Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Axon guidance wikipedia , lookup
Multielectrode array wikipedia , lookup
Caridoid escape reaction wikipedia , lookup
Donald O. Hebb wikipedia , lookup
Biological neuron model wikipedia , lookup
Metastability in the brain wikipedia , lookup
Types of artificial neural networks wikipedia , lookup
State-dependent memory wikipedia , lookup
Development of the nervous system wikipedia , lookup
Neural coding wikipedia , lookup
Biochemistry of Alzheimer's disease wikipedia , lookup
Adult neurogenesis wikipedia , lookup
Stimulus (physiology) wikipedia , lookup
Neural oscillation wikipedia , lookup
Mirror neuron wikipedia , lookup
Limbic system wikipedia , lookup
Long-term depression wikipedia , lookup
Central pattern generator wikipedia , lookup
Environmental enrichment wikipedia , lookup
Apical dendrite wikipedia , lookup
Synaptogenesis wikipedia , lookup
Hippocampus wikipedia , lookup
Memory consolidation wikipedia , lookup
Long-term potentiation wikipedia , lookup
Chemical synapse wikipedia , lookup
Endocannabinoid system wikipedia , lookup
Spike-and-wave wikipedia , lookup
Circumventricular organs wikipedia , lookup
Nervous system network models wikipedia , lookup
Molecular neuroscience wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Neuroanatomy wikipedia , lookup
Premovement neuronal activity wikipedia , lookup
Nonsynaptic plasticity wikipedia , lookup
Feature detection (nervous system) wikipedia , lookup
Activity-dependent plasticity wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Eyeblink conditioning wikipedia , lookup
Optogenetics wikipedia , lookup
Pre-Bötzinger complex wikipedia , lookup
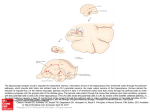
Learning-related postburst afterhyperpolarization reduction in CA1 pyramidal neurons is mediated by protein kinase A M. Matthew Oha,1, Bridget M. McKaya,b, John M. Powerc, and John F. Disterhofta,b,1 aDepartment of Physiology and bNeuroscience Program, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611-3008; and cQueensland Brain Institute, The University of Queensland, Brisbane, OLD 4072, Australia Edited by Richard F. Thompson, University of Southern California, Los Angeles, CA, and approved December 9, 2008 (received for review August 5, 2008) Learning-related reductions of the postburst afterhyperpolarization (AHP) in hippocampal pyramidal neurons have been shown ex vivo, after trace eyeblink conditioning. The AHP is also reduced by many neuromodulators, such as norepinephrine, via activation of protein kinases. Trace eyeblink conditioning, like other hippocampus-dependent tasks, relies on protein synthesis for consolidating the learned memory. Protein kinase A (PKA) has been shown to be a key contributor for protein synthesis via the cAMP-response element-binding pathway. Here, we have explored a potential involvement of PKA and protein kinase C (PKC) in maintaining the learning-related postburst AHP reduction observed in CA1 pyramidal neurons. Bath application of isoproterenol (1 !M), a "-adrenergic agonist that activates PKA, significantly reduced the AHP in CA1 neurons from control animals, but not from rats that learned. This occlusion suggests that PKA activity is involved in maintaining the AHP reduction measured ex vivo after successful learning. In contrast, bath application of the PKC activator, (–) indolactam V (0.2 !M), significantly reduced the AHP in CA1 neurons from both control and trained rats, indicating that PKC activity is not involved in maintaining the AHP reduction at this point after learning. hippocampus ! protein kinase C ! trace eyeblink T he postburst afterhyperpolarization (AHP) has been repeatedly demonstrated ex vivo to be reduced in hippocampal pyramidal neurons after hippocampus-dependent learning, such as the trace eyeblink conditioning (EBC) task (1). Trace EBC is a hippocampus-dependent task (2–4) that, like others (5, 6), requires protein synthesis for learning and consolidation of the memory (7), yet the molecular cascade that underlies this learning-related AHP alteration after trace EBC has not been studied or identified. The postburst AHP is predominantly a Ca2!-dependent K! current with 2 distinct components (8–10). The SK2 channels underlie the apamin-sensitive medium AHP (11–13) and have been shown to be intimately involved in regulating synaptic plasticity in dendritic spines along with NMDA receptors (14). The channel underlying the later, slow AHP has yet to be discovered; however, it is thought to be localized to the apical and basal dendrites in close proximity to the soma (15, 16). Because of its somatic localization, the slow AHP may play a significant role in the final somatic integration of synaptic inputs. Unless specified, we will be referring to both the medium and slow components of the AHP throughout the text. Activation of protein kinases via various neuromodulators reduces the AHP (10, 17). Specifically, activation of cholinergic and metabotropic glutamate receptors have been shown to reduce the AHP via protein kinase C (PKC) and Ca2!/calmodulin-dependent protein kinase II (CaMKII) (18–22). Activation of monoamine receptors reduces the AHP via protein kinase A (PKA) (21, 23). In addition to reducing the AHP, all 3 kinases have been implicated in long-term potentiation (LTP), with PKC and CaMKII being involved in the early, induction phase and 1620 –1625 ! PNAS ! February 3, 2009 ! vol. 106 ! no. 5 PKA demonstrated to be crucial for the late, protein synthesisdependent phase (6, 24–26). One of the signaling pathways that leads to protein synthesis involves PKA-mediated activation of MAPK, and subsequently, cAMP-response element-binding proteins (CREB) (6, 25, 27). Transgenic mice with a constitutively active form of CREB have recently been shown to have CA1 pyramidal neurons with significantly reduced AHPs (28). Interestingly, EBC is also significantly impaired by disruption of the cAMP/PKA pathway in the cerebellum (29), a structure known to be critically involved in learning this task (30). PKC levels in the hippocampus have been shown to be modulated by various learning tasks. Increase in membraneassociated PKC has been demonstrated in the hippocampus and cerebellum of rabbits after learning EBC (31–33). However, a decrease in membrane-associated PKC has also been shown in the hippocampus after spatial learning in rats and mice (34–36). PKC activation is also thought to underlie odor-discrimination learning, as a PKC activator occluded the learning-related AHP reduction in piriform cortical neurons evaluated ex vivo (37). Although PKC activation has been shown to be capable of activating MAPK (38), numerous studies have demonstrated that the cAMP/PKA pathway activation in the hippocampus is also essential for long-term memory and consolidation of hippocampus-dependent tasks (5, 6, 25). It remains to be determined if PKC and PKA are involved in maintaining the learningrelated AHP reduction in CA1 hippocampal pyramidal neurons after trace EBC. We report here that learning the trace EBC task occludes the PKA-, but not PKC-mediated AHP reduction in CA1 pyramidal neurons at a timepoint after rats have achieved a behavioral criterion indicating that they have learned trace EBC well. This occlusion strongly suggests that PKA activity and its associated signaling cascade are involved in maintaining the learningrelated AHP reduction observed in CA1 neurons ex vivo at a timepoint when the initial consolidation of the memory and related protein synthesis have occurred. Results Postburst AHP Is Reduced in CA1 Neurons from Rats that Learned Trace EBC. All trained rats used for the biophysical experiments reached a minimum of 60% conditioned responses (CRs) by the final training session and exhibited significantly more CRs than the pseudoconditioned animals throughout the training sessions Author contributions: M.M.O. and J.F.D. designed research; M.M.O. and B.M.M. performed research; J.M.P. contributed new reagents/analytic tools; M.M.O. and B.M.M. analyzed data; and M.M.O., B.M.M., and J.F.D. wrote the paper. The authors declare no conflict of interest. This article is a PNAS Direct Submission. 1To whom correspondence may be addressed. E-mail: [email protected] or [email protected]. © 2009 by The National Academy of Sciences of the USA www.pnas.org"cgi"doi"10.1073"pnas.0807708106 (Fig. 1B). The day after the last training session, postburst AHP was measured from visually identified CA1 pyramidal neurons in whole-cell current clamp mode using a 50-Hz train of 8 antidromic action potentials (APs). Similar to other studies that examined the AHP after trace EBC (39–41), CA1 pyramidal neurons from trained animals had significantly reduced AHPs ex vivo (Fig. 2). The AHP peak amplitude (see Fig. 2B) (F2,105 " 9.919, P " 0.0001), the AHP amplitude measured 1 sec after the end of the last AP (see Fig 2C) (1 sec AHP: F2,105 " 6.688, P " 0.0018) and the integrated AHP area (see Fig. 2D) (F2,105 " 6.607, P " 0.0020) from trained rats were significantly smaller than those from pseudoconditioned and naïve animals. Additionally, nearly 30% of CA1 neurons from trained animals had AHP measures smaller than one standard deviation from the mean of the naïve neurons (see Fig. 2 B–D). Similar to previous reports, the AHP measurements from pseudoconditioned and naïve rats were nearly identical (Fisher’s PLSD p’s all #0.2), and thus, were combined as controls for the rest of the analyses. No significant learning-related differences were observed for other membrane properties (Table 1). Learning Occluded the PKA-Induced AHP Reduction. Isoproterenol (Iso) is a !-adrenergic agonist that is commonly used to activate PKA (21, 42, 43) and has been used to suppress the postburst AHP (21, 39). Thus, we tested for the potential PKA involvement in maintaining the learning-related AHP reduction by changing the perfusate to an aCSF with 1 "M Iso and repeating the biophysical measurements after the baseline measurements were recorded. We chose 1 "M Iso because we wanted to reduce, not abolish, the postburst AHP. Repeated ANOVA, using data from all of the neurons in both training groups to which Iso was applied, revealed that Iso significantly reduced the AHP in CA1 neurons from control animals and made these values very similar to those from trained animals (Fig. 3). Iso had no impact on the AHP measures in CA1 neurons from trained rats (see Fig. 3). To verify that Iso was reducing the AHP via a PKA-mediated mechanism, we measured the AHP before bath application of the PKA inhibitor H 89 (5–10 "M), 10 min after H 89, and 10 min after H 89 and Iso in a few neurons (n " 5) from control animals. H 89 reduced the peak AHP (6.57 $ 1.25 to 5.78 $ 1.24 mV; paired t test, P % 0.05) but did not affect the 1 sec AHP nor Oh et al. NEUROSCIENCE Fig. 1. Young adult F344xBN rats readily learn trace EBC. (A) Rats were given 2 training sessions a day over 3 days. Hippocampal slices and biophysical recordings were made the day after the fifth training session. (B) Trained (Trace, n " 18) rats exhibited significantly more CRs than pseudoconditioned (Pseudo, n " 10) rats during all training sessions (repeated ANOVA: F(1,5,130) " 10.729, P % 0.0001; unpaired t test: P % 0.05 for session 1, p’s %0.0005 for sessions 2–5). Additionally, the trained rats exhibited greater CRs over the training sessions (F4,68 " 13.738, P % 0.0001), whereas the pseudoconditioned rats remained unchanged throughout the 5 sessions (F4,36 " 0.743, P " 0.569). There was no difference between the groups in the spontaneous blinks during the habituation session (P # 0.4). Fig. 2. Postburst AHP in CA1 pyramidal neurons is significantly reduced after learning trace EBC. (A) Examples of antidromically evoked AHP are illustrated. The APs have been truncated for illustration purposes. (B–D Left) Learning had a significant impact on the peak AHP amplitude (F2,105 " 9.919, P " 0.0001), amplitude measured 1 sec after the last AP (F2,105 " 6.688, P " 0.0018), and integrated AHP area (F2,105 " 6.607, P " 0.0020). All 3 measures were significantly reduced in CA1 neurons from trained rats. (B–D Right) In addition to the reductions, the distribution of the AHP measures were dramatically shifted to smaller values than one standard deviation from the mean of the naïve neurons. Of the CA1 neurons, 51% (21/41) from trained rats had smaller AHP peak amplitudes as compared to 15% and 25% of CA1 neurons from naïve (7/47) and pseudoconditioned (5/20) rats. Forty-four percent of CA1 neurons (18/41) from trained rats had smaller 1 sec AHP amplitude as compared to 6% and 10% from naïve (3/47) and pseudoconditioned (2/20) rats. Finally, 42% of CA1 neurons from trained (17/41) rats had smaller AHP area as compared to 9% and 10% from naïve (4/47) and pseudoconditioned (2/20) rats. N, naïve; P, pseudoconditioned; T, trace EBC. Numbers in parentheses represent the number of neurons recorded from the respective groups. Fisher’s PLSD *P % 0.05, **P % 0.001, ***P % 0.0001. the AHP area (p’s #0.8). More importantly, H 89 prevented Iso from reducing the AHP measures (p’s #0.11). The occluding effect of learning on the action of Iso strongly suggests that the PNAS ! February 3, 2009 ! vol. 106 ! no. 5 ! 1621 Table 1. Basic membrane properties of recorded CA1 neurons Duration (s) Latency (ms) IR (M') Sag (mV) RMP (mV) Vh (mV) Naïve (n " 47) Pseudo (n " 20) Trace (n " 41) 3.93 $ 0.13 112.5 $ 7.3 65.1 $ 2.8 5.87 $ 0.22 (68.0 $ 0.8 (68.3 $ 0.2 4.04 $ 0.20 145.7 $ 14.5 74.3 $ 4.1 6.27 $ 0.41 (67.2 $ 1.0 (68.1 $ 0.3 3.55 $ 0.19 119.0 $ 8.6 74.8 $ 3.6 5.48 $ 0.21 (69.0 $ 0.6 (68.0 $ 0.2 Note: Duration, AHP duration; Latency, latency to AHP peak amplitude; IR, input resistance; RMP, resting membrane potential; Vh, membrane holding potential. PKA signaling cascade is involved in maintaining the learningrelated AHP reduction and is active for at least one day ex vivo after the last training session. Learning Did not Occlude PKC-Mediated AHP Reduction. The post- burst AHP is reduced by various PKC activators (17, 19, 21). We chose to use 0.2 "M (–) indolactam V (Indo), as Wheal and colleagues effectively demonstrated that this concentration would reduce &50% of the slow AHP current (21). Thus, after the initial baseline measurements, Indo was added to the perfusate, and the biophysical properties were remeasured. Indo significantly reduced the AHP in neurons from both the control and trained groups (Fig. 4). Our analyses did not reveal a significant drug-by-behavior interaction (repeated ANOVAs for peak AHP, 1 sec AHP, and AHP area: F’s %2.001, P’s #0.17). Indo did significantly reduce the peak AHP amplitude, 1 sec AHP, and AHP area (see Fig. 4, repeated ANOVAs comparing initial measures vs. post Indo) (F1,22 # 43, P’s %0.0001). Thus, unlike the PKA activator Iso, PKC activation was not occluded from further reducing the AHP in CA1 neurons from animals that learned the trace EBC task. Discussion The main finding of the present study is that learning the hippocampus-dependent trace EBC task occluded the PKAmediated AHP reduction in CA1 pyramidal neurons. Additionally, the learning-related AHP reduction in CA1 neurons was again demonstrated ex vivo. This reproducible AHP reduction in CA1 neurons after hippocampus-dependent learning establishes it as a cellular hallmark of learning-related intrinsic plasticity. Thus, the question remains, how is PKA recruited to cause or maintain this cellular feature of learning? PKA is a cAMP-dependent protein kinase that has been extensively studied for its role in the late phase of long-term potentiation (L-LTP) and long-term memory (6, 25, 44). PKA has also been shown to effectively reduce the AHP in hippocampal pyramidal neurons (23, 45). Recently, Lin et al. (46) demonstrated that activated PKA induces internalization of the apamin-sensitive AHP channels, SK2, in the dendritic spines of hippocampal neurons. In addition, compounds that reduce the AHP facilitate LTP induction (15, 47–49). Thus, an interesting dynamic exists between the two: small AHP facilitates LTP, and a large AHP impairs LTP. Therefore, it is very possible that PKA activation bridges the AHP reduction and LTP facilitation. Both L-LTP and long-term memory consolidation of tasks including trace EBC have been shown to be protein synthesisdependent (6, 7, 25, 27). There are multiple ways of triggering protein synthesis, including the cAMP/PKA pathway (Fig. 5) (6). Furthermore, transgenic mice that express a dominant negativeform of the PKA regulatory subunit (50) or that have target mutations of CREB (51) have significant long-term memory deficits on the spatial water maze and contextual fear1622 ! www.pnas.org"cgi"doi"10.1073"pnas.0807708106 Fig. 3. Learning occluded the PKA mediated AHP reduction. Examples of 1 "M Iso’s effect on a CA1 neuron from a naïve (A) and a trace EBC (B) rat are illustrated. Note that Iso reduced the postburst AHP without abolishing it in the CA1 neuron from the naïve animal. The APs have been truncated for illustration purposes. (C) Significant drug-by-behavior interactions were revealed with repeated ANOVAs for the peak AHP amplitude (F1,30 " 4.241, P " 0.0482), 1 sec AHP (F1,30 " 6.314, P " 0.0176), and AHP area (F1,30 " 7.092, P " 0.0123). Further analyses revealed that Iso (1 "M) reduced the peak AHP (8.37 $ 0.61 to 6.67 $ 0.79 mV, P % 0.005), 1 sec AHP and AHP area measures in CA1 neurons (n " 17) from control rats. Iso did not have an impact on the peak (5.46 $ 0.52 to 5.01 $ 0.70mV, P # 0.12), 1 sec AHP (P # 0.60) and area (P # 0.33) measures in CA1 neurons (n " 15) from trained rats. Thus, the resulting post-Iso AHP measures were nearly identical between the 2 groups (unpaired t test: p’s #0.12). The impact of Iso on AHP peak amplitude is similar to that shown for the AHP area, and thus is not illustrated. Pre, initial baseline measures; Iso, post isoproterenol measures. Unpaired t test: *P % 0.01, **P % 0.005. Paired t test: †P % 0.005, ††P % 0.001. conditioning tasks. Interestingly, EBC is also significantly impaired by disruption of the cAMP/PKA pathway in the cerebellum (29), a structure known to be critically involved in learning this task (30). In addition, extinction of delay EBC, which depends on an intact hippocampus (52), is impaired by lesion of the locus coeruleus (53). CREB activation and the AHP have been linked in a study by Barco and colleagues, who demonstrated that activation of CREB causes a significant AHP reduction in CA1 pyramidal neurons (28). Thus, it is possible Oh et al. Fig. 4. Learning did not occlude the PKC-mediated AHP reduction. Examples of 0.2 "M (–) indolactam V’s (Indo) effect on CA1 neurons from naïve (A) and trace EBC (B) rats are illustrated. (C) Indolactam significantly reduced the AHP peak amplitude, amplitude measured at 1 sec (1s), and the integrated area (Area) in CA1 neurons from both the control and trained rats. The impact of Indo on AHP peak amplitude is similar to that shown for the AHP area, and thus is not illustrated. Pre, initial baseline measures; Indo, post indolactam measures. Unpaired t test: *P % 0.05, **P % 0.01. Paired t test: †P % 0.05, ††P % 0.01, †††P % 0.001. that the activation of the PKA signaling pathway led to the activation of CREB, which could help maintain the learningrelated AHP reduction in hippocampal pyramidal neurons after the subject has learned a hippocampus-dependent task, such as trace EBC. In addition to having the same protein synthesis requirement, LTP and trace EBC have another interesting parallel. DelgadoGarcia and colleagues have recently demonstrated that inducing LTP in the CA3-CA1 synapse just before or concurrently with training mice on trace EBC prevents learning; whereas, LTP induced a month before training has no effect on learning (54). Furthermore, learning trace EBC enhanced synaptic transmission between the CA3-CA1 synapses (55) and prevented subsequent LTP (54). Our present results do not rule out the involvement of other signaling cascades, such as those mediated by PKC. In the present study, we measured the AHP after the rats had consolOh et al. idated the memory for trace EBC (see Fig. 1) and found that the PKC activator, (–) Indo, reduced the AHP similarly in CA1 neurons from both trained and control animals. The slow AHP in neurons from both trained and control rats was reduced by &50% with 0.2 "M Indo, as previously described by Wheal and colleagues (21) (see Fig. 4). Notably, 1 "M Iso reduced the slow AHP in neurons from control rats by 40% (see Fig. 3). The similar slow AHP reduction by both compounds in neurons from control rats further suggests that the differential involvement of PKC and PKA that we observed is not a nonspecific consequence of differentially effective doses of drugs used to activate the 2 pathways. However, this does not rule out PKC involvement in the learning-related AHP reduction. It is possible that PKC activity may have played a critical role in the early phase of learning and the resulting AHP changes, similar to that observed for LTP induction (5, 56). These other signaling pathways could be involved in modulating intrinsic plasticity and memory formation in other regions of the brain. For example, the AHP reduction in layer II piriform cortical neurons after odor discrimination learning (57) has been shown to be mediated by PKC (37). It is possible that PKA is not a viable option for maintaining the learning-related AHP reduction after odor discrimination tasks, as the adenylyl cyclase (AC) critically involved for odor learning has been shown to be tightly temporally regulated for successfully learning this task (58). Cui et al. (59) recently demonstrated that a temporally specific increase and decrease in cAMP levels in the olfactory bulb are important for successfully learning odor preference. These temporally regulated cAMP levels could lead to a tight regulation of PKA activity, making PKC activity a better regulator of long-lasting memory for odor-discrimination tasks. Learning may not engage all kinase cascades in every cell in the hippocampus. Indeed, a detailed study of the cAMP/PKA and ERK/MAP kinase signal cascade revealed that only about 10% of cells in CA1 show coactivation of AC, PKA, MAPK, and the CREB kinase MSK1 after learning a contextual fearPNAS ! February 3, 2009 ! vol. 106 ! no. 5 ! 1623 NEUROSCIENCE Fig. 5. Highly simplified schematic of PKA involvement in the AHP reduction after learning. Under normal condition, the Ca2! influx into the cytosol, via the L-type Ca2! channel and internal Ca2! stores, activates the AHP. During learning, activation of monoamine receptors (e.g., noradrenaline, dopamine, serotonin) along with a rise in internal Ca2! levels activate adenylyl cyclase, causing an increase in cAMP levels leading to PKA activation (reviewed in (5, 6, 25, 27). Activated PKA leads to MAPK/ERK activation of the CREB signaling pathway, ultimately leading to gene transcription and translation. The activated CREB and PKA also lead to a reduction in the AHP. This reduced AHP channel activity, in turn, would lead to a longer duration depolarization, resulting in a longer time window for the continuation of the cAMP/PKA signaling cascade. conditioning task (60). Similarly, not all hippocampal pyramidal neurons undergo the dramatic AHP reduction after learning, as reported here (see Fig. 2 B–D) and previously (40, 41). In addition to reducing the AHP, PKA activity has also been implicated in reducing another potassium current, the Kv4.2mediated A-type potassium current (61, 62). Recently, Hoffman and colleagues demonstrated that PKA activation led to Kv4.2 channels being removed from the surface of the cell membrane (62), similar to the SK2 apamin-sensitive AHP channels (46). The internalization of the Kv4.2 via PKA activity results in a reduced A-type potassium current (62). Given the similar PKA action on the SK2 and Kv4.2 channels, it is possible that the enhanced neuronal excitability observed in hippocampal pyramidal neurons ex vivo after learning (via reductions in both the medium and slow AHPs) is a result of internalization of both the apamin-sensitive SK2 and channels that underlie the slow AHP by PKA activity. However, it will not be possible to confirm this until the molecular identity of the slow AHP is discovered. Age-related enlargement of the postburst AHP in hippocampal pyramidal neurons have been demonstrated and postulated to contribute to the normal aging-related learning impairments (63, 64). Age-related spatial learning impairments have been linked to reduced levels of AC type 1 in the hippocampus of aged mice (65) and can be ameliorated by compounds that enhance cAMP/PKA signaling (66). Interestingly, !-amyloid has been shown to inactivate PKA in hippocampal neurons (67). AC type 1 has also been shown to be reduced in hippocampus of Alzheimer’s disease patients (68). Taken together, the normal age- and Alzheimer disease-related learning impairments may result from not only the enlarged AHP with aging, but also from the lack of proper AHP modulation by PKA. In summary, the present results strongly suggest that PKA activity is one mechanism by which the learning-related AHP reduction is maintained in CA1 pyramidal neurons. It is noteworthy that L-LTP, protein synthesis, long-term memory, and now maintenance of learning-related AHP reduction all have PKA activity as a common substrate. Given this and the inverse relationship between AHP and LTP, the molecular mechanisms and signaling-cascades that underlie the learning-related AHP reduction may be similar to that found for LTP. Materials and Methods training session consisted of 30 CS-US pairings with an intertrial interval of 20 to 40 sec (30 sec average). A pseudoconditioning session consisted of explicitly unpaired presentation of 30 CS and 30 US with a 10 to 20 sec intertrial interval (15 sec average). Whole-cell current clamp recordings were made from visually identified CA1 pyramidal neurons using an Axoclamp 2A amplifier, as previously described (69). Briefly, 1 day after the last training session, hippocampal slices (300 "m) were cut using a Leica vibratome in an ice-cold ACSF (124 NaCl, 26 NaHCO3,3 KCl, 2.4 CaCl2, 2 MgSO4, 1.25 NaH2PO4, and 25 D-glucose, gassed with 95% O2-5% CO2). The slices were incubated for &30 min at 34 °C and allowed to cool down to room temperature for at least another 30 min before being transferred to a submersion chamber on an upright Leica DMLFS microscope. The patch electrodes (3– 6M') were filled with (in mM) 120 KMeSO4, 10 KCl, 10 Hepes, 4 Mg2ATP, 0.4 NaGTP, 10 Na2 phosphocreatine, 0.5% neurobiotin, pH adjusted to 7.3 with KOH, 280 $ 5 mOsm. No correction was made for a &10-mV liquid junction potential. All measurements were made #5 min after membrane rupture to allow for adequate solution equilibration with the neuron held near – 67 mV (65 ( 70 mV) at 34 to 35 °C. A potential error in membrane potential recording may occur if the current injection and membrane potential recording are performed with the same patch electrode (70). Thus, AHP measurements were obtained by a train of 8 antidromic pulses (50 Hz) using a concentric bipolar stimulating electrode placed on the alveus near the recorded CA1 neuron. Similar AHP values were observed with a train of 8 APs (50 Hz) evoked by direct somatic current injections (data not shown). More importantly, the observed AHP correlated well with learning irrespective of the mode used to trigger the AHP. Thus, a train of 8 APs at 50 Hz was chosen to ensure a sufficiently large AHP to easily observe learning-related AHP changes, as previous study illustrated that the size of the AHP is dependent on the frequency and number of APs used to evoke it (71). Included in the aCSF were 50 "M D-AP5, 10 "M NBQX, and 12.5 "M gabazine to eliminate NMDA-, AMPA-, and GABAA-mediated synaptic responses. Biophysical measures were repeated after a 10-min bath application of drug compounds used throughout the experiments. A few experiments were carried out to ensure that neither switching the drug perfusate line nor the duration of the biophysical recordings significantly impacted the AHP measures (n " 5; paired t-tests of peak AHP, 1 sec AHP, AHP area, AHP duration, sag, and holding potential; p’s #0.13). Notably, the input resistance did increase over the duration of the recordings (P % 0.05), similar to that previously reported for internal pipette solutions containing KMeSO4 (72). However, no significant drug-by-group interaction was observed for the input resistance measures with any of the compounds used (F’s %4.2, P’s #0.05). Thus, it is unlikely that the effects of Iso or Indo on the AHP measures were significantly impacted by the increase in input resistance over time. D-AP5, NBQX, gabazine, Iso, and H 89 were obtained from Tocris Bioscience. KMeSO4 was obtained from ICN. (–) Indo was obtained from Axxora LLC. All other compounds were obtained from Sigma. Subjects were young adult (2–3 months old) male F1 hybrid Fischer 344 ) Brown Norway rats that were group housed (3 per cage) in a climatecontrolled vivarium on a 12:12 light:dark cycle with ad libitum access to food and water. Animal care and experimental protocols were done following National Institutes of Health guidelines and approved by the Northwestern University Institutional Animal Care and Use Committee. Trace EBC was performed as previously described (39). Briefly, using sterile surgical techniques and under isofluorane inhalation anesthesia, the rats were implanted with a headgear to deliver the airpuff and to record EMG activity from the upper right eyelid, and allowed to recover for a minimum of 3 days before training. Fig. 1 illustrates the training schedule. Rats were trained individually in a sound-attenuating chamber with a tone conditioned stimulus (CS; 250 ms, 8 kHz, 85 dB free field) and a corneal airpuff unconditioned stimulus (US; 100 ms, 4.5 psi) separated by a 250-ms blank trace interval. A Data Acquisition and Analysis. The biophysical recordings and analyses were performed blind to the behavioral status of the animal. The behavioral and biophysical data were digitized and analyzed on-line using custom software routines written in LabVIEW (National Instruments). Complete analyses were performed off-line using procedures developed with LabVIEW. Statistical analyses were performed using StatView. Significant main effects were evaluated using Fisher’s PLSD post hoc tests. All data are reported as the means $ SE. 1. Disterhoft JF, Oh MM (2006) Learning, aging and intrinsic neuronal plasticity. Trends Neurosci 29:587–599. 2. Kim JJ, Clark RE, Thompson RF (1995) Hippocampectomy impairs the memory of recently, but not remotely, acquired trace eyeblink conditioned responses. Behav Neurosci 109:195–203. 3. McGlinchey-Berroth R, Carrillo MC, Gabrieli JD, Brawn CM, Disterhoft JF (1997) Impaired trace eyeblink conditioning in bilateral, medial-temporal lobe amnesia. Behav Neurosci 111:873– 882. 4. Moyer JR, Jr, Deyo RA, Disterhoft JF (1990) Hippocampectomy disrupts trace eye-blink conditioning in rabbits. Behav Neurosci 104:243–252. 5. Izquierdo I, Medina JH (1997) Memory formation: the sequence of biochemical events in the hippocampus and its connection to activity in other brain structures. Neurobiol Learn Mem 68:285–316. 6. Kandel ER (2001) The molecular biology of memory storage: a dialogue between genes and synapses. Science 294:1030 –1038. 7. Inda MC, Delgado-Garcia JM, Carrion AM (2005) Acquisition, consolidation, reconsolidation, and extinction of eyelid conditioning responses require de novo protein synthesis. J Neurosci 25:2070 –2080. 8. Sah P, Faber ES (2002) Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol 66:345–353. 9. Storm JF (1990) Potassium currents in hippocampal pyramidal cells. Prog Brain Res 83:161–187. 10. Wu WW, Oh MM, Disterhoft JF (2002) Age-related biophysical alterations of hippocampal pyramidal neurons: implications for learning and memory. Ageing Res Rev 1:181–207. 11. Stocker M (2004) Ca2!-activated K! channels: molecular determinants and function of the SK family. Nat Rev Neurosci 5:758 –770. 1624 ! www.pnas.org"cgi"doi"10.1073"pnas.0807708106 ACKNOWLEDGMENTS. The authors thank John Linardakis and Elizabeth Matthews for help with the behavioral experiments, and Elizabeth Matthews for critical reading and helpful discussion of the manuscript. This work was supported by National Institutes of Health Grants AG08796 and AG20506 (to J.F.D.). Oh et al. Oh et al. 43. Huang YY, Kandel ER (1996) Modulation of both the early and the late phase of mossy fiber LTP by the activation of beta-adrenergic receptors. Neuron 16:611– 617. 44. Bailey CH, Giustetto M, Huang YY, Hawkins RD, Kandel ER (2000) Is heterosynaptic modulation essential for stabilizing Hebbian plasticity and memory? Nat Rev Neurosci 1:11–20. 45. Lancaster B, Hu H, Gibb B, Storm JF (2006) Kinetics of ion channel modulation by cAMP in rat hippocampal neurones. J Physiol 576:403– 417. 46. Lin MT, Lujan R, Watanabe M, Adelman JP, Maylie J (2008) SK2 channel plasticity contributes to LTP at Schaffer collateral-CA1 synapses. Nat Neurosci 11:170 –177. 47. Cohen AS, Coussens CM, Raymond CR, Abraham WC (1999) Long-lasting increase in cellular excitability associated with the priming of LTP induction in rat hippocampus. J Neurophysiol 82:3139 –3148. 48. Kramar EA, et al. (2004) A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci 24:5151– 5161. 49. Norris CM, Halpain S, Foster TC (1998) Reversal of age-related alterations in synaptic plasticity by blockade of L-type Ca2! channels. J Neurosci 18:3171–3179. 50. Abel T, et al. (1997) Genetic demonstration of a role for PKA in the late phase of LTP and in hippocampus-based long-term memory. Cell 88:615– 626. 51. Bourtchuladze R, et al. (1994) Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79:59 – 68. 52. Schmaltz LW, Theios J (1972) Acquisition and extinction of a classically conditioned response in hippocampectomized rabbits (Oryctolagus cuniculus). J Comp Physiol Psychol 79:328 –333. 53. McCormick DA, Thompson RF (1982) Locus coeruleus lesions and resistance to extinction of a classically conditioned response: involvement of the neocortex and hippocampus. Brain Res 245:239 –249. 54. Madronal N, Delgado-Garcia JM, Gruart A (2007) Differential effects of long-term potentiation evoked at the CA3 CA1 synapse before, during, and after the acquisition of classical eyeblink conditioning in behaving mice. J Neurosci 27:12139 –12146. 55. Gruart A, Munoz MD, Delgado-Garcia JM (2006) Involvement of the CA3-CA1 synapse in the acquisition of associative learning in behaving mice. J Neurosci 26:1077–1087. 56. Huang YY, Kandel ER (1994) Recruitment of long-lasting and protein kinase A-dependent long-term potentiation in the CA1 region of hippocampus requires repeated tetanization. Learn Mem 1:74 – 82. 57. Saar D, Grossman Y, Barkai E (1998) Reduced after-hyperpolarization in rat piriform cortex pyramidal neurons is associated with increased learning capability during operant conditioning. Eur J Neurosci 10:1518 –1523. 58. Wang H, Storm DR (2003) Calmodulin-regulated adenylyl cyclases: cross-talk and plasticity in the central nervous system. Mol Pharmacol 63:463– 468. 59. Cui W, Smith A, Darby-King A, Harley CW, McLean JH (2007) A temporal-specific and transient cAMP increase characterizes odorant classical conditioning. Learn Mem 14:126 –133. 60. Sindreu CB, Scheiner ZS, Storm DR (2007) Ca2!-stimulated adenylyl cyclases regulate ERK-dependent activation of MSK1 during fear conditioning. Neuron 53:79 – 89. 61. Hoffman DA, Magee JC, Colbert CM, Johnston D (1997) K! channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature 387:869 – 875. 62. Hammond RS, Lin L, Sidorov MS, Wikenheiser AM, Hoffman DA (2008) Protein kinase A mediates activity-dependent Kv4.2 channel trafficking. J Neurosci 28:7513–7519. 63. Disterhoft JF, Oh MM (2006) Pharmacological and molecular enhancement of learning in aging and Alzheimer’s disease. J Physiol Paris 99:180 –192. 64. Thibault O, Gant JC, Landfield PW (2007) Expansion of the calcium hypothesis of brain aging and Alzheimer’s disease: minding the store. Aging Cell 6:307–317. 65. Mons N, Segu L, Nogues X, Buhot MC (2004) Effects of age and spatial learning on adenylyl cyclase mRNA expression in the mouse hippocampus. Neurobiol Aging 25:1095–1106. 66. Bach ME, et al. (1999) Age-related defects in spatial memory are correlated with defects in the late phase of hippocampal long-term potentiation in vitro and are attenuated by drugs that enhance the cAMP signaling pathway. Proc Natl Acad Sci USA 96:5280 –5285. 67. Vitolo OV, et al. (2002) Amyloid beta-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci USA 99:13217–13221. 68. Yamamoto M, et al. (2000) Hippocampal level of neural specific adenylyl cyclase type I is decreased in Alzheimer’s disease. Biochim Biophys Acta 1535:60 – 68. 69. Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF (2003) Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol 90:2171–2179. 70. Hu H, Vervaeke K, Storm JF (2007) M-channels (Kv7/KCNQ channels) that regulate synaptic integration, excitability, and spike pattern of CA1 pyramidal cells are located in the perisomatic region. J Neurosci 27:1853–1867. 71. Wu WW, Chan CS, Disterhoft JF (2004) Slow afterhyperpolarization governs the development of NMDA receptor-dependent afterdepolarization in CA1 pyramidal neurons during synaptic stimulation. J Neurophysiol 92:2346 –2356. 