, 2004; Gadsby et al., 2009), thereby allowing passage of monovalent ions and molecules smaller than 180 Da, in the order of K+ ≥ Na+ > choline >> inositol > sucrose (Chatwal et al., 1983). An open PTX “pump-channel” conducts 13 even larger organic cations such as N-methyl-D-glucamine (NMDA) but 50-fold more slowly than Na+ ions (Artigas and Gadsby, 2004; Gadsby et al., 2009). At a saturating concentration of only 100 nM, PTX transforms thousands of pump molecules into channels in the plasma membrane of a single cell (Gadsby et al., 2009). Despite its powerful effect at low concentration, the pump-channel does not stay permanently open while PTX is bound. Rather, it alternates between open and closed positions throughout the bound period (Gadsby et al., 2009). The duration of time a pump-channel remains open (i.e., the open probability) is affected by ATP and other ion concentrations. For instance, the open probability is five to six-fold higher in the presence of cytoplasmic ATP than without (Gadsby et al., 2009). The effect of palytoxin transforming a single Na+/K+ ATPase molecule into a channel cause a 106-fold gain in dissipative cation flow. Thus, in contrast to the Na+/K+ ATPase which transports about 100 ions/s, an open pump conveys perhaps a million Na+ and K+ ions per second at resting membrane potential. Therefore, it is estimated that even a single open pump channel could overwhelm a single blood cell to the point of death (Gadsby et al., 2009). 1.7 Background of Experimental Techniques Utilized 1.7.1 Brain Slice Technique The use of the in vitro brain slice preparation for electrophysiological recording has grown dramatically since its development (Teyler, 1980) in 1966, when Yamamoto and McIlwain (1966) first recorded electrical activity from cortical slices comparable to that from an intact preparation. By 1980, spontaneous or evoked electrical activity had been demonstrated in slices prepared from human and animal neocortex, olfactory bulb, spinal cord, thalamus and hypothalamus. In vitro live brain slices offer numerous experimental advantages over intact preparation (Teyler, 1980): 1. Brain slices maintain intimate neuron-glia associations (Andrew and Macvicar, 14 1994). 2. Brain slices display visual landmarks of different brain structures and allow expansive areas of the rodent brain to be examined simultaneously. 3. In comparison to in vivo and intact preparation, live slices allow more control and consistency over the chemical and physical environment of the slice. 4. Movements experienced in vivo from heart beat and respiration are reduced and differential blood flow to various areas is absent. 5. One brain can yield numerous slices, allowing consistency in terms of the animal’s genetics and experience. 6. Finally, anaesthetics may not be required in brain tissue preparations. 1.7.2 Light Transmittance Imaging Light transmittance (LT) imaging measures changes in tissue translucence, one component of intrinsic optical signals, in real time. The swelling of neurons and/or glial cells (from excitatory synaptic input, action potential discharge or hypo-osmolality) is detected as a local increase in light transmittance (Andrew and Macvicar, 1994) because swollen cells scatter less light as membranes become more planar and light refraction is reduced. During a depolarization caused by OGD or ouabain (to be discussed below), the neurons and glia that depolarize swell as water follows the influx of ions intracellularly, which is detected as an increase in LT. A decrease in LT may be observed within minutes in dendritic regions (Obeidat et al., 2000) because the dendritic beads that form are the ideal diameter of 2-5 µm to scatter the light (Jarvis et al., 2001; Malm, 1999; Obeidat et al., 2000). As a real-time signal that can detect neuronal activation and damage, LT imaging can also be recorded simultaneously with other techniques such as patch clamping or extracellular recording. Brain slices are particularly useful when used in LT imaging, as multiple regions can be monitored simultaneously (Brisson et al., 2014). 1.7.3 Recording the Negative Shift 15 One of the key characteristics of spreading depolarization is the negative voltage shift recorded in the extracellular space. The shift ranges from 15-35 mV in magnitude in vivo and is generated by the simultaneous depolarization of nearly all cells near the recording electrode (Somjen et al., 1992). As the depolarizing wave front passes the recording electrode, the collective intracellular depolarization of the neurons and astrocytes generates a negative-going shift in the extracellular potential. Although the shift may drift back to baseline, unlike in SD the evoked field potential is permanently lost following AD in slices as the result of damage to the neurons (Jarvis et al., 2001). 1.7.4 Fluorescence Microscopy and 2PLSM Live tissue strongly scatters light at visible wavelengths, making it challenging to image with traditional optical microscopes (Denk and Svoboda, 1997; Svoboda and Yasuda, 2006). In conventional fluorescence microscopy, light shines evenly to excite the entire specimen. The objective collects the fluorescence, which is passed through dichroic and emission filters before it forms an image that can be viewed through an eyepiece or a camera (Nwaneshiudu et al., 2012; White and Errington, 2005). The scattering caused by neural tissue under conventional fluorescence microscopy degrades the resolution and contrast of the image (Denk and Svoboda, 1997) and limits the imaging depth that can be achieved (Denk et al., 1994). Also, because the entire specimen is exposed to the excitation light, the tissue is at risk of photobleaching and phototoxicity (Denk and Svoboda, 1997; Svoboda and Yasuda, 2006). Photobleaching is the photochemical destruction of the fluorophore and primarily occurs when excited electrons in the fluorophore react with dissolved oxygen instead of emitting energy as fluorescence. This produces a highly reactive oxygen species (ROS) responsible for causing damage to the cell and renders the fluorophore irreversibly bleached (Fischer et al., 2011). In confocal scanning microscopy a pinhole aperture is placed in front of the light detector to remove the off-focus light, and thereby increase the resolution and contrast (Denk and 16 Svoboda, 1997). In contrast to conventional microscopy where the entire specimen is exposed to light, in confocal scanning microscopy light is focused on a single point in the sample. The pinhole aperture is scanned across the field in the X-Y plane, allowing horizontal optical sectioning of the specimen (Yuste, 2005; Nwaneshiudu et al., 2012). Unfortunately, confocal microscopy is still subject to several shortcomings, such as its limited imaging depth (<20 µM) (Lichtman et al., 1987) and excitation of the tissue above and below the image plane. Also, the pinhole aperture rejects potentially useful light that has been scattered from the tissue. Compensating for lost light by increasing illumination is limited by the corresponding increase in phototoxicity (Mainen et al., 1999; Denk and Svoboda, 1997; Svoboda and Yasuda, 2006). A serious limitation of both conventional and confocal microscopy is that the tissue must usually be fixed. Conventional microscopy requires that the specimen is thinly cut and fixed with antibleaching agents and/or immunofluorescence labeling (Nwaneshiudu et al., 2012; Bacallao et al., 1995). Two photon laser scanning microscopy (2PLSM) imaging addresses these limitations. Invented twenty-five years ago (Denk et al., 1990), 2PLSM combines advantages of conventional and confocal fluorescence microscopy without the disadvantages single photon excitation. 2PLSM uses a high frequency pulsing (~100 MHz) and scanning laser to bring two long- wavelength photons together in space and time to excite a fluorophore (e.g. yellow fluorescent protein, YFP) that emits a photon in the visible spectrum. The objective collects the emitted (“fluorescent”) photon. The non-linear relationship between the excitation rate and light intensity as well as the longer wavelengths used in 2PLSM have advantages. First, the excitation is localized to the tiny focal volume being observed, with the intensity decreasing quadratically in the volume surrounding the focus, producing three-dimensional, diffraction-limited resolution and sectioning, even deep in tissue (Svoboda and Yasuda, 2006). This localization of the excitatory photons has benefits. First, the tissue outside of the focal slice will not be subject to photodamage (Denk et al., 1990, Denk et al., 1994; Stelzer et al 1994). Second, due to the nature 17 of non-linear excitation, the scattered excitation photons are too dilute to excite off-focus (Svoboda and Yasuda, 2006). Third, the longer wavelengths used in 2PLSM penetrate tissue further (in comparison to the shorter wavelengths used in 1 photon microscopy). Finally, to compensate for the signal lost from scattering, the laser intensity can be increased without phototoxicity occurring outside of the plane being imaged (Denk et al., 1994). These advantages make 2PLSM an attractive method to use with live tissue, in time-lapse studies and during in vivo experiments. Thus, in addition to increased resolution and optical sectioning abilities of 2PLSM relative to traditional fluorescent microscopes, a greater depth in imaging can be achieved (up to 1 mm) in live tissue (Beaurepaire et al., 2001; Theer et al. 2003). In summary, two photon imaging enables high-resolution and high-contrast fluorescence imaging, even deep within tissue. With the ability to visualize dendritic spines, soma swelling and dendritic beading of individual neurons (Svoboda et al., 1997; Svoboda et al.,1998), it can be used to quantify the extent of damage in tissue at a subcellular level as it develops in real time (Douglas et al., 2011). Similar to confocal fluorescence microscopy, it can image optical sections of a specimen (a stack), providing the option of stacking multiple scanned optical planes and thereby generating a three-dimensional reconstruction of the specimen (Denk and Svoboda, 1997). 2PLSM can be useful in various experiments, from observing calcium dynamics in dendritic spines to in vivo functional imaging (Denk and Svoboda, 1997; Murphy et al., 2008). 1.8 Objectives The molecular mechanism linking ischemic failure of the Na+/K+ pump to the onset of a large inward current in neurons is still unknown. Conventional voltage- and ligand-gated channel blockers do not prevent ischemic or “anoxic” depolarization (AD), but the rapid ionic movement characterizing the onset of AD points towards a channel or pore-like structure facilitating this event. Furthermore, some drugs are effective in blocking or delaying AD without affecting baseline excitability, which may mean that the currents driving AD act beyond the standard 18 channels and neurotransmitter receptors generating the normal physiology of neurons. The possibility of a pathological channel or pore forming to drive AD is plausible with the discovery of the mitochondrial permeability transition pore (mPTP) and pannexins that open during AD. Thus, we asked whether AD initiation may involve the formation of a pore or pathological channel that enables ions to rapidly flow down their gradients, and whether the Na+/K+ ATPase is responsible. If the poison palytoxin causes rapid depolarization in cells by acting on the Na+/K+ pump, then exposing brain slices to palytoxin should recapitulate the effects of OGD on slices. We also investigate if palytoxin`s effect is delayed by the same drugs that delay OGD-induced AD as indirect support for the idea that AD during ischemia cold be generated by the conversion of the Na+/K+ ATPase into a non-selective cationic channel. In the present study, we set out to answer the following questions: I. Does PTX induce a spreading depolarization similar to that imaged during AD? II. Does PTX induce a negative shift similar to that recorded during AD? III. Using 2PLSM, does PTX induce pyramidal neuron injury similar to that imaged postAD? IV. Is the onset of the PTX- induced depolarization delayed by drugs that delay AD onset? V. Is PTX-induced depolarization weaker in lower brain gray matter vs. higher brain gray matter, as we have reported with AD (Brisson et al., 2014)? 19 Figures Phosphorylation opens outer gate P ATP -> ADP + P K+ binding closes outer gate Na+ binding closes inner gate ATP binding opens inner gate Figure 1. Alternative-access model of the Na+/K+ pump. In each cycle, using the energy released from the hydrolysis of adenosine triphosphate, the enzyme transports sodium and potassium ions across the membrane against their respective concentration gradients. Access to the binding sites is controlled by two “gates” illustrated by the small horizontal dark bars, one for the extracellular side and one for the cytoplasmic side. Conformations with the two gates closed correspond to states in which Na+ or K+ ions are “occluded.” We propose that subtle changes in this cycle caused by lack of ATP or of inorganic phosphate (both the result of ischemia) could lead to one or both gates undergoing delayed closing or early opening. With both gates momentarily open, the pump would essentially act as a channel. Modified from Horisberger (2004). 20 Figure 2. The subunits of the Na+/K+ pump. A) Side-view of the pump embedded in plasma membrane. B) Face-on view of the pump showing potential channel (*) (figure modified from Dobretsov and Stimers, 2005). 21 Chapter 2 Methods 2.1 Live Brain Slice Preparation All procedures were carried out under protocols submitted by Dr. David Andrew and approved by the Queen’s University Animal Care Committee. Male Sprague-Dawley (age 3-10 weeks; Charles River, St. Constant, PQ) or C57B mice of either sex (Charles River) were housed in a controlled environment (25°C, 12 h light/dark cycle) and fed Purina lab chow and water ad libitum. Each animal was placed in a plastic rodent restrainer (DecapiCone; Braintree Scientific, Braintree, MA) and decapitated using a guillotine. The brain was excised and immersed in icecold high sucrose artificial cerebrospinal fluid (aCSF) gassed with 95% O2/5% CO2. The composition is described below. Coronal slices (400 µM) of neocortex with underlying striatum or hippocampus, or of brainstem were cut using a Leica 1000-T vibratome. The slices were incubated in regular aCSF (equimolar NaCl replacing sucrose) at room temperature for 1-6 hours before being transferred to the recording chamber where they were submerged in flowing oxygenated aCSF (3 ml/min) at 32-34°C. For 2PLSM experiments, slices (350 µm thick) were prepared as above from >30 day old C57 black mice of the BG.Cg-Tg (Thy1-YFP) 16Jrs/J strain. A portion of this mouse strain’s projection neurons express yellow fluorescent protein (YFP) driven by the thy1 gene promoter (Feng et al., 2000), which can be visualized using the 2PLSM. There is also a proportion of strongly fluorescent neurons in the mesencephalic nucleus of the midbrain (Brisson et al., 2014). The slices were prepared in the same manner as the rat slices but using mouse aCSF. 22 2.2 Experimental Solutions High sucrose aCSF for dissection and slicing was composed of (in mM) 240 sucrose, 3.3 KCl, 26 NaHCO3, 1.3 MgSO4.7H2O, 1.23 NaH2PO4, 11 D-glucose and 1.8 CaCl2. Regular rat aCSF was composed of 120 NaCl, 3.3 KCl, 26 NaHCO3, 1.3 MgSO4.7H2O, 1.23 NaH2PO4, 11 Dglucose and 1.8 CaCl2. Mouse aCSF was rat aCSF with the addition of 20 mM mannitol. Oxygen/glucose deprivation (OGD) aCSF which simulates ischemia in vitro was made by decreasing aCSF glucose from 11 to 1 mM and gassing the aCSF with 95% N2/5% CO2. Palytoxin (Wako Chemicals USA, Richmond, VA), dibucaine (Sigma-Aldrich, St. Louis, MO) and carbetapentane (Sigma-Aldrich) were added to aCSF as required. All solutions were made using distilled water supplied by Botterel Hall, Queen’s University. 2.3 Imaging Changes in Light Transmittance (∆LT) Imaging was used to detect the changes in light transmittance caused by alterations in scattering or absorbance within live tissue in real time (Figure 3). A cerebral or brainstem slice was placed in an imaging chamber with a glass coverslip base and held down with small pieces of silver wire at the slice edges. The slice was superfused with aCSF (3 ml/min) before recording and was maintained at 32-34°C. The solution superfusing the slice was switched to OGD or palytoxin (PTX) aCSF at the start of each recording session. The slice was illuminated using light from a broadband halogen light source (Carl Zeiss SNT 12V 100W) on an upright microscope (Axioscope 2FS plus, ZEISS; Examiner D1, ZEISS). The images were visualized using a 2.5 X objective and the video frames were captured with a 12-bit digital camera (Hamamatsu C474252) at 30 Hz using Imaging Workbench 6 software (INDEC Biosystems, Sanata Clara, CA). The transmittance value (T) of the first image (Tcont) was subtracted from each subsequent image (Texpt) of the series, so the difference image (Texpt - Tcont) revealed areas where LT changed over time. To account for intrinsic regional differences in Tcont (e.g. white vs grey matter), the difference signal was normalized by dividing by Tcont and converted to a percentage to represent 23 the digital intensity of the control image. Thus the change in light transmittance or ΔLT = [(Texp - Tcont)/ Tcont ] x 100 = [ΔT/T] %. The ΔLT was depicted using a pseudocolour intensity scale. Changes in LT through the tissue was measured from baseline, with increased LT represented by blue-green-yellow-red pseudocolouring and decreased transmittance indicated by magenta pseudocolouring. Latency to depolarization was determined as the lapsed time between the aCSF change and the first visually detected focal increase in LT relative to the LT in surrounding tissue that proceeds to propagate into adjacent gray matter. Increased LT primarily indicates tissue swelling and decreased LT indicates primarily dendritic beading (which scatters light reaching the detector) (Davies et al., 2007). Thus as a depolarization propagates through gray matter it is visualized as a propagating increase in LT which may be followed by a decrease in LT over the ensuing 10-15 minutes. 2.4 Recording the Negative Shift The negative DC shift is the electrical signature of the sudden depolarization of cells near the recording electrode. As the cells go positive intracellularly, the extracellular space goes negative with respect to a distant ground electrode. The glass micropipette recording electrode was pulled from thin-walled capillary glass and was filled with aCSF before it was mounted on a 3-D micromanipulator. As described for LT imaging, each slice was placed in the recording chamber and superfused with aCSF at 32-34°C. The ground electrode was placed in the bath and the recording micropipette was placed 50-100 µM deep in layer II/III of the cortex or in layer CA1 of the hippocampus. Following exposure to OGD or PTX aCSF, a negative DC shift in the extracellular potential was usually recorded as the front of elevated LT passed by the recording micropipette. The digitized data were acquired and plotted using Axon Imaging Workbench software (Axon Instruments). 24 2.5 Two-Photon Laser Scanning Microscopy (2PLSM) Transgenic mice expressing YFP in neurons were imaged in 2PLSM experiments. A slice was placed in the imaging chamber, held down with netting and superfused with flowing aCSF at 32-34°C. The imaging chamber was mounted on a fixed stage of an upright Axioscope II FS microscope (Carl Zeiss, Jena, Germany). YFP+ neurons were imaged using a Zeiss x40 or x63 water-immersion objective with appropriate filter sets and a Zeiss LSM 710 NLO meta multiphoton system coupled to a Coherent Ti:sapphire laser (Brisson and Andrew, 2012). Threedimensional image stacks were taken usually at 3µm increments, allowing optical sectioning of each neuronal cell body at several depths. The section with the largest area was determined visually and the cell body periphery outlined using Zeiss LSM software which then calculated the cross-sectional area. 2.6 Pretreatment of Slices with Dibucaine and Carbetapentane Slices were placed on netting in a beaker bubbled with 95% O2/5% CO2 at room temperature. Dibucaine (DIB) experiments consisted of pretreating slices with 10 µM or 1 µM DIB for 35-45 minutes. Carbetapentane (CP) experiments consisted of pretreating slices with 30 µM CP for 40±5 minutes. Slices were pretreated at concentrations and pretreatment times based on previous studies (White et al., 2012; Douglas et al., 2011; Anderson et al., 2005). Depolarization latency of the slices pretreated with DIB or CP under OGD or PTX aCSF was captured using LT imaging (as outlined above). The onset time of anoxic depolarization (AD) or palytoxin-induced depolarization (PD) in each drug-treated slice was calculated as a percentage of the onset time of the subsequent untreated (control) slice in order to accommodate for the hourly variation in AD onset time. In general, the latency to AD and PD onset increased throughout each day which is typically observed in our laboratory. 25 2.7 Statistical Analysis Recordings were terminated if slices appeared visibly damaged from dissection or if slices from the same animal consistently did not display AD under OGD exposure within a window of AD onset times ranging from 4 to 8 minutes. To analyze statistical significance, unpaired t-tests and one-way repeated-measures ANOVA followed by Tukey’s post hoc method were employed using SPSS Statistics. Data were deemed statistically significant at p<0.05. All data are presented as mean ± standard error. 26 Figures Figure 3. A schematic of the equipment used to image changes in light transmittance. A broadband halogen light source is shone through the brain slice. When the light reaches the tissue it is scattered, absorbed or transmitted. The transmitted light is collected and digitized by a 27 charge-coupled device (CCD). Each digitized image is processed with a frame grabber board controlled by imaging software. Image modified from Anderson and Andrew (2002). Note that an infrared filter was not used in the study. 28 Chapter 3 Results 3.1 Light Transmittance (LT) Imaging of Anoxic and Palytoxin-induced Depolarization We initially carried out experiments exposing cerebral slices to OGD to induce AD, first to replicate previous laboratory results and second to provide a basis for comparison with PTX exposure. Light transmittance (LT) imaging revealed AD induced by 10 minutes of OGD. At AD onset, one or more focal increases(s) of elevated LT (representing cell swelling) appeared and spread through neocortical gray matter as front(s) of increased LT, leaving damage in its wake which was imaged as a reduction in LT (Figure 4). The OGD-induced neocortical AD onset time was 289±10 s (n=48). To conserve expensive PTX and initially estimate a concentration that would induce a similar front of spreading depolarization, we first applied a small bolus of concentrated drug into the imaging chamber where each neocortical slice was placed with the aCSF flow stopped. Imaging was started immediately after PTX application to the static bath to a final concentration of 100 or 10 nM. As PTX slowly diffused around the slice, spreading depolarization was reliably induced, initiating as one or more focal increases in LT with each proceeding to spread across the neocortex as a front of elevated LT. The onset of palytoxin-induced depolarization (PD) was significantly earlier using 100 nM PTX vs 10 nM PTX (F(2,29)=47.45, p=0.005) (Table 1). Often during or subsequent to PD in the neocortex, elevated LT fronts were generated in the thalamus, striatum and/or hippocampus (Table 2). To ensure that the static bath itself did not induce anoxia that might trigger AD, several slices were imaged for 10 minutes in the static bath containing only aCSF. In a few slices AD was induced (usually in neocortex, Table 2) but it was significantly later than in slices where the front was induced by PTX (p<0.001) (Table 1). 29 To determine a low but effective concentration of PTX that evoked PD using a superfusion application, 10 or 1 nM PTX in aCSF flowed over slices for 10 minutes (8 rats). At 10 nM PTX the neocortical PD onset time was 246±14 s (n=45) (Figure 5). Compared to 10 nM PTX, 1 nM PTX time was significantly later and more variable at 402±49 s (n=16; F(2,119)=14.36, p<0.001) (Figures 6, 7) . Based on these results we used 10 nM PTX in a flowing bath for the rest of the study. In addition to the neocortex (and like OGD), PTX superfusion induced depolarization(s) in the hippocampus, striatum and thalamus (Table 3). The neocortex was depolarized in the highest proportion of slices in response to OGD or to PTX as measured by LT imaging. In neocortex superfusion of 10 nM PTX induced depolarization earlier and in a higher proportion of slices compared to 1 nM PTX which demonstrated a dose effect (Figure 4). Latency to depolarization in the hippocampus was significantly shorter with 10 nM PTX compared to 1 nM PTX (F(2,40)=13.59, p=0.04). No significant difference in the latency to depolarization between 10 nM and 1 nM PTX was found in the striatum (F(2,41)=6.20, p=0.9) and thalamus (F(2,33)=1.42, p=0.89). To compare with PD, we also superfused OGD for 10 minutes onto slices (Figure 4). The OGD-induced AD latency in the neocortex (289±10s) was significantly shorter than the 1 nM PD latency in neocortex (p<0.001). OGD-induced AD latency was longer than the latency to 10 nM neocortical PD but this was barely significant (p=0.09). The OGD-induced latencies to depolarization in the hippocampus and striatum were significantly longer than the 10 nM PD latencies induced in the same regions (p<0.001; p=0.004), while there was no significant difference between OGD and 10 nM PTX in the latency to depolarization in the thalamus (p=0.23). Meanwhile, the latency to depolarization in the hippocampus (p=0.16), thalamus (p=0.7) and striatum (p=0.442) were not significantly different between OGD and 1 nM PTX. The LT imaging characteristics of PD in the neocortex and subcortical regions (Figures 5, 6) were strikingly similar to AD induced by OGD (Figure 4). Both OGD and PD generated 30 focal depolarization(s) in the neocortex and often in the surrounding “higher” brain structures including the thalamus, striatum, and hippocampus. These focal depolarizations propagated into adjacent gray matter. A decrease in LT was then often detected in these regions where a LT front had passed through. The decrease developed slowly over the ensuing 10-15 minutes. The maximum ∆LT was similar between OGD (n=8) and 10 nM PTX (n=8) (t(8)= -0.23, p=0.83), although PTX displayed a greater minimum ∆LT than OGD (Figure 8) (t(14)=2.47, p=0.03). Like AD, when PD was observed in the striatum, hippocampus or thalamus, these areas also displayed decreased LT following the front of spreading depolarization (Figures 5, 6). 3.2 LT Imaging of OGD- and Palytoxin-Induced Depolarization in the Brainstem Our imaging of AD induced by 10 minutes of OGD in the brainstem extends a preliminary study in our lab examining the acute effects of OGD in brainstem. Swelling, often followed by AD (the dramatic spreading increase in LT) was observed in the superficial regions of superior colliculus and inferior colliculus, in periaqueductal gray and in substantia nigra reticulata. The medial geniculate nucleus (the most caudal part of the thalamus) also usually displayed AD as expected. Similarly, brainstem slices superfused with 10 nM PTX for 10 minutes exhibited swelling often accompanied by PD in the same brainstem regions affected by OGD (Table 4). The red nucleus in midbrain did not show increased LT under OGD or PTX. Also, the substantia nigra compacta displayed no notable increase in LT before exhibiting decreased light transmittance in response to OGD or PTX exposure. In contrast, the substantia nigra reticulata displayed only an increase but no decrease in LT when imaged for 10-40 minutes after exposure to 10 minutes of OGD or PTX. These observations suggested minimal damage in the red nucleus and nigra, although this requires electrophysiological confirmation and additional imaging. 31 3.3 Extracellular Recording of Anoxic- and Palytoxin-Induced Depolarization To confirm that the moving front of elevated LT represents a regional depolarization of neurons, during OGD we measured the change in the extracellular voltage within the CA1 hippocampal region while simultaneously imaging LT changes in the same region. We first obtained a baseline recording before switching to OGD-aCSF. During the first few minutes of OGD there was no change in the baseline potential. At the moment the AD front reached the recording electrode, there was a distinct negative-going shift in the field potential (Figure 9). The sudden drop in the voltage displayed a mean amplitude of 4.2 ± 1.0 mV (n=9) for OGD. With superfusion of PTX, the results using this simultaneous recording technique were similar (Figure 9). The mean voltage amplitude was 4.1±0.8 mV (n=14) during PTX exposure (Figure 10) which was not significantly different from OGD (t(21)=0.06, p=0.9). The key finding for OGD or PTX exposure was the coincidence of the negative shift onset as the LT front reached the micropipette tip. Both parameters are well established indicators of spreading depolarization. 3.4 Two-Photon Laser Scanning Microscopy (2PLSM) To confirm that, like OGD, PTX caused cellular swelling and dendritic beading as implicated by our LT imaging findings, we used 2PLSM to image pyramidal and mesencephalic neurons in cortical and brainstem coronal slices, respectively. Slices were imaged before and immediately after exposure to 10 minutes of OGD or PTX. In the neocortex OGD caused pyramidal cell bodies to swell by 53 ± 3% (n=68), calculated by measuring the largest crosssectional area of each soma (Figure 11). Similarly, PTX exposure caused pyramidal cell bodies to swell by 47±3% (n=106) (Figures 12, 13). The somata swelling caused by OGD and PTX were not significantly different (t(172)= -1.45, p=0.1). Both PTX and OGD caused the disappearance of dendritic spines and subsequent swelling of dendrites which often formed chains of dilations or “beads”. 32 We did not image the effect of OGD on brainstem mesencephalic (MES) neurons because Brisson et al. (2014) previously detected no significant swelling of MES cell bodies after exposure to 15 minutes of OGD. They noted only minor swelling of the primary processes. In the current study 10 minutes of PTX caused MES somata to swell by 14 ± 13% (n=13 cells from 2 mice) (Figures 14, 15, 16). This is significantly less than the 47% observed with pyramidal neurons noted above (t(31)=7.45, p<0.001). Mesencephalic neurons are pseudounipolar, with secondary and tertiary dendrites located far from the cell body. Thus we could not observe if dendritic spines disappear or form beads; however the primary processes were not obviously dysmorphic (Figure 14). So while these midbrain neurons showed some PTX-induced swelling, it was minimal compared to the neocortical neurons. This small effect on brainstem neurons compared to neocortical neurons was similar to OGD treatment. 3.5 LT Imaging of AD and PD in Slices Pretreated with Dibucaine and Carbetapentane To further explore similarities between OGD and PTX we tested two drugs that are known to delay AD to observe whether either of these drugs also delay PD (Table 5, Figure 17). Pretreatment of slices with dibucaine at 1 or 10 M or carbetapentane at 30 M caused a significantly longer AD and PD latency compared to no pretreatment (refer to “mean depol latency” and “control vs. drug pretx depol latency: t value, p value” columns in Table 5). Pretreatment with 10 M dibucaine caused a more prolonged delay of AD than 1 M dibucaine but the delay in PD by 10 M dibucaine and 1 M dibucaine was similar. At 30 M, carbetapentane was more than twice as effective in delaying PD than AD (t(26)= -2.36, p=0.026). Thus carbetapentane and dibucaine are individually effective in delaying both AD and PD onset. 33 Tables # of slices # of slices displaying no displaying depolarization depolarization mean depolarization latency (s) 100 nM PTX 2 16 60 ± 18 10 nM PTX 4 8 205 ± 48 control aCSF (static bath) 14 8 473 ± 36 Table 1. Latency of spreading depolarization induced in slices using a static bath and measured by LT imaging. 100 nM PTX induced depolarization earliest and with more consistency. The latency delay for 10 nM PTX was shorter and depolarization was less likely, demonstrating a dose effect. The static bath condition itself could induce spreading depolarization in a few cases, but only after a prolonged period of no flow. 34 Neocortex Striatum Hippocampus Thalamus 100 nM PTX 16/18 12/15 5/6 6/11 10 nM PTX 8/12 4/10 1/6 4/6 control 8/14 2/19 0/11 0/13 Table 2. Regional frequency of spreading depolarization induced in slices using a static bath. Induction of spreading depolarization was monitored with LT imaging in four higher brain regions. 100 nM PTX consistently induced depolarization in the neocortex, striatum and hippocampus. 10 nM PTX application also reliably induced depolarization in the neocortex but less reliably in the striatum and hippocampus. 35 Depol (n) OGD Depol Latency (s) PTX 10 nM Depol. Depol Latency DND (n) (s) (n) Depol (n) PTX 1 nM Depol Latency (s) DND (n) DND (n) Neocortex 48 289 ± 10 0 45 246 ± 14 0 16 402 ± 49 2 Striatum 27 237 ± 16 2 15 158 ± 10 5 2 173 ± 3 5 Hippocampus 14 352 ± 27 6 20 175 ± 17 1 9 274 ± 44 2 Thalamus 17 296 ± 17 3 14 241 ± 24 5 5 263 ± 64 5 Table 3. Regional generation of OGD- or palytoxin-induced spreading depolarization in higher brain gray matter using LT imaging of superfused slices. The neocortex depolarized in the highest proportion of slices in response to OGD or to PTX as measured by LT imaging. 10 nM PTX induced depolarization earlier (in neocortex) and in a higher proportion of slices compared to 1 nM PTX. DND=slices that did not display a spreading depolarization. 36 Medial Geniculate Nucleus (Thalamus) Substantia Nigra Pars Reticulata Superior Colliculus Periaqueductal Gray Inferior Colliculus Red nucleus n 6 7 11 18 8 4 OGD Spread Depol Swelling 5 1 4 3 11 0 13 5 8 0 0 0 n 7 7 10 12 4 4 PTX Spread Depol Swelling 4 3 0 7 7 3 6 6 3 1 0 0 Table 4. Regional generation of 10 nM palytoxin-induced spreading depolarization in brainstem gray matter. The medial geniculate nucleus of the thalamus (belonging to the higher brain) and various brainstem structures displayed either a propagating signal or swelling alone in response to OGD or PTX. The red nucleus was the only structure examined that did not show any response. 37 1 µM dibucaine 10 µM dibucaine 30 µM carbetapentane control vs drug pretreatment (µM) no. of slices (n) drug mean mean % delay drug pretx treatment depol depol latency: mean % by pretx drug of duration latency delay AD vs PD: t t value, p (m) treatment (s) value (%) value, p value control 10 - 1 Dib 10 45±5 control 12 - 1 Dib 12 45±5 control 16 - 10 Dib 17 35±5 control 28 - 10 Dib 29 35±5 control 14 - 30 CP 12 40±5 control 21 - 30 CP 19 40±5 OGD 190 ± 14 t(18)= -3.023, 44 ± 11 267 ± 21 p=0.007 PTX 246 ± 37 t(22)= -1.935, 61 ± 20 342 ± 33 p=0.066 OGD 200 ± 13 t(31)= -6.303, 72 ± 19 315 ± 13 p<0.001 PTX 217 ± 17 t(55)= -3.417, 62 ± 17 286 ± 11 p=0.001 OGD 206 ± 20 t(24)= -2.801, 51 ± 16 288 ± 20 p=0.01 PTX 207 ±21 t(38)= -6.402, 129 ± 29 398 ± 21 p<0.001 t(17)= -0.721, p=0.481 t(42)= 0.395, p=0.695 t(26)= -2.360, p=0.026 Table 5. Spreading depolarization latency in slices pretreated with dibucaine or carbetapentane. Slices were pretreated with either dibucaine or carbetapentane for 35-45 minutes before superfusion with OGD or PTX treatment. The latency to depolarization was compared to a subsequent untreated control slice superfused with the OGD or PTX to determine the % delay caused by the pretreatment drug. All drug pretreatment conditions caused significant delay in AD and in PD. Depol=depolarization, pretx=pretreatment. 38 Figures Figure 4. Light transmittance (LT) imaging reveals damage in higher gray matter following AD induced by 10 minutes of oxygen-glucose deprivation (OGD). A) and B) depict two separate neocortical slices that were superfused with OGD-aCSF solution. The front of elevated LT (representing cell swelling during AD onset) arises and propagates as distinct waves coursing through neocortical gray matter (arrows), leaving damage that is imaged as light scatter (purple pseudocolouring) in their wake. In both A and B, AD does not involve the striatum (Str) which remains swollen but not terminally injured as is the neocortical gray by 24 minutes. 39 Figure 5. Palytoxin at 10 nM induces an OGD-like sequence of LT change. Arrows show fronts of highly elevated LT (red pseudocolouring) arising focally and coursing independently through the neocortical and striatal gray (Str). The PTX is washed off after 10 minutes. These regions are subsequently damaged as shown by the reduced light transmittance at 33 minutes. 40 Figure 6. Palytoxin at 1 nM superfusion induces an OGD-like sequence of LT change. Arrows show fronts of elevated LT arising focally and coursing first through the neocortical gray and then hippocampus (Hipp). Note the spread of the signal through the CA1 cell body layer (red pseudocolouring). The PTX is washed off after 10 minutes. These regions are subsequently damaged over the ensuing 20 minutes. 41 Figure 7. Distribution of palytoxin-induced depolarization (PD) latency measured by LT imaging. The distribution of depolarization latencies induced by superfusion of 10 or 1 nM PTX in neocortex. At 1 nM PTX, PD occurred later over a wider range of times. At 10 nM PTX, the PD onset time is significantly earlier (p<0.001) and within a narrower time range, demonstrating a dose effect. 42 70 OGD 10 nM PTX 60 50 40 ∆LT% 30 20 10 0 -10 -20 -30 -40 * Figure 8. Maximal and minimal LT changes in the neocortex induced by OGD or by PTX. Comparison of the responses to OGD and 10 nM PTX in terms of spreading depolarization strength (peak LT increase) and subsequent injury (maximal LT decrease). Maximum increase in LT values were comparable between OGD and PTX (t(8)= -0.228, p=0.826), however PTX showed a significantly greater decrease in LT compared to OGD (t(14)=2.474, p=0.027). 43 Figure 9. Negative shift detected when elevated LT front passes recording electrode. A) OGD evokes anoxic depolarization that propagates across the CA1 region of the hippocampus, recorded both as an elevated LT front (arrow) and a negative voltage shift as the front passes the tip of the recording micropipette (white dot). B) Bath application of 10 nM palytoxin evokes a similar negative shift in the extracellular voltage recorded in layers II/III of the neocortex at the moment the LT front (arrow) passes the recording pipette (white dot). These recordings are similar to AD evoked by OGD or 100 µM ouabain. 44 Negative Shift Amplitude (mV) 12 10 8 OGD 6 10 nM PTX 4 2 0 0 5 10 15 Slice number Figure 10. Amplitudes of the negative-shift in extracellular voltage during depolarization induced by OGD or by palytoxin. The amplitudes of the negative-going shift in the extracellular voltage measured during OGD or PTX-aCSF superfusion. There is no significant difference in response between the two treatments (t(21)=0.949, p=0.709). 45 Layer V Neocortical Pyramidal Neurons Pre-OGD Post-OGD A) 25 m B) 25 m C) 15 m Figure 11. Two-photon laser scanning microscopy (2PLSM) images of pyramidal neurons before and after OGD-aCSF. A small proportion of pyramidal neurons in all regions of neocortex are YFP-positive in YFP+ transgenic mice. Immediately following AD induced by 10 minutes of OGD, layer V neocortical pyramidal neuron cell bodies swell and dendrites become beaded, forming a background of fluorescent dots as shown in Figures A and B. In C, the cell body field is shown at higher magnification to illustrate the evoked neuronal swelling. 46 Layer V Neocortical Pyramidal Neurons Pre-PTX Post-PTX A) 25 m B) 15 m Figure 12. 2PLSM images of pyramidal neurons before and after 10 nM palytoxin aCSF exposure. Similar to the previous figure showing pyramidal cell responses to OGD, neocortex layer V pyramidal neuron cell bodies swell and their dendrites bead upon depolarization induced by PTX. As previously detailed with OGD by Andrew et al. (2006), as dendrites swell in response to PTX they lose their dendritic spines (white arrows) and form dysmorphic and beaded dendrites. 47 Figure 13. Distribution of pyramidal neuron soma swelling induced by OGD or PTX. The change in the cross-sectional (XS) area of the cell body was measured to determine neocortex layer V pyramidal neuron soma swelling or shrinking in response to 10 minutes of OGD-aCSF or 10 nM PTX-aCSF. Both treatments showed a similar wide range of irreversible swelling. 48 Midbrain Mesencephalic Neurons 10 min PTX Pre-PTX 15 min PTX A) 60 m B) 60 m C) 60 m Figure 14. 2PLSM images of midbrain mesencephalic neurons before and after exposure to 10 nM PTX. Exposure of YFP-positive pseudounipolar neurons to 15 minutes of 10 nM PTX in the mesencephalic nucleus of the brainstem elicited minor swelling of cell bodies without major dendritic beading developing in the background or along primary processes (green arrows). 49 Midbrain Mesencephalic Neurons Pre-PTX Post-PTX A) 35 m B) 20 m C) 20 m Figure 15. 2PLSM images of midbrain mesencephalic neurons before and after 10 nM PTX. Higher magnification optical slices reveal some swelling of mesencephalic neuronal cell bodies after 15 minutes of PTX exposure. Also common with PTX is loss of the YFP dye from some cells, implicating some neuronal damage. While the swelling is less than that induced in pyramidal neurons by PTX, it is more than what was observed with OGD by Brisson et al. (2014). A) Of cells that swell in PTX, 3 show dye loss (red arrows). B) The ghost of one of three swollen cells can be seen following dye loss (red arrow). C) The outline of two neuron cell bodies pre-PTX exposure (right) is juxtaposed directly onto the cell bodies post-PTX exposure (left), visualizing the degree of swelling that has occurred during PTX exposure. 50 Figure 16. Distribution of swelling by mesencephalic neuronal cell bodies induced by OGD or by 10 nM PTX. Change in cross-sectional (XS) area of MES cell bodies was measured to determine soma swelling or shrinking in response to 15 minutes of exposure to 10 nM PTXaCSF. MES neurons swelled on average of 14 ±3% in response to 15 minutes of PTX. A second population of MES neurons better resisted OGD-induced swelling when exposed to 15 minutes of OGD-aCSF (Brisson et al., 2014). 51 mean depolarization time as % of control 300 * 19 250 12 200 17 29 12 10 150 OGD ≥10 ≥12 PTX 100 50 0 No Pretx 1µM Dib 10µM Dib 30µM CP drug pretreatment Figure 17. A comparison of the spreading depolarization latency in slices pretreated with dibucaine or carbetapentane to untreated slices for 35-45 minutes before being superfused with OGD or PTX (figure derived from data presented in Table 5). The latency to depolarization of a drug pretreated slice was compared to a subsequent untreated control slice superfused with the same PTX or OGD to determine the % delay caused by the pretreatment drug. 30 µM carbetapentane caused a PD delay that was significantly greater than the delay of AD, as indicated by the asterisk (*) (t(26)= -2.36, p=0.026). The difference in delay between AD and PD was not significantly different with 1 µM dibucaine (t(17)= -0.72, p=0.48) and 10 µM dibucaine (t(42)=0.39, p=0.69) pretreatment conditions. Pretx=pretreatment. 52 Chapter 4 Discussion The purpose of our study was to investigate the effects of the potent marine poison palytoxin (PTX) on mammalian gray matter. Specifically, we investigated if PTX induces a spreading depolarization of neurons in a manner similar to ischemia-induced anoxic depolarization (AD). Can the actions of PTX provide insight as to how ischemia induces AD? We found that palytoxin induces a depolarization (PD) in the neocortex detected by LT imaging and exhibiting characteristics often indistinguishable from anoxic depolarization. Simultaneous recording and imaging confirmed that the increased LT front was a collective neuronal depolarization within the recorded region. Furthermore as imaged using 2PLSM, palytoxin treatment evoked significant pyramidal cell body swelling and dendritic beading with the disappearance of dendritic spines. This neuronal injury was indistinguishable from OGD-induced injury. LT imaging in brainstem slices revealed that PD affected the same brainstem structures and in a similar manner to OGD-induced AD. Brainstem mesencephalic neurons under 2PLSM imaging displayed only moderate swelling from PTX exposure which was significantly less than swelling measured in pyramidal neurons. Again this was similar to the effects of AD. Finally, the AD-delaying drugs dibucaine and carbetapentane were found to also significantly delay PD onset. 4.1 Anoxic Depolarization in Cortical Slices Slice exposure to 10 minutes of OGD reliably evoked AD in the neocortex at approximately 5 minutes, replicating the well documented findings of other studies (Jarvis et al., 2001; Andrew et al., 1999; Obeidat et al., 2000; Douglas et al., 2011). Variation could occur due to quality in dissection, age of rat, age of slice and most importantly, slight differences in temperature. Like previous studies, in coronal slices under OGD we observed a pronounced wave of increased LT followed by decreased LT in the neocortex, striatum, hippocampus and thalamus. 53 Less dramatic LT changes developed in brainstem gray matter (Brisson et al., 2014; Jarvis et al., 2001; Anderson et al., 2005; Brisson et al., 2013; Brisson and Andrew, 2012). Using extracellular voltage recordings, we measured a negative shift in extracellular field potential when an OGD-induced propagating wavefront of increased LT passed the recording electrode. Replicating previous studies measuring AD-associated changes in field recordings, this negative-going extracellular potential shift coincided with the increased LT front at the recording electrode and so represents a collective depolarization of neurons near the electrode. The consistent detection of a negative shift associated with the LT wavefront demonstrated that the front represents spreading depolarization. Imaging of neocortical pyramidal neurons in layers V with 2PLSM after exposure to 10 minutes of OGD reproduced our previous observations of neuron injury in the form of dendritic beading, neuron cell body swelling and loss of dendritic spines (Jarvis et al., 2001; Douglas et al., 2011; Davies et al., 2007). Our 2PLSM confirmed that while the cell bodies showed damage in the form of swelling, there was also dendritic beading which accounts for the decreased LT that follows areas that undergo a wave of increased LT. Neocortical neurons that do not undergo AD despite exposure to OGD retain their responsiveness and show minimal signs of damage when observed in 2PLSM imaging (Jarvis et al., 2001). On the other hand, OGD exposure that induces AD consistently causes permanent depolarization and neuronal injury with no return of function. Our procedure in the current study consisted of 10 minutes of OGD because AD irreversibly damages neurons in cortical brain slices, causing the permanent depolarization and loss of the evoked field potential (Jarvis et al., 2001; Obeidat and Andrew, 1998; Taylor et al., 1999; Weber and Taylor, 1994; Anderson et al., 2005). Overall, our findings here reproduced these previously published observations for OGDinduced AD and provided us with a standard experimental condition to compare with our novel palytoxin observations. 