Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Evolution of human intelligence wikipedia , lookup
Environmental enrichment wikipedia , lookup
Donald O. Hebb wikipedia , lookup
Biochemistry of Alzheimer's disease wikipedia , lookup
Development of the nervous system wikipedia , lookup
Human multitasking wikipedia , lookup
Blood–brain barrier wikipedia , lookup
Premovement neuronal activity wikipedia , lookup
Human brain wikipedia , lookup
Neurolinguistics wikipedia , lookup
Neurophilosophy wikipedia , lookup
Neurogenomics wikipedia , lookup
Brain morphometry wikipedia , lookup
Selfish brain theory wikipedia , lookup
Activity-dependent plasticity wikipedia , lookup
Neuroeconomics wikipedia , lookup
Synaptic gating wikipedia , lookup
Molecular neuroscience wikipedia , lookup
Artificial general intelligence wikipedia , lookup
Neuroinformatics wikipedia , lookup
Aging brain wikipedia , lookup
Holonomic brain theory wikipedia , lookup
Mind uploading wikipedia , lookup
Circumventricular organs wikipedia , lookup
Optogenetics wikipedia , lookup
Cognitive neuroscience wikipedia , lookup
Brain Rules wikipedia , lookup
Channelrhodopsin wikipedia , lookup
Neuroplasticity wikipedia , lookup
Haemodynamic response wikipedia , lookup
History of neuroimaging wikipedia , lookup
Nervous system network models wikipedia , lookup
Neuropsychology wikipedia , lookup
Impact of health on intelligence wikipedia , lookup
Neuroanatomy wikipedia , lookup
Metastability in the brain wikipedia , lookup
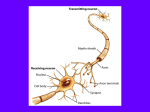
AAAS Summary PERINATAL DRUG/ALCOHOL EXPOSURE AND NEURONAL SUICIDE PUBLIC HEALTH IMPLICATIONS John W. Olney Drug-Induced Neuronal Suicide in the Developing Brain In a series of recent studies (1-5), it has been shown that several major classes of drugs, when administered to immature rodents during the period of synaptogenesis, trigger widespread neuronal suicide throughout the developing brain. The period of synaptogenesis, also known as the brain growth spurt period, occurs in different species at different times relative to birth. In rats and mice it is a postnatal event, but in humans it extends from the sixth month of gestation to several years after birth. Thus, it appears that there is a period of several years, encompassing portions of both pre and postnatal human development, during which immature neurons are prone to commit suicide if exposed to certain drugs that interfere with their developmental program. Converting Natural Neuroapoptosis into Pathological Neuroapoptosis In the developing nervous system of all species, including humans, neurons live or die at the mercy of a gene regulated program. For example, during synaptogenesis, the program requires that neurons form synaptic connections and become integrated into complex neural networks; and, for any neuron that fails to become properly integrated, the program dictates that it shall commit suicide (undergo “programmed cell death”). Programmed cell death (currently referred to as “apoptosis”) is recognized as a natural spontaneous process whereby redundant or unsuccessful neurons commit suicide and are deleted from the developing brain. Under normal circumstances, only a small percentage of neurons are deleted by this natural process. The above scenario begs an important question - what happens if environmental factors intercede and cause neurons to fail to make the appropriate connections? Presumably, this could cause large numbers of neurons that would otherwise have survived, to receive a suicide signal, causing them to self-destruct and be removed from the brain. In recent research, we are beginning to identify powerful environmental factors that can act in this manner and cause millions of immature neurons to commit suicide, thus converting a natural process for keeping the brain tidy, into a pathological process that rids the brain of millions of neurons that were otherwise destined to become successfully integrated into the developing nervous system and make positive contributions to its functional capacity. Drugs that Trigger Neuroapoptosis What types of drugs can cause developing neurons to commit suicide? Those that have been identified thus far act by several different mechanisms but have a single property in common - they all suppress neuronal activity. Four separate classes of drugs (NMDA glutamate antagonists, GABAmimetics, sodium channel blockers, and alcohol) have been shown to trigger neuroapoptosis in the developing animal brain. The activity level of a neuron is determined by the amount of excitatory versus inhibitory transmitter input it receives. NMDA glutamate antagonists and sodium channel blockers reduce neuronal activity by decreasing the amount of glutamate excitatory transmitter input; GABAmimetics reduce neuronal activity by increasing the amount of GABA inhibitory transmitter input. Alcohol is unique in having both NMDA antagonist and GABAmimetic properties. Thus, alcohol suppresses neuronal activity by a dual mechanism, and this makes it particularly effective in triggering neuroapoptosis in the developing brain (2, 4). Under what circumstances might the developing human nervous system come in contact with these apoptosis-promoting agents? Many drugs that trigger developmental neuroapoptosis in animal brain are used as sedatives, anticonvulsants or anesthetics in obstetric and pediatric medicine, and many also are drugs that are sometimes abused by pregnant women. Alcohol Alcohol deserves special consideration because of the frequency with which it is used/abused by pregnant mothers, and because it is well established that alcohol can have serious deleterious effects on the developing human brain (fetal alcohol syndrome, FAS) (6, 7). Although the devastating effects of alcohol on the human fetal brain have been recognized for 3 decades (6), the mechanism(s) underlying these effects have remained shrouded in mystery. The recent observation (2, 4) that a single alcohol intoxication episode can cause millions of neurons to commit suicide and be deleted from the developing rodent brain, provides a much more promising explanation than has heretofore been advanced, for the micro-encephaly (reduced brain size) and lifelong neurobehavioral disturbances associated with FAS. Other Drugs of Abuse Alcohol, although the most frequently abused drug in human society, is not the only drug with apoptogenic properties that is abused by pregnant women. Other examples, include phencyclidine (PCP, angel dust), ketamine (Special K), nitrous oxide (laughing gas) and many drugs classified as barbiturates and benzodiazepines. It is a common practice in a club drug setting to imbibe alcohol while abusing various other drugs, such as ketamine. This seemingly harmless recreational behavior is ill advised for pregnant women, because we have found in animal studies that alcohol, even at low doses, substantially enhances the apoptosis-promoting activity of ketamine. Use of Apoptogenic Drugs in Obstetric/Pediatric Medicine: Numerous drugs that have recently been shown to trigger neuroapoptosis in the developing animal brain have been used in obstetric and pediatric medicine for many years. These drugs are considