Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Gel electrophoresis of nucleic acids wikipedia , lookup
SNP genotyping wikipedia , lookup
Expanded genetic code wikipedia , lookup
Holliday junction wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Transcription factor wikipedia , lookup
Human genome wikipedia , lookup
Epigenetics of human development wikipedia , lookup
DNA vaccination wikipedia , lookup
Short interspersed nuclear elements (SINEs) wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Long non-coding RNA wikipedia , lookup
Molecular cloning wikipedia , lookup
RNA interference wikipedia , lookup
History of genetic engineering wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Nucleic acid tertiary structure wikipedia , lookup
Epigenomics wikipedia , lookup
DNA polymerase wikipedia , lookup
Frameshift mutation wikipedia , lookup
RNA silencing wikipedia , lookup
Nucleic acid double helix wikipedia , lookup
Microevolution wikipedia , lookup
DNA supercoil wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Genetic code wikipedia , lookup
Non-coding DNA wikipedia , lookup
History of RNA biology wikipedia , lookup
Point mutation wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Polyadenylation wikipedia , lookup
Non-coding RNA wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
RNA-binding protein wikipedia , lookup
Helitron (biology) wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Messenger RNA wikipedia , lookup
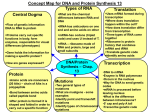
Hyde Chapter 10—Solutions 67 10 GENE EXPRESSION: TRANSCRIPTION CHAPTER SUMMARY QUESTIONS 2. The central dogma of molecular biology describes the flow of genetic information from DNA to RNA to protein. DNA controls its own replication. The transfer of information from DNA to RNA is termed transcription. The RNA is then converted into proteins via a process called translation. 4. Both transcription and translation occur in the cytoplasm of prokaryotes. Therefore, the mRNA can be translated before completion of its synthesis. However, the situation in eukaryotes is different: transcription occurs in the nucleus and translation in the cytoplasm. Therefore, these two processes are not coupled. 6. The complete bacterial RNA polymerase is termed the holoenzyme. It consists of the core enzyme plus the sigma factor (). The core enzyme is composed of four subunits: two , a , and a '. 8. A stem-loop structure can form when a single strand of DNA or RNA has a double helical section (see figure 10.11). An inverted repeat is a sequence read outward on both strands of a double helix from a central point (see figure 10.11). A tandem repeat is a segment of nucleic acid repeated consecutively; that is, the same sequence repeats in the same direction on the same strand: 5’-TCCGGTCCGGTCCGG-3’ 3’-AGGCCAGGCCAGGCC-5’ A DNA sequence with a seven-base inverted repeat is 5’-ATTACCGCGGTAAT-3’ 3’-TAATGGCGCCATTA-5’ 10. The two main types of transcription terminators in bacteria are termed rho-dependent and rho-independent. Both contain an inverted repeat sequence that generates a stemloop structure necessary to terminate transcription. In rho-independent terminators, the stem-loop structure is followed by a run of U’s which is enough to terminate transcription (U:A bonds are very weak and will denature spontaneously). Rhodependent terminators, on the other hand, lack the U-rich sequence. They rely 68 Hyde Chapter 10—Solutions instead on a protein called rho (), which uses ATP to denature the RNA–DNA hybrid, thereby causing termination of transcription. 12. There are three different types of RNA polymerase III promoters. Type 1 and Type 2 promoters are rather unusual because they require sequences that are located between 55 and 80 bases downstream of the transcription initiation point and within the gene. Please refer to figure 10.21. 14. As a result of posttranscriptional modifications, the mature eukaryotic mRNA has features lacking in the primary transcript: (1) 5'-CAP (a 7-methyl guanosine added in the “wrong” direction—5' 5'—by the enzyme guanylyl transferase). (2) 3'-polyA tail [a string of A residues added to the cleaved 3' end of the primary transcript by the enzyme poly(A) polymerase]. (3) Introns excised and exons spliced (by a protein– RNA complex termed the spliceosome). (4) RNA editing (by either guide RNAs inserting or deleting specific U’s, or through deamination of specific C’s or A’s). 16. a. The spliceosome, which is composed of several small nuclear ribonucleoproteins, functions to excise introns and splice exons in the primary mRNA transcript. It recognizes conserved sequences that flank and are within the intron. b. Guide RNA is an RNA molecule that guides the process of mRNA editing. It has a sequence that is complementary to the final mRNA, thereby aiding in the addition or deletion of specific U residues. c. Argonaute proteins bind to mature 22-nucleotide microRNAs to generate a complex termed microribonucleoprotein. This miRNP can perform three different functions: (1) it can bind mRNA and block its translation, (2) it can bind mRNA and result in its cleavage by endonucleases, or (3) it can enter the nucleus and repress gene transcription, a phenomenon termed gene silencing. EXERCISES AND PROBLEMS 18. A consensus sequence is made up of the nucleotides that appear in a significant proportion of cases when similar sequences are aligned. The best way to get to the consensus sequence is to follow these steps: 1. Consider each position of the sequences separately. 2. Count the number of different bases found at that position. 3. The base with the highest number is the one that will be included in the consensus sequence. For example, in position 1, there are 5 G’s and only 1 C and 1 A. Therefore, the consensus sequence will have a G in position 1. Similar logic provides the consensus sequence: G A T C T A G. Hyde Chapter 10—Solutions 69 20. With no sigma factors, there would be random starts of transcription; with no rho factor, there would be a failure to properly terminate transcription at rho-dependent termination sites. 22. Hybridization of nucleic acids is carried out between species to determine the extent to which it occurs. Presumably, the greater the amount of hybridization, the more similar the genomes of two species and therefore the more closely they are related in an evolutionary sense (chapter 25). 24. a. The mRNA is transcribed from the template strand based on the rules of complementarity: C (mRNA) is transcribed from G (DNA), G (mRNA) from C (DNA), A (mRNA) from T (DNA), and U (mRNA) from A (DNA). These percentages should be equivalent, and therefore the template strand of DNA would have the following percentages: 18% T, 27% G, 33% C, and 22% A. b. The nontemplate strand, which is the complement of the template strand, will therefore contain 18% A, 27% C, 33% G, and 22% T. Alternatively, these percentages can be derived directly by remembering that the nontemplate strand is the coding strand. Therefore, it will have the same sequence as the mRNA, substituting U for T. c. The base composition of the entire molecule will be the average base percentages of the two strands, or 20% A, 30% C, 30% G, and 20% T. 26. Top strand: 5' ––– 3' Bottom: 3' ––– 5' (template, or transcribed strand) Begin by writing the RNA that could be transcribed from each strand. Since the DNA represents the beginning portion of the gene, the RNA must have an AUG to start protein synthesis. Unfortunately, both strands yield RNAs with one or more AUGs. The RNA from the top strand has AUGs in both directions, but in each case the AUG is followed by a termination signal, UAA, or UAG. This RNA could not make a protein. The bottom strand produces an RNA with only one AUG. Since transcription and protein synthesis both proceed 5' 3', the left end of the bottom strand must be 3'. 28. If transcription of the genes is rho-dependent, the RNAs made at 40oC will be longer than those made at 30oC. Since the rho cannot function at the high temperature, RNA polymerase will read past the termination region. If transcription is rho-independent, a rho mutant will have no effect on transcription, and hence, the size of the RNAs. 70 Hyde Chapter 10—Solutions 30. 32. Enhancers bind activator proteins that also bind proteins of the RNA polymerase at the promoter. In some eukaryotic genes, there are many enhancers, allowing numerous levels of control and forcing enhancers further and further upstream. For activators bound to enhancers to bind polymerase proteins, the DNA must loop around. 