54 4.2 Palytoxin Causes Spreading Depolarization in the Neocortex When palytoxin at concentrations of 1, 10 or 100 nM were applied to coronal brain slices we observed one or more robust spreading fronts of increased LT in the neocortex, striatum, hippocampus and/or thalamus. The images resembled AD induced by OGD as described above. The neocortex was the most consistent region to depolarize when exposed to PTX or OGD. A higher concentration of PTX in both static bath and superfusion conditions induced depolarization earlier and more consistently. A 10 nM PTX superfusion induced depolarization in the neocortex at ~4 minutes while a 1 nM PTX superfusion was ~7 minutes, indicating a concentrationdependent latency for PD. Given that the delivery of PTX via the static bath was less uniform than superfusion, we measured depolarization times of the higher brain structures based on lower doses of PTX-aCSF superfusion. During 10 and 1 nM PTX superfusion, the higher brain structures depolarized earlier and more consistently under 10 nM PTX, especially neocortex and hippocampus. Although the striatum and hippocampus displayed more sensitivity to 10 nM PTX as indicated with a significantly earlier depolarization latency compared to OGD, the depolarization times of the neocortex and thalamus were comparable between 10 nM PTX and OGD. Overall, of the higher brain structures we examined, the neocortex and thalamus showed similar vulnerability to 10 nM PTX and to OGD-mediated depolarization. Neocortical gray superfused with 1 nM PTX depolarized anytime between 3-10 minutes while 10 nM PTX was 3-5 minutes, so we opted to use 10 nM for the remainder of the study. In the neocortex, depolarization induced by 10 nM PTX superfusion displayed a similar peak %∆LT as did OGD-induced AD. However PTX displayed a subsequent greater minimum %∆LT with a mean -25% ∆LT compared to the -13% measured after OGD exposure. This suggest more acute injury induced by PTX vs OGD. However Brisson et al. (2014) reported a reduction of -25to -30% ∆LT in the neocortex after OGD exposure which corresponds with our 55 PTX value here. Also, dendritic beading may take longer than the mean 23 minutes we spent imaging slices under OGD. Douglas et al. (2011) found approximately -25% ∆LT after a 10 minute recovery from 10 minutes OGD in control aCSF. We conclude that the level of injury from OGD or from PTX are comparable. Previous work by others showed that PTX depolarizes the membrane potential of skeletal, cardiac and smooth muscle well as spinal cord tissue and nerve fibers (see Rossini and Bigiani, 2011). As yet there are no intracellular recordings from neurons directly showing the actions of PTX. As well, no work has yet been published studying PTX in relation to ischemia. Here we show that PTX induces a propagating increased LT front in all higher brain structures and initiates this wavefront earlier at higher concentrations. This signal indicates that neurons not only depolarize but do so in a propagating manner in response to PTX. This is not unexpected given that PTX converts the Na+/K+ pump into a channel to cause cellular depolarization. The molecular interaction between PTX and the pump leads to the disruption of ion gradients, which in turn disrupts the function of other ion-transport systems such as voltage-gated Ca2+ channels, Na+/Ca2+ exchangers and the Na+/H+ antiport. These events downstream further impair ion equilibria (Rossini and Bigiani, 2011) and likely also occur with AD. Palytoxin has been studied in concert with ouabain and other cardiac glycosides. The context has been to understand PTX`s molecular action on the Na+/K+ pump (Ozaki et al., 1985; Habermann and Chhatwal, 1982) as it compares to simple blockade by ouabain. Ouabain and digitoxin have been well studied in relation to cardiac ischemia (Rose and Valdes, 1994). Like OGD-induced AD, it is not understood what triggers the large inward current once a cardiac glycoside binds to the Na+/K+ pump. We propose that a percentage of pump molecules actually open when bound by cardiac glycosides which would link ouabain binding to the pump with the sudden generation of a strong inward current. This requires electrophysiological studies. 56 4.3 The Negative Shift Confirms a Population Depolarization A negative shift in DC potential accompanying spreading depression in the cerebral cortex was first described by Leao as a surface negative wave or negative “slow voltage variation” that coincided with the spreading depression reaching the region of the electrode placed within the pia mater (Leao, 1947; Leao, 1951; Marshall,1959). Brisson et al. (2014) imaged the spreading increase in LT induced by OGD while simultaneously recording intracellularly and found when the increased LT front passed the recording electrode, the recorded cell depolarized rapidly. The coordinated depolarization of local neurons as induced by spreading depression leads to a local negative potential extracellularly, which can be detected by a recording electrode in the vicinity. We confirmed the consistent presence of a sudden negative shift concurrent with the PTX-induced propagating wave at the recording electrode in the neocortex. The negative shift examples recorded during PD in the neocortex were similar in waveform to the negative shifts we obtained from OGD-induced AD recordings in layer CA1 of the hippocampus. Their amplitudes were indistinguishable. The consistent negative shift as the increased LT wavefront passes the recording electrode indicates a synchronized depolarization of the local neurons induced by palytoxin. 4.4 2PLSM Confirms Similar Neuronal Injury Induced by OGD or Palytoxin Using 2PLSM, we imaged acute neuronal injury evoked by OGD or PTX on layer V pyramidal cells in real time. Like OGD, 10 minutes of PTX caused cell body swelling, dendritic beading and the loss of dendritic spines. This confirms what we detected with LT imaging following PD: the cell body swelling corresponds to the increased LT and the decreased LT that follows corresponds to the generation of dendritic beads. Our findings of OGD-induced neuronal swelling replicates several studies conducted previously in our laboratory using 2PLSM (Brisson et al., 2014; Douglas et al., 2011) and in vivo (Murphy et al., 2008). 57 So as with OGD, PTX leaves pyramidal neurons permanently damaged in the wake of the spreading depolarization it evokes, with dendritic beading and cell body swelling serving as indicators of neuronal injury (Jarvis et al., 2001; Obeidat et al., 2000; Tanaka et al., 1999), driving home the point that ischemia is as deadly as this potent toxin. It is firmly established that PTX at even picomolar concentrations can open the Na+/K+ ATPase to form a channel that allows Na+ and K+ to flow down their concentration gradients, evoking a strong inward current. Given the thousands of pump molecules in a neuron’s plasma membrane, this current would be formidable even though the conductance across each channel is relatively small (Gadsby et al., 2009). The simultaneous opening of the Na+ and K+ gates causing the conductance is both molecularly “subtle” and reversible (Gadsby et al., 2009). Just the right concentration allows the channel to flicker between open and closed states (Figure 18d) (Gadsby et al., 2009; Artigas and Gadsby, 2002). It is possible that cardiac glycosides at higher concentrations might act in a similar way to cause an AD-like response. Such a possibility has not been tested. The most convincing experiment would involve patching oocytes with Na+/K+ pump molecules in their plasma membrane and then evoking a PTX-like channel conductance (Figure 18) with micromolar concentrations of ouabain or digitoxin. The logical extension of this experiment is to substitute the drug with simulated ischemia, for example by using OGD. The opening of PTXlike channels by OGD (with other Na+ and K+ channels blocked) would provide direct evidence that AD is generated by ischemia, converting the Na+/K+ ATPase into an open channel. All of our data accumulated to date points to this possibility. 4.5 Brainstem LT Imaging Reveals PTX Affects the Same Structures as OGD Previous unpublished LT imaging studies of AD in the brainstem in our laboratory found that certain structures are more susceptible to AD. These include the superior and inferior colliculi, tegmental nucleus and periaqueductal gray (PAG) area. However most of the ventral brainstem regions displayed minimal changes in LT following AD (Brisson et al., 2014). This 58 was because Brisson et al. (2014) imaged brainstem regions in the same frame with cerebral or cerebellar gray matter which are more translucent. The result was very low levels of light passing through the ventral brainstem and consequently ∆LT values were also very low. In the present study, we imaged only brainstem regions with stronger illumination to increase resultant ∆LT values in those regions. As a result we confirmed a spreading signal in the dorsal structures above and also detected ∆LT in ventral brainstem nuclei evoked by OGD. The substantia nigra reticulata underwent AD and/or swelling, but the typical decrease in LT following the AD and/or swelling was not observed. The substantia nigra pars compacta exhibited decreased LT shortly after OGD application, without showing increased LT preceding it. Furthermore, upon PTX application to brainstem slices, the same structures displayed sensitivity to PTX. Both PD and AD associated increases in LT were followed by decreased LT, often only covering part of the structure imaged. Generally brainstem regions were less damaged by PTX than higher brain regions, as also observed with OGD. So the selective response of structures within the brainstem to PTX mimicked the selectivity we observed with OGD. In contrast to higher gray matter, the “lower” hypothalamus and brainstem demonstrate greater resistance to depolarization and better survive global ischemia in patients as follows sudden cardiac arrest (Brisson et al., 2014). Patch recordings of neurons in the higher brain display sudden AD onset with a rapid depolarization to near zero millivolts, immediate AP inactivation, and irreversible swelling and dendritic beading of pyramidal neurons in the higher brain (Brisson et al., 2013; Brisson et al., 2014; Brisson and Andrew, 2012; Davies et al., 2007). In contrast, in the lower brain slices there is a long latency to AD onset with a gradual depolarization and very slow AP inactivation. Furthermore, the depolarization in lower brain neurons may not reach zero millivolts, and neurons always recover in control aCSF (Brisson et al., 2013; Brisson et al., 2014; Brisson and Andrew, 2012). 59 Our finding that the brainstem shows some resilience (relative to higher brain) to such a deadly pump poison like palytoxin demonstrates an additional similarity between the action of this drug and the action of both ouabain and ischemia (Balestrino, 1995; Brisson et al., 2014). Andrew and colleagues have proposed that higher expression of the 13 pump isoform by brainstem neurons confers neuroprotection. Our unpublished data indicate that higher brain regions strongly express the 1α1 isoform, while lower brain regions express a higher proportion of the 1α3 isoform (Figure 19).Whereas 1α1 has been found to be important in maintaining the ionic gradient during basal conditions, 1α3 is vital in maintaining the gradient in non-basal conditions such as a high [Na+]i (Azarias et al., 2013). The 1α3 has a greater affinity for ATP relative to 1α1 and should therefore be more resistant to hypoxia and ischemia (Blanco et al., 1995). In addition, studies have shown that 1α3 isoform is required for the rapid restoration of large transient increases in [Na+]i and has a lower Na+ affinity than 1α1 (Munzer et al., 1994; Zahler et al., 1997; Azarias et al., 2013). So the 1α3 isoform functions more efficiently under ischemic conditions. If the pump channel is more resistant to opening in the 13 form, it could explain why palytoxin and ischemia are less damaging to brainstem neurons. 4.6 2 Photon Laser Scanning Microscopy of Mesencephalic Neurons After Palytoxin Exposure To support our brainstem LT imaging we used 2PLSM imaging to demonstrate moderate neuronal injury at the subcellular level caused by acute PTX exposure. Mesencephalic (MES) neurons are primary sensory neurons derived from neural crest cells that migrate into the central nervous system (CNS) during development (Zhang et al., 2012). Neural crest cells that remain in the peripheral nervous system (PNS) form the dorsal root ganglia, which may explain the dorsal root ganglia-like resiliency MES neurons demonstrate against metabolic stress (Burchiel, 1984; Utzschneider et al., 1991). We examined MES neurons in the trigeminal nucleus of YFP+ mouse brainstem, which is a neuron population in this strain with sufficient fluorescence to be imaged. 