72. Kaczorowski CC, Disterhoft J, Spruston N (2007) Stability and plasticity of intrinsic membrane properties in hippocampal CA1 pyramidal neurons: effects of internal anions. J Physiol 578:799 – 818. PNAS ! February 3, 2009 ! vol. 106 ! no. 5 ! 1625 NEUROSCIENCE 12. Oh MM, Power JM, Thompson LT, Disterhoft JF (2000) Apamin increases excitability of CA1 hippocampal pyramidal neurons. Neurosci Res Comm 27:135–142. 13. Stocker M, Krause M, Pedarzani P (1999) An apamin-sensitive Ca2!-activated K! current in hippocampal pyramidal neurons. Proc Natl Acad Sci USA 96:4662– 4667. 14. Ngo-Anh TJ, et al. (2005) SK channels and NMDA receptors form a Ca2!-mediated feedback loop in dendritic spines. Nat Neurosci 8:642– 649. 15. Sah P, Bekkers JM (1996) Apical dendritic location of slow afterhyperpolarization current in hippocampal pyramidal neurons: implications for the integration of longterm potentiation. J Neurosci 16:4537– 4542. 16. Bekkers JM (2000) Distribution of slow AHP channels on hippocampal CA1 pyramidal neurons. J Neurophysiol 83:1756 –1759. 17. Nicoll RA (1988) The coupling of neurotransmitter receptors to ion channels in the brain. Science 241:545–551. 18. Pineda JC, Bargas J, Flores-Hernandez J, Galarraga E (1995) Muscarinic receptors modulate the afterhyperpolarizing potential in neostriatal neurons. Eur J Pharmacol 281:271–277. 19. Malenka RC, Madison DV, Andrade R, Nicoll RA (1986) Phorbol esters mimic some cholinergic actions in hippocampal pyramidal neurons. J Neurosci 6:475– 480. 20. Dutar P, Nicoll RA (1988) Classification of muscarinic responses in hippocampus in terms of receptor subtypes and second-messenger systems: electrophysiological studies in vitro. J Neurosci 8:4214 – 4224. 21. Grabauskas G, Lancaster B, O’Connor V, Wheal HV (2007) Protein kinase signalling requirements for metabotropic action of kainate receptors in rat CA1 pyramidal neurones. J Physiol 579:363–373. 22. Pedarzani P, Storm JF (1996) Evidence that Ca/calmodulin-dependent protein kinase mediates the modulation of the Ca2!-dependent K! current, IAHP, by acetylcholine, but not by glutamate, in hippocampal neurons. Pflugers Arch 431:723–728. 23. Pedarzani P, Storm JF (1993) PKA mediates the effects of monoamine transmitters on the K! current underlying the slow spike frequency adaptation in hippocampal neurons. Neuron 11:1023–1035. 24. Huang YY, Nguyen PV, Abel T, Kandel ER (1996) Long-lasting forms of synaptic potentiation in the mammalian hippocampus. Learn Mem 3:74 – 85. 25. Impey S, Obrietan K, Storm DR (1999) Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron 23:11–14. 26. Impey S, et al. (1998) Stimulation of cAMP response element (CRE)-mediated transcription during contextual learning. Nat Neurosci 1:595– 601. 27. Deisseroth K, Mermelstein PG, Xia H, Tsien RW (2003) Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol 13:354 –365. 28. Lopez de Armentia M, et al. (2007) cAMP response element-binding protein-mediated gene expression increases the intrinsic excitability of CA1 pyramidal neurons. J Neurosci 27:13909 –13918. 29. Cartford MC, Samec A, Fister M, Bickford PC (2004) Cerebellar norepinephrine modulates learning of delay classical eyeblink conditioning: evidence for post-synaptic signaling via PKA. Learn Mem 11:732–737. 30. Christian KM, Thompson RF (2003) Neural substrates of eyeblink conditioning: acquisition and retention. Learn Mem 10:427– 455. 31. Van der Zee EA, Kronforst-Collins MA, Maizels ET, Hunzicker-Dunn M, Disterhoft JF (1997) #Isoform-selective changes in PKC immunoreactivity after trace eyeblink conditioning in the rabbit hippocampus. Hippocampus 7:271–285. 32. Olds JL, Anderson ML, McPhie DL, Staten LD, Alkon DL (1989) Imaging of memoryspecific changes in the distribution of protein kinase C in the hippocampus. Science 245:866 – 869. 33. Freeman JH, Jr, Scharenberg AM, Olds JL, Schreurs BG (1998) Classical conditioning increases membrane-bound protein kinase C in rabbit cerebellum. Neuroreport 9:2669 –2673. 34. Nogues X, Micheau J, Jaffard R (1994) Protein kinase C activity in the hippocampus following spatial learning tasks in mice. Hippocampus 4:71–77. 35. Olds JL, et al. (1990) Discrimination learning alters the distribution of protein kinase C in the hippocampus of rats. J Neurosci 10:3707–3713. 36. Golski S, Olds JL, Mishkin M, Olton DS, Alkon DL (1995) Protein kinase C in the hippocampus is altered by spatial but not cued discriminations: a component task analysis. Brain Res 676:53– 62. 37. Seroussi Y, Brosh I, Barkai E (2002) Learning-induced reduction in post-burst afterhyperpolarization (AHP) is mediated by activation of PKC. Eur J Neurosci 16:965–969. 38. Sweatt JD (2004) Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol 14:311–317. 39. Kuo AG, Lee G, McKay BM, Disterhoft JF (2008) Enhanced neuronal excitability in rat CA1 pyramidal neurons following trace eyeblink conditioning acquisition is not due to alterations in IM. Neurobiol Learn Mem 89:125–133. 40. Moyer JR, Thompson LT, Disterhoft JF (1996) Trace eyeblink conditioning increases CA1 excitability in a transient and learning-specific manner. J Neurosci 16:5536 –5546. 41. Thompson LT, Moyer JR, Disterhoft JF (1996) Transient changes in excitability of rabbit CA3 neurons with a time course appropriate to support memory consolidation. J Neurophysiol 76:1836 –1849. 42. Thomas MJ, Moody TD, Makhinson M, O’Dell TJ (1996) Activity-dependent betaadrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron 17:475– 482.