34. The RNA is processed differently in the different tissues. In the pituitary, we see five introns and six coding regions, but in the adrenal, we see only four introns and five coding regions. The second DNA loop in the adrenal is larger than either the second or third loop in the pituitary. The third coding region must be removed in the adrenal, making the large second loop. 36. Prions are proteins that change the shape of proteins, not unlike many enzymes. The fact that they are mutant forms of a protein that change their unmutated equivalents into the mutated forms only makes them an oddity rather than a forbidden transfer in the central dogma. They do not represent a protein self-replication loop. 38. a. b. c. The (A + G)/(T + C) ratio of any double-stranded DNA molecule is 1.0. After all, the percentage of purines (A + G) should be equal to that of pyrimidines (T + C). The two mRNA transcripts that result would be complementary to each other and near identical copies of the double-stranded DNA molecule that they resulted from (with U substituted for T). Therefore, the (A + U)/(G + C) ratio in mRNA would be equal to the (A + T)/(G + C) DNA ratio of 1.4. The (A + U)/(G + C) ratio would still be 1.4, regardless of which DNA strand is transcribed! The easiest way to visualize this is to draw a double-stranded DNA molecule with an (A + T)/(G + C) ratio of 1.4 (that is, 14 AT pairs, and 10 CG pairs). If you transcribe either DNA strand, you will find that each RNA will have A + U = 14 and C + G = 10. Note that this will be true regardless of how you draw your DNA molecule. In other words, you could have the 14 AT pairs distributed as 14 A’s on one strand and 14 T’s on the other strand, or 7 A’s and 7 T’s on each strand, and so forth. In every single case where one strand is transcribed, the (A + U)/(G + C) ratio in the mRNA will be 1.4. Try it and see! Hyde Chapter 10—Solutions d. 71 As we discussed in part (b), the two mRNA transcripts would be near identical copies of the double-stranded DNA molecule (with U substituted for T), and so the (A + G)/(U + C) ratio in mRNA would be 1.0. Because it is not, the mRNA is transcribed from one strand only. This 1.2 ratio reflects the relative proportions of (A + G) and (T + C) on the transcribed DNA strand. Consider, for example, this DNA molecule which has the requisite (A + T)/(G + C) ratio of 1.4: 5'-TTTTTTAAAAAAAAGGGGGCCCCC-3' 3'-AAAAAATTTTTTTTCCCCCGGGGG-5' Transcription of the bottom strand yields an mRNA with the sequence 5'-UUUUUUAAAAAAAAGGGGGCCCCC-3' and an (A + G)/(U + C) ratio of 1.18 (or approximately 1.2). 40. Transcription of all four exons produces a primary transcript that is 1000 + 500 + 1000 + 800 = 3300 nucleotides in length. If this primary transcript is cleaved 50 nucleotides before the end of the fourth exon and then a 250 nucleotide poly(A) tail is added, the size of the mature mRNA transcript will be 3300 – 50 + 250 = 3500 nucleotides long. This corresponds to the larger transcript found in unaffected individuals. The shorter transcript is 3000 nucleotides long. The shortening of this transcript by 500 nucleotides can be explained by an alternative splicing event in which exon 2 is omitted from the shorter transcript. This would result in a transcript that is 1000 + 1000 + 800 – 50 + 250 = 3000 nucleotides in length. Another possible explanation is that exon 4 in the affected individuals might contain an alternative polyadenylation site. To produce a 3000 nucleotide transcript including exon 2, this site would have to be located 550 nucleotides before the end of exon 4. In this case, the transcript would be 1000 + 500 + 1000 + 800 – 550 + 250 = 3000 nucleotides long. The affected individuals in this family could have two types of mutations: (1) mutation of one of the splice junctions surrounding exon 2 or (2) a mutation in exon 4 creating a new polyadenylation site. CHAPTER INTEGRATION PROBLEM a. The total length of the mRNA transcript is 350 bases. However, the 50 A residues constitute the poly(A) tail which is added after transcription from the DNA. Therefore, the double-stranded DNA that codes for this mRNA would have to be 300 bp long. B-DNA contains 10 base pairs per 3.4 nm (one helical turn), or 1 bp per 0.34 nm. The length of the DNA would be 300 bp 0.34 nm/bp = 102 nm. b. The DNA coding strand of the laf+ gene is the nontemplate strand. It is equivalent to the mRNA, with T’s instead of U’s. It will also lack the string of A’s at the end. 72 c. Hyde Chapter 10—Solutions Replication requires a primer for initiation; transcription initiation does not require a primer. Replication of the entire DNA initiates from specific sequences termed replication origins; each gene, including the laf+ gene, is transcribed separately from its own promoter. Replication occurs in two directions using both strands of DNA as a template, while transcription is unidirectional and uses only one strand of DNA per gene. d. The promoter for the laf+ gene lacks a TATA-box. TATA-less promoters typically have a downstream promoter element (DPE) located at positions +28 to +34 and having the consensus sequence AGACGTG (in the coding strand). This sequence will show up in the mRNA as AGACGUG, and it does show in the laf+ mRNA. e. The laf+ gene is transcribed by RNA polymerase II. RNA pol II promoters typically have an initiator region (InR) located at –3 to +5. The +1 nucleotide is usually an adenine flanked by pyrimidines. This is the case here. In addition, RNA pol II transcripts are cleaved about 15–30 nucleotides after the sequence 5'-AUAAA-3', and they pick up the poly(A) tail at the cleavage site. This is also true of the laf+ gene. f. The presence of two Laf proteins can be explained by alternative splicing of the laf+ mRNA. So let’s first try to figure out the coding region of the laf+ gene. The initiation codon will have to be an AUG. There are two codons early on: at positions 83–85 and 118–120. The latter is almost immediately followed by a termination signal, UAA. Therefore, the initiation codon is located at positions 83–85. Now let’s look for potential introns. Remember the consensus sequences of introns: GU(AAGU) at the 5' end, CAG at the 3' end, and UACUAAC in the middle. Scanning of the laf+ gene reveals two introns. A termination signal, UAA, is found at position 252–254. The start and stop signals, and introns are highlighted here: 1 AUCCAGUAAC UUGAUACUGA ACGAAACAGA CGUGGCCGAA CCGUACACCU 51 ACCGACUGCC UUCACGUUAC CGCGAUUAAC GAAUGAAUUA UCAUCAUUUC 101 UUUCCCGUAA GUAAACCAUG UACUAACAAU UAUCUCUCUA UCCAGGGAUU 151 CUUCGUGGAU AUGUUAAUCA CCUGGUGCGU AAGUCAGAUA UUGACUUUUA 201 UAUUUAUUCG AUUCAACGAU ACUAACUUGA CAGAAAAGUG CAAAACCUAA 251 GUAACUAUCU UCACUGCAUU AUUUGUGAAA UAAACUUCGU AAUGGGUAGC 301 AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA AAAAAAAAAA The two introns split the laf+ gene into three exons. Exon 1 extends from bases 83 to 106 and encodes 8 amino acids; exon 2 is from bases 146 to 178 and encodes 11 amino acids; and exon 3, bases 234 to 251 and 6 amino acids. The laf+ mRNA that contains all three exons will be translated to produce LafL which consists of Hyde Chapter 10—Solutions 73 8 + 11 + 6 = 25 amino acids. The laf+ mRNA can be alternatively spliced to contain exons 1 and 3. This mRNA will produce LafS which consists of 8 + 6 = 14 amino acids. g. Mutant 1. The failure to produce an mRNA transcript indicates that the mutation must have occurred in the promoter sequence. Mutant 2. The failure to produce a Laf protein from a normal-length mRNA transcript indicates that the mutation likely occurred in the ribosome-binding site or in the start codon. Mutant 3. The presence of a larger than normal protein points to translation beyond the normal termination signal. Therefore, the point-mutation converted the UAA at positions 252–254 into a triplet that codes for an amino acid. Indeed, eight codons further downstream is a UGA triplet, which is a termination codon. Therefore, the LafXL would be 25 + 8 = 33 amino acids. Mutant 4. The LafXS is the hardest to explain. It is most likely the result of a mutation that generated a new initiation codon, which is located 18 bases upstream from a termination codon. Because initiation codons are AUG, we need to look for triplets that are AUX (where X = A, C, or U), AYG (where Y = A, C, or G), or ZUG (where Z = C, G, or U). A systematic scanning of the laf+ mRNA reveals a potential mutation site at the CUG in position 56–58. If the coding strand of DNA had a C to A mutation at that position, the mRNA will have an A, and a translation initiation codon. This is followed by a stretch of six codons that ends with a UAA termination signal. This mRNA would be translated into a LafXS protein of seven amino acids. h. The laf+ allele encodes the normal wild-type Laf+ protein which is responsible for small feet. A heterozygous individual (laf+laf) possesses one laf+ allele which is transcribed into normal mRNA, and then translated into the normal Laf+ protein. Therefore, one copy of the wild-type allele is sufficient to produce enough of the normal protein that is responsible for the wild-type phenotype of small feet. In homozygous recessive (laf- laf-) individuals, there are two mutant alleles and no wild-type allele. The absence of laf+ mRNA means only nonfunctional product is produced. This leads to the mutant form of feet, or large feet.