60 PTX exposure caused MES somata swelling, although significantly less than by cortical pyramidal neurons evoked by a similar dose of PTX. In a previous study of MES neurons exposed to OGD, there was no detectable swelling (Brisson et al., 2014). MES neurons are the most resistant neurons to OGD and ouabain exposure (Brisson et al., 2014), recovering 80-100% of their membrane potential, input resistance and action potential amplitude after 10 minutes of OGD. Some MES neurons can easily recover from multiple OGD exposures (Brisson et al., 2014). MES neurons in the previous OGD study did not swell and here we detected only 15% swelling in response to 10 nM PTX. It was surprisingly less than the 40% swelling of neocortical pyramidal cells evoked by that same PTX concentration. The neurons seem impervious to OGD as well as PTX, except for some swelling. Again a more resilient 13 form of the pump might explain the minimal neuronal injury. Like OGD and ouabain treatment, we have found that PTX causes less acute damage to brainstem. PTX targeting the 1α3 isoform would explain why the higher brain with its lower levels of 1α3 is more incapacitated after PTX exposure than the brainstem. In the brainstem, proportionally more 1α3 would help maintain Na+/K+ gradients in non-basal states as during PTX exposure assuming that like OGD, the 1α3 isoforms functioned better than 1α1 during exposure to ouabain. Furthermore, Brisson and colleagues (2014) found with whole cell recordings that MES neurons would hyperpolarize during or immediately after OGD and these hyperpolarizations were blocked by ouabain. They theorized that hyperpolarization is generated in response to OGD by up-regulating the Na+/K+ pump given that the pump operates at only 50% capacity in basal conditions (Gottron and Lo, 2009). The brainstem may resist AD and PD because there is a higher proportion of 1α3 pump isoforms which are better able to upregulate and are affected less by OGD, ouabain or PTX. This is conjecture until the proper experiments are carried out. 61 4.7 Palytoxin Delay Using Dibucaine and Carbetapentane In this study both 1 and 10 µM dibucaine as well as 30 µM carbetapentane were effective in significantly delaying PD and AD. Given the potent action of PTX, 10 µM dibucaine was used initially in the current study to assess whether it could delay PD. Once we observed a significant delay, we pretreated neurons with 1 µM dibucaine and also observed a delay in PD latency. Unlike 10 µM dibucaine, 1 µM dibucaine does not affect the normal functioning of neocortical neurons, preserving the evoked field potential amplitude at ~90% of its initial value (Douglas et al., 2011). Slice pretreatment with 30 µM carbetapentane also delayed PD. At 10-50 µM, carbetapentane (CP) pretreatment for 30-35 minutes delayed OGD-induced AD in neocortical slices (Anderson et al., 2005). At 10 µM, CP (30 minutes) blocked or delayed AD without altering the evoked synaptic response in layers II/III in the neocortex and in layer CA1 of hippocampal slices (Anderson et al., 2005). Our studies confirm previous studies that found 1-10 µM dibucaine or 10-50 µM carbetapentane pretreatment effective in delaying AD induced by OGD. We now show that these drugs also effectively delay palytoxin-induced depolarization. Ouabain has been reported to inhibit PTX by preventing PTX from binding to Na+/K+ pump (they share similar but not identical binding sites). However it is not known if CP and dibucaine act on the Na+/K+ pump, but this is a possibility. In addition, CP and dibucaine delay AD by decreasing neuronal excitability but in different ways. Dibucaine raises the action potential (AP) threshold by blocking AP-associated sodium channels, while CP reduces excitability by increasing the slow afterhyperpolarization (sAHP) (White et al., 2012), which occurs in many neurons when they fire repetitively (Larsson, 2013). The sigma receptor ligands carbetapentane, dextromorphan and 4-IBP N-(Nbenzylpiperidine-4-yl)-4-iodobenzamide have been found to block normoxic SD (Anderson and Andrew, 2002) as do a number of caines (Douglas et al., 2011). It is interesting that SD and AD likely share the same mechanism that causes pump failure but that with SD the metabolic load is 62 less (which allows tissue that undergo SD to recover (Anderson et al., 1999)). These drugs may all act on the Na+/K+ pump to delay failure and possibly opening of the pump channel. 4.8 Potential Significance of Our Findings This study provides evidence that PTX evokes a strong spreading depolarization in higher brain gray matter with several properties strikingly similar to anoxic depolarization elicited by ischemia-like conditions. Two facts are well established. First, that PTX converts the Na+/K+ pump into an open cationic channel. Second, that such a conversion will promote spreading depolarization in gray matter because of the double jeopardy of pump failure with the generation of inward current driven by simultaneous Na+ influx and K+ efflux. The logical possibility is that ischemia evokes spreading depolarization in the brain by holding open the gates of the Na+/K+ ATPase pump. This would not only hold true for patients undergoing stroke onset, but for the innumerable patients experiencing global ischemia from sudden heart failure or from massive head trauma. In all these clinical situations recurring spreading depolarizations in the higher gray matter are known to worsen outcome (Drier, 2011; Brisson et al., 2014). Could a repetitive opening of the Na+/K+ pump initiate and drive each event? 4.9 Future Directions Using live brain slices, our study demonstrates dramatically similar effects shared by PTX treatment and simulated ischemia. Of course correlation does not prove causality. There is a direct experimental technique to prove that ischemia itself turns the Na+/K+ ATPase into an open cationic channel. The technique is not yet available in Dr. Andrew’s laboratory but we will instigate the approach with the purchase of a higher quality membrane patch amplifier. The procedure involves patching membrane on pyramidal neurons in neocortical slices. Standard blockers of voltage- and ligand-gated channels are superfused on the preparation to reduce background channel noise. PTX is then applied at picomolar concentrations and the characteristic 63 pump channel current recorded in voltage clamp (Figure 18). A similar current will then be evoked (hopefully) without PTX under OGD conditions. A more immediate experiment involves imaging PTX-induced depolarization in the presence of the voltage-sensitive Na+ channel blocker TTX. Before the open pump itself was considered a channel, a number of studies showed that the PTX-induced inward current was unaffected by 1 µM TTX (Karaki et al., 1998; Yoshizumi et al., 1991). This proved that standard voltage-dependent sodium channels were not involved. OGD disables the pump and other energy-dependent functions to cause first a slow, then rapid depolarization that characterizes AD. Standard voltage-dependent channels seem to contribute some inward current, but they can be blocked which only delays AD onset. Ouabain in the presence of glucose and oxygen similarly incapacitates the pump causing an AD-like response. So failure of the Na+/K+ pump is critical as shown by ouabain and PTX. Yet the rapid depolarization is an active process and not a slow, passive event which is expected by simply blocking the pump. At a meeting examining spreading depolarization this past summer (Brennan et al., 2014) it was clear that computer models could account for depolarization by neurons to approximately -30 to -20 mV but not to zero millivolts as recorded in real neurons. This was because most voltage-sensitive Na+ channels inactivate when depolarized. A formidable number of channels conducting Na+ are required that remain open as the membrane potential approaches zero. The pump channel satisfies this criterion. A larger pore-type channel was suggested to conduct molecules of several hundred daltons during AD in 1999 (Tanaka et al., 1999). This proposition was shortly followed by the discovery of the mitochondrial transition pore and the pannexin hemichannel (Ricchelli et al., 2003; Bennett et al., 2003), both passing molecules at least 1200 Da. Neurotoxins commonly target ion channels or ion transporters that drive normal physiological function and thereby shut 64 down the nervous system (Wang, 2008). Palytoxin may take advantage the pump`s vulnerability by holding open the channel in a similar manner to ischemia. Overall this study documents several striking similarities between the effects of PTX and of OGD throughout brain gray matter. The possibility that the two treatments share a similar molecular mechanism deserves further study because this could supply the link between pump failure and brain depolarization. 65 Figures Figure 18. PTX transforms the Na+/K+ pump into a channel, one ATPase at a time. In recordings of outside-out patches from guinea-pig ventricular myocytes, just the right concentration of PTX can open the pump to evoke (a,b) macroscopic currents, (c) a single channel event that remains open for many seconds (arrow), and (d) a single channel event that flickers open and closed. From Gadsby et al. (2009). 66 Figure 19. Proportions of the different Na+/K+ pump isoforms in the cortex and brainstem. Real-time qPCR measurements of neocortex (black) and whole brainstem (white) mRNA expression of the Na+/K+ ATP pump isoforms reveal the neocortex and brainstem express different levels of the 1α1 and 1α3 isoforms. The ischemia sensitive 1α1 isoform is strongly expressed in the cortex but not in the brainstem, while the ischemia-resistant 1α3 is proportionally more predominant in the brainstem. The 1β1 is equally expressed in the neocortex and brainstem and is not related to ischemia. From Andrew (2013). 67 References Alonso G, Phan V, Guillemain I, Saunier M, Legrand A, Anoal M, Maurice T (2000) Immunocytochemical localization of the sigma 1 receptor in the adult rat central nervous system. Neuroscience 97: 155-170. Anderson TR, Andrew RD (2002) Spreading depression: Imaging and blockade in the rat neocortical brain slice. J Neurophysiol 88: 2713-2725. Anderson TR, Jarvis CR, Biedermann AJ, Molnar C, Andrew RD (2005) Blocking the anoxic depolarization protects without functional compromise following simulated stroke in cortical brain slices. Journal of Neurophysiology 93: 963-979. Andrew RD, Jarvis CR, Obeidat AS (1999) Potential sources of intrinsic optical signals imaged in live brain slices. Methods 18: 185-196. Andrew RD (2013) Higher brain susceptibility and lower brain resiliency to ischemia. CIHR grant 00208256. Andrew RD, Macvicar BA (1994) Imaging cell volume changes and neuronal excitation in the hippocampal slice. Neuroscience 62: 371-383. Andrew RD, Labron MW, Boehnke SE, Carnduff L, Kirov SA (2007) Physiological evidence that pyramidal neurons lack functional water channels. Cerebral Cortex 17: 787-802. Artigas P, Gadsby DC (2002) Ion channel—like properties of the Na/K pump. Ann N Y Acad Sci 976: 31-40. 68 Artigas P, Gadsby DC (2006) Ouabain affinity determining residues lie close to the Na/K pump ion pathway. Proceedings of the National Academy of Sciences 103: 12613-12618. Artigas P, Gadsby DC (2004) Large diameter of palytoxin-induced Na/K pump channels and modulation of palytoxin interaction by Na/K pump ligands. The Journal of General Physiology 123: 357-376. Ashton D, Willems R, Wynants J, Van Reempts J, Marrannes R, Clincke G (1997) Altered Na+ channel function as an in vitro model of the ischemic penumbra: Action of lubeluzole and other neuroprotective drugs. Brain Res 745: 210-221. Azarias G, Kruusmagi M, Connor S, Akkuratov EE, Liu XL, Lyons D, Brismar H, Broberger C, Aperia A (2013) A specific and essential role for Na, K-ATPase alpha3 in neurons coexpressing alpha1 and alpha3. J Biol Chem 288: 2734-2743. Bacallao R, Kiai K, Jesaitis L (1995) Guiding principles of specimen preservation for confocal fluorescence microscopy. Handbook of biological confocal microscopy. pp. 311-325. Springer US. Balestrino M, Somjen GG (1986) Chlorpromazine protects brain tissue in hypoxia by delaying spreading depression-mediated calcium influx. Brain Res 385: 219-226. Balestrino M (1994) Studies on anoxic depolarization. J Neurosci Methods 52: A1. Balestrino M (1995) Pathophysiology of anoxic depolarization: New findings and a working hypothesis. J Neurosci Methods 59: 99-103. 69 Bargiotas P, Krenz A, Hormuzdi SG, Ridder DA, Herb A, Barakat W, Penuela S, von Engelhardt J, Monyer H, Schwaninger M (2011) Pannexins in ischemia-induced neurodegeneration. Proc Natl Acad Sci 108(51): 20772-20777. Beaurepaire E, Oheim M, Mertz J (2001) Ultra-deep two-photon fluorescence excitation in turbid media. Opt Commun 188: 25-29. Bennett MV, Contreras JE, Bukauskas FF, Sáez JC (2003) New roles for astrocytes: Gap junction hemichannels have something to communicate. Trends Neurosci 26: 610-617. Blanco G (2005) Na, K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. In Seminars in nephrology 25(5): 292-303. Branston NM, Ladds A, Symon L, Wang AD (1984) Comparison of the effects of ischaemia on early components of the somatosensory evoked potential in brainstem, thalamus, and cerebral cortex. Journal of Cerebral Blood Flow & Metabolism 4: 68-81. Brennan KC, Dahlem MA, Huang H, Miura RM (July 2014) Workshop on cortical spreading depression (CSD) and related neurological phenomena. (Fields Institute, University of Toronto). Bretón RR, Rodríguez JCG (2012) Excitotoxicity and oxidative stress in acute ischemic stroke. Stroke 8: 9. Brisson CD, Lukewich MK, Andrew RD (2013) A distinct boundary between the higher brain’s susceptibility to ischemia and the lower brain’s resistance. PloS One 8(11): e79589. 70 Brisson CD, Hsieh Y, Kim D, Jin AY, Andrew RD (2014) Brainstem neurons survive the identical ischemic stress that kills higher neurons: Insight to the persistent vegetative state. PloS One 9(5): e96585. Brisson CD, Andrew RD (2012) A neuronal population in hypothalamus that dramatically resists acute ischemic injury compared to neocortex. J Neurophysiol 108: 419-430. Brown M, Markus H, Oppenheimer S (2006) Stroke medicine. Neurol Sci 27: 205. Burchiel KJ (1984) Spontaneous impulse generation in normal and denervated dorsal root ganglia: Sensitivity to alpha-adrenergic stimulation and hypoxia. Exp Neurol 85: 257-272. Bureš J, Burešová O (1981) Cerebral [K+]e increase as an index of the differential susceptibility of brain structures to terminal anoxia and electroconvulsive shock. J Neurobiol 12: 211-220. Busl KM, Greer DM (2010) Hypoxic-ischemic brain injury: Pathophysiology, neuropathology and mechanisms. Neurorehabilitation 26: 5-13. Cheng Z, Lan D, Wu P, Zhu Y, Dong Y, Ma L, Zheng P (2008) Neurosteroid dehydroepiandrosterone sulphate inhibits persistent sodium currents in rat medial prefrontal cortex via activation of sigma-1 receptors. Exp Neurol 210: 128-136. Chhatwal G, Hessler H, Habermann E (1983) The action of palytoxin on erythrocytes and resealed ghosts. Naunyn Schmiedebergs Arch Pharmacol 323: 261-268. Cobos EJ, Entrena JM, Nieto FR, Cendan CM, Del Pozo E (2008) Pharmacology and therapeutic potential of sigma(1) receptor ligands. Curr Neuropharmacol 6:344-366. 71 Creveling CR, McNeal ET, Daly JW, Brown GB (1983) Batrachotoxin-induced depolarization and [3H]batrachotoxinin-a 20 alpha-benzoate binding in a vesicular preparation from guinea pig cerebral cortex. Mol Pharmacol 23: 350-358. Davies ML, Kirov SA, Andrew RD (2007) Whole isolated neocortical and hippocampal preparations and their use in imaging studies. J Neurosci Methods 166: 203-216. Deeds JR, Handy SM, White KD, Reimer JD (2011) Palytoxin found in palythoa sp. zoanthids (anthozoa, hexacorallia) sold in the home aquarium trade. Plos One 6(4): e18235. Denk W, Delaney K, Gelperin A, Kleinfeld D, Strowbridge B, Tank D, Yuste R (1994) Anatomical and functional imaging of neurons using 2-photon laser scanning microscopy. J Neurosci Methods 54: 151-162. Denk W, Strickler JH, Webb WW (1990) Two-photon laser scanning fluorescence microscopy. Science 248: 73-76. Denk W, Svoboda K (1997) Photon upmanship: Why multiphoton imaging is more than a gimmick. Neuron 18: 351-357. Denk W, Svoboda K (1997) Photon upmanship: Why multiphoton imaging is more than a gimmick. Neuron 18: 351-357. Deshpande JK, Wieloch T (1986) Flunarizine, a calcium entry blocker, ameliorates ischemic brain damage in the rat. Anesthesiology 64: 215-224. Dirnagl U, Iadecola C, Moskowitz MA (1999) Pathobiology of ischaemic stroke: An integrated view. Trends Neurosci 22: 391-397. 72 Dobretsov M, Stimers JR (2005) Neuronal function and alpha3 isoform of the Na/K-ATPase. Front Biosci 10: 2373-2396. Douglas HA, Callaway JK, Sword J, Kirov SA, Andrew RD (2011) Potent inhibition of anoxic depolarization by the sodium channel blocker dibucaine. J Neurophysiol 105: 1482-1494. Dreier JP (2011) The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Nat Med 17: 439-447. Falini A, Barkovich AJ, Calabrese G, Origgi D, Triulzi F, Scotti G (1998) Progressive brain failure after diffuse hypoxic ischemic brain injury: A serial MR and proton MR spectroscopic study. AJNR Am J Neuroradiol 19: 648-652. Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR (2000) Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron 28: 41-51. Fischer RS, Wu Y, Kanchanawong P, Shroff H, Waterman CM (2011) Microscopy in 3D: A biologist's toolbox. Trends Cell Biol 21: 682-691. Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE (2009) The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 323:934-937. Gadsby DC, Takeuchi A, Artigas P, Reyes N (2009) Peering into an ATPase ion pump with single-channel recordings. Philos Trans R Soc Lond B Biol Sci 364: 229-238. Ginsberg MD (1997) The new language of cerebral ischemia. Am J Neuroradiol 18: 1435-1446. 73 Gottron MA, Lo DC (2009) The Na/K-ATPase as a drug target for ischemic stroke. In New Strategies in Stroke Intervention. Pp. 129-151. Humana Press. Guitart X, Codony X, Monroy X (2004) Sigma receptors: Biology and therapeutic potential. Psychopharmacology 174: 301-319. Habermann E, Chhatwal G (1982) Ouabain inhibits the increase due to palytoxin of cation permeability of erythrocytes. Naunyn Schmiedebergs Arch Pharmacol 319: 101-107. Hadjikhani N, Sanchez Del Rio M, Wu O, Schwartz D, Bakker D, Fischl B, Kwong KK, Cutrer FM, Rosen BR, Tootell RB, Sorensen AG, Moskowitz MA (2001) Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci 98: 4687-4692. Hartings JA, Rolli ML, Lu XC, Tortella FC (2003) Delayed secondary phase of peri-infarct depolarizations after focal cerebral ischemia: Relation to infarct growth and neuroprotection. J Neurosci 23: 11602-11610. Hayashi T, Maurice T, Su T (2000) Ca2+ signaling via σ1-receptors: Novel regulatory mechanism affecting intracellular Ca2 concentration. J Pharmacol Exp Ther 293: 788-798. Hilgemann DW (2003) From a pump to a pore: How palytoxin opens the gates. Proceedings of the National Academy of Sciences 100: 386-388. Hirota K, Browne T, Appadu BL, Lambert DG (1997) Do local anaesthetics interact with dihydropyridine binding sites on neuronal L-type Ca2+ channels?. British journal of anaesthesia 78: 185-188. Hirsh JK, Wu CH (1997) Palytoxin-induced single-channel currents from the sodium pump synthesized by in vitro expression. Toxicon 35: 169-176. 74 Holler M, Dierking H, Dengler K, Tegtmeier F, Peters T (1986) Effect of flunarizine on extracellular ion concentration in the rat brain under hypoxia and ischemia. Acute brain ischemia, medical and surgical therapy (Eds. N. Battistina, P. Fiorani, R. Courbier, F. Plum, and C. Fieshi). Raven Press, New York, 229-236. Horisberger J (2004) Recent insights into the structure and mechanism of the sodium pump. Physiology 19: 377-387. Hossmann K (1994) Viability thresholds and the penumbra of focal ischemia. Ann Neurol 36: 557-565. Ishihara K, Akbar M, Sasa M (1999) Effects of OPC-24439, a sigma ligand, on neuronal activities in the hippocampus. Nihon Yakurigaku Zasshi 114: 204P-208P. Ishikawa M, Hashimoto K (2010) The role of sigma-1 receptors in the pathophysiology of neuropsychiatric diseases. J Receptor Ligand Channel Res 3:25-36. Jarvis CR, Anderson TR, Andrew RD (2001) Anoxic depolarization mediates acute damage independent of glutamate in neocortical brain slices. Cereb Cortex 11: 249-259. Jing J, Aitken PG, Somjen GG (1994) Interstitial volume changes during spreading depression (SD) and SD-like hypoxic depolarization in hippocampal tissue slices. J Neurophysiol 71: 2548-2551. Jones TH, Morawetz RB, Crowell RM, Marcoux FW, FitzGibbon SJ, DeGirolami U, Ojemann RG (1981) Thresholds of focal cerebral ischemia in awake monkeys. J Neurosurg 54:773-782. Karaki H, Nagase H, Ohizumi Y, Satake N, Shibata S (1988) Palytoxin‐induced contraction and release of endogenous noradrenaline in rat tail artery. Br J Pharmacol 95:183-188. 75 Katnik C, Guerrero WR, Pennypacker KR, Herrera Y, Cuevas J (2006) Sigma-1 receptor activation prevents intracellular calcium dysregulation in cortical neurons during in vitro ischemia. J Pharmacol Exp Ther 319: 1355-1365. Kaufmann AM, Firlik AD, Fukui MB, Wechsler LR, Jungries CA, Yonas H (1999) Ischemic core and penumbra in human stroke. Stroke 30: 93-99. Kume T, Nishikawa H, Taguchi R, Hashino A, Katsuki H, Kaneko S, Minami M, Satoh M, Akaike A (2002) Antagonism of NMDA receptors by σ receptor ligands attenuates chemical ischemia-induced neuronal death in vitro. Eur J Pharmacol 455: 91-100. Kuroda Y, Miyamoto K, Tanaka K, Maeda Y, Ishikawa J, Hinata R, Otaka A, Fujii N, Nakagawa T (2000) Interactions between local anesthetics and Na channel inactivation gate peptides in phosphatidylserine suspensions as studied by 1H-NMR spectroscopy. Chemical and Pharmaceutical Bulletin-Tokyo- 48: 1293-1298. Larsson HP (2013) What determines the kinetics of the slow afterhyperpolarization (sAHP) in neurons? Biophys J 104: 281-283. Lauritzen M (1994) Pathophysiology of the migraine aura: The spreading depression theory. Brain 117: 199-210. Leão AA (1944) Spreading depression of activity in the cerebral cortex. J Neurophysiol . (Doctoral Dissertation, Harvard University). Leão AA (1951) The slow voltage variation of cortical spreading depression of activity. Electroencephalogr Clin Neurophysiol 3: 315-321. 76 Leão AA (1947) Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol 10: 409-414. Leonard B (2004) Sigma receptors and sigma ligands: Background to a pharmacological enigma. Pharmacopsychiatry 37: 166-170. Lichtman JW, Magrassi L, Purves D (1987) Visualization of neuromuscular junctions over periods of several months in living mice. J Neurosci 7: 1215-1222. Lipton P (1999) Ischemic cell death in brain neurons. Physiol Rev 79: 1431-1568. Liu RR, Murphy TH (2009) Reversible cyclosporin A-sensitive mitochondrial depolarization occurs within minutes of stroke onset in mouse somatosensory cortex in vivo: A two-photon imaging study. J Biol Chem 284: 36109-36117. Luigetti M, Goldsberry GT, Cianfoni A (2012) Brain MRI in global hypoxia–ischemia: A map of selective vulnerability. Acta Neurol Belg 112: 105-107. Madry C, Haglerod C, Attwell D (2010) The role of pannexin hemichannels in the anoxic depolarization of hippocampal pyramidal cells. Brain 133: 3755-3763. Mainen Z, Maletic-Savatic M, Shi S, Hayashi Y, Malinow R, Svoboda K (1999) Two-photon imaging in living brain slices. Methods 18: 231-239. Malm WC (1999) Introduction to visibility. Cooperative Institute for Research in the Atmosphere, NPS Visibility Program, Colorado State University. Marchal G, Benali K, Iglesias S, Viader F, Derlon JM, Baron JC (1999) Voxel-based mapping of irreversible ischaemic damage with PET in acute stroke. Brain 122 (12): 2387-2400. 77 Marshall WH (1959) Spreading cortical depression of leao. Physiol Rev 39: 239-279. Martin RL, Lloyd HGE, Cowan AI (1994) The early events of oxygen and glucose deprivation: Setting the scene for neuronal death? Trends Neurosci 17: 251-257. Matsumoto RR, McCracken KA, Pouw B, Zhang Y, Bowen WD (2002) Involvement of sigma receptors in the behavioral effects of cocaine: Evidence from novel ligands and antisense oligodeoxynucleotides. Neuropharmacology 42: 1043-1055. Maurice T, Su T (2009) The pharmacology of sigma-1 receptors. Pharmacol Ther 124: 195-206. Moore RE, Scheuer PJ (1971) Palytoxin: A new marine toxin from a coelenterate. Science 172: 495-498. Moustafa R, Baron J (2008) Pathophysiology of ischaemic stroke: Insights from imaging, and implications for therapy and drug discovery. Br J Pharmacol 153: S44-S54. Müller M, Somjen GG (1998) Inhibition of major cationic inward currents prevents spreading depression-like hypoxic depolarization in rat hippocampal tissue slices. Brain Res 812: 1-13. Muller M, Somjen GG (2000) Na+ dependence and the role of glutamate receptors and Na+ channels in ion fluxes during hypoxia of rat hippocampal slices. J Neurophysiol 84: 18691880. Munzer JS, Daly SE, Jewell-Motz EA, Lingrel JB, Blostein R (1994) Tissue- and isoformspecific kinetic behavior of the na,K-ATPase. J Biol Chem 269: 16668-16676. 78 Murphy TH, Li P, Betts K, Liu R (2008) Two-photon imaging of stroke onset in vivo reveals that NMDA-receptor independent ischemic depolarization is the major cause of rapid reversible damage to dendrites and spines. J Neurosci 28: 1756-1772. Murray CJ, Lopez AD (1997) Global mortality, disability, and the contribution of risk factors: Global burden of disease study. The Lancet 349: 1436-1442. Nwaneshiudu A, Kuschal C, Sakamoto FH, Anderson RR, Schwarzenberger K, Young RC (2012) Introduction to confocal microscopy. J Invest Dermatol 132: e3. Obeidat AS, Andrew RD (1998) Spreading depression determines acute cellular damage in the hippocampal slice during oxygen/glucose deprivation. Eur J Neurosci 10: 3451-3461. Obeidat AS, Jarvis CR, Andrew RD (2000) Glutamate does not mediate acute neuronal damage after spreading depression induced by O2/glucose deprivation in the hippocampal slice. Journal of Cerebral Blood Flow & Metabolism 20: 412-422. Obrenovitch TP, Urenjak J (1997) Altered glutamatergic transmission in neurological disorders: From high extracellular glutamate to excessive synaptic efficacy. Prog Neurobiol 51: 39-87. Obrenovitch T (2001) Excitotoxicity in neurological disorders: An alternate viewpoint. In Neuroprotection: Basic and clinical aspects (Lo E, Scottsdale M, eds), pp353-377. Prominent Press. Obrenovitch T, Urenjak J, Zilkha E, Jay T (2000) Excitotoxicity in neurological disorders—the glutamate paradox. International Journal of Developmental Neuroscience 18: 281-287. Oda M, Yoshida A, Ikemoto Y (1992) Blockade by local anaesthetics of the single Ca2 ‐activated K channel in rat hippocampal neurones. Br J Pharmacol 105: 63-70. 79 Ozaki H, Nagase H, Urakawa N (1985) Interaction of palytoxin and cardiac glycosides on erythrocyte membrane and Na/K ATPase. European Journal of Biochemistry 152: 475-480. Pietrobon D, Moskowitz MA (2014) Chaos and commotion in the wake of cortical spreading depression and spreading depolarizations. Nature Reviews Neuroscience 15: 379-393. Prenen GH, Go KG, Postema F, Zuiderveen F, Korf J (1988) Cerebral cation shifts in hypoxicischemic brain damage are prevented by the sodium channel blocker tetrodotoxin. Exp Neurol 99: 118-132. Quirion R, Bowen WD, Itzhak Y, Junien JL, Musacchio J, Rothman RB, Tsung-Ping S, Tam SW, Taylor DP (1992) A proposal for the classification of sigma binding sites. Trends Pharmacol Sci 13: 85-86. Ragsdale DS, McPhee JC, Scheuer T, Catterall WA (1994) Molecular determinants of statedependent block of Na+ channels by local anesthetics. Science 265: 1724-1728. Ricchelli F, Beghetto C, Gobbo S, Tognon G, Moretto V, Crisma M (2003) Structural modifications of the permeability transition pore complex in resealed mitochondria induced by matrix-entrapped disaccharides. Arch Biochem Biophys 410: 155-160. Riobó P, Franco JM (2011) Palytoxins: Biological and chemical determination. Toxicon 57: 368375. Riobó P, Paz B, Franco JM, Vázquez JA, Murado MA, Cacho E (2008) Mouse bioassay for palytoxin. Specific symptoms and dose-response against dose–death time relationships. Food and Chemical Toxicology 46: 2639-2647. 80 Risher WC, Lee MR, Fomitcheva IV, Hess DC, Kirov SA (2011) Dibucaine mitigates spreading depolarization in human neocortical slices and prevents acute dendritic injury in the ischemic rodent neocortex. PloS One 6: e22351. Rose AM, Valdes R,Jr (1994) Understanding the sodium pump and its relevance to disease. Clin Chem 40: 1674-1685. Rossi DJ, Oshima T, Attwell D (2000) Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nature 403: 316-321. Rossini GP, Bigiani A (2011) Palytoxin action on the Na+K+-ATPase and the disruption of ion equilibria in biological systems. Toxicon 57: 429-439. Scheiner-Bobis G, Meyer zu Heringdorf D, Christ M, Habermann E (1994) Palytoxin induces K+ efflux from yeast cells expressing the mammalian sodium pump. Mol Pharmacol 45: 11321136. Senda T, Matsuno K, Okamoto K, Kobayashi T, Nakata K, Mita S (1996) Ameliorating effect of SA4503, a novel σ 1 receptor agonist, on memory impairments induced by cholinergic dysfunction in rats. Eur J Pharmacol 315: 1-10. Seshadri S, Beiser A, Kelly-Hayes M, Kase CS, Au R, Kannel WB, Wolf PA (2006) The lifetime risk of stroke: Estimates from the framingham study. Stroke 37: 345-350. Singhal AB, Lo EH, Dalkara T, Moskowitz MA (2011) Ischemic stroke: Basic pathophysiology and neuroprotective strategies. In Acute ischemic stroke Ischemic stroke: Basic pathophysiology and neuroprotective strategies. pp1-24. Springer Berlin Heidelberg. 81 Somjen GG (2004) Ions in the brain: Normal function, seizures, and stroke. Oxford University Press USA. Somjen GG (2002) Ion regulation in the brain: Implications for pathophysiology. Neuroscientist 8: 254-267. Somjen G, Müller M (2000) Potassium-induced enhancement of persistent inward current in hippocampal neurons in isolation and in tissue slices. Brain Res 885: 102-110. Somjen G, Aitken P, Czeh G, Herrearas O, Jing J, Young J (1992) Mechanisms of spreading depression: A review of recent findings and a hypothesis. Can J Physiol Pharmacol 70: S248S254. Somjen GG (2001) Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiological Reviews 81: 1065-1096. Stafstrom CE, Schwindt PC, Chubb MC, Crill WE (1985) Properties of persistent sodium conductance and calcium conductance of layer V neurons from cat sensorimotor cortex in vitro. J Neurophysiol 53:153-170. Stelzer EHK, Hell S, Lindek S, Stricker R, Pick R, Storz C, Ritter G, Salmon N (1994) Nonlinear absorption extends confocal fluorescence microscopy into the ultra-violet regime and confines the illumination volume. Opt Commun 104: 223-228. Svoboda K, Tank DW, Denk W (1996) Direct measurement of coupling between dendritic spines and shafts. Science-New York then Washington-: 716-718. Svoboda K, Denk W, Kleinfeld D, Tank DW (1997) In vivo dendritic calcium dynamics in neocortical pyramidal neurons. Nature 385: 161-165. 82 Svoboda K, Yasuda R (2006) Principles of two-photon excitation microscopy and its applications to neuroscience. Neuron 50: 823-839. Tanaka E, Yamamoto S, Inokuchi H, Isagai T, Higashi H (1999) Membrane dysfunction induced by in vitro ischemia in rat hippocampal CA1 pyramidal neurons. J Neurophysiol 81: 18721880. Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H (1997) Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. Journal of Neurophysiology 78: 891-902. Taylor CP, Meldrum BS (1995) Na channels as targets for neuroprotective drugs. Trends Pharmacol Sci 16: 309-316. Taylor CP, Weber ML, Gaughan CL, Lehning EJ, LoPachin RM (1999) Oxygen/glucose deprivation in hippocampal slices: Altered intraneuronal elemental composition predicts structural and functional damage. J Neurosci 19: 619-629. Teyler TJ (1980) Brain slice preparation: Hippocampus. Brain Res Bull 5: 391-403. Theer P, Hasan MT, Denk W (2003) Two-photon imaging to a depth of 1000 μm in living brains by use of a Ti: Al2O3 regenerative amplifier. Opt Lett 28: 1022-1024. Thompson RJ, Zhou N, MacVicar BA (2006) Ischemia opens neuronal gap junction hemichannels. Science 312: 924-927. Tosteson M (2000) Mechanism of action, pharmacology, and toxicology. Food Science and Technology-New York-Marcel Dekker-: 549-566. 83 Toyoshima C, Kanai R, Cornelius F (2011) First crystal structures of Na+/K+ ATPase: New light on the oldest ion pump. Structure 19: 1732-1738. Urenjak J, Obrenovitch TP (1996) Pharmacological modulation of voltage-gated Na+ channels: A rational and effective strategy against ischemic brain damage. Pharmacological Reviews 48: 21-67. Utzschneider DA, Kocsis JD, Waxman SG (1991) Differential sensitivity to hypoxia of the peripheral versus central trajectory of primary afferent axons. Brain Res 551: 136-141. Vinogradova L, Koroleva V, Bures J (1991) Re-entry waves of Leao's spreading depression between neocortex and caudate nucleus. Brain Res 538: 161-164. Wang D (2008) Neurotoxins from marine dinoflagellates: A brief review. Marine Drugs 6: 349371. Weber ML, Taylor CP (1994) Damage from oxygen and glucose deprivation in hippocampal slices is prevented by tetrodotoxin, lidocaine and phenytoin without blockade of action potentials. Brain Res 664: 167-177. White NS, Errington RJ (2005) Fluorescence techniques for drug delivery research: Theory and practice. Adv Drug Deliv Rev 57: 17-42. White SH, Brisson CD, Andrew RD (2012) Examining protection from anoxic depolarization by the drugs dibucaine and carbetapentane using whole cell recording from CA1 neurons. J Neurophysiol 107: 2083-2095. 84 Wilke RA, Mehta RP, Lupardus PJ, Chen Y, Ruoho AE, Jackson MB (1999) Sigma receptor photolabeling and sigma receptor-mediated modulation of potassium channels in tumor cells. J Biol Chem 274: 18387-18392. Wong BY, Coulter DA, Choi DW, Prince DA (1988) Dextrorphan and dextromethorphan, common antitussives, are antiepileptic and antagonize N-methyl-D-aspartate in brain slices. Neurosci Lett 85: 261-266. Woodruff TM, Thundyil J, Tang S, Sobey CG, Taylor SM, Arumugam TV (2011) Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol Neurodegener 6: 11. Wytrzes LM, Chatrian G, Shaw C, Wirch AL (1989) Acute failure of forebrain with sparing of brain-stem function: Electroencephalographic, multimodality evoked potential, and pathologic findings. Arch Neurol 46: 93-97. Xie Y, Zacharias E, Hoff P, Tegtmeier F (1995) Ion channel involvement in anoxic depolarization induced by cardiac arrest in rat brain. Journal of Cerebral Blood Flow & Metabolism 15: 587-594. Xie Y, Dengler K, Zacharias E, Wilffert B, Tegtmeier F (1994) Effects of the sodium channel blocker tetrodotoxin (TTX) on cellular ion homeostasis in rat brain subjected to complete ischemia. Brain Res 652: 216-224. Yamada A, Tanaka E, Niiyama S, Yamamoto S, Hamada M, Higashi H (2004) Protective actions of various local anesthetics against the membrane dysfunction produced by in vitro ischemia in rat hippocampal CA1 neurons. Neurosci Res 50: 291-298. 85 Yamamoto C, McIlwain H (1966) Electrical activities in thin sections from the mammalian brain maintained in chemically‐defined media in vitro. J Neurochem 13: 1333-1343. Yamamoto S, Tanaka E, Shoji Y, Kudo Y, Inokuchi H, Higashi H (1997) Factors that reverse the persistent depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. J Neurophysiol 78: 903-911. Yoshizumi M, Houchi H, Ishimura Y, Masuda Y, Morita K, Oka M (1991) Mechanism of palytoxin-induced Na+ influx into cultured bovine adrenal chromaffin cells: Possible involvement of Na+ H+ exchange system. Neurosci Lett 130: 103-106. Yuste R (2005) Fluorescence microscopy today. Nature Methods 2: 902-904. Zahler R, Zhang ZT, Manor M, Boron WF (1997) Sodium kinetics of Na,K-ATPase alpha isoforms in intact transfected HeLa cells. J Gen Physiol 110: 201-213. Zhang B, Zhang XY, Luo PF, Huang W, Zhu FP, Liu T, Du YR, Wu QH, Lu J, Xiu Y, Liu LN, Huang HP, Guo S, Zheng H, Zhang CX, Zhou Z (2012) Action potential-triggered somatic exocytosis in mesencephalic trigeminal nucleus neurons in rat brain slices. J Physiol 590: 753762. Zhang H, Cuevas J (2005) Sigma receptor activation blocks potassium channels and depresses neuroexcitability in rat intracardiac neurons. J Pharmacol Exp Ther 313: 1387-1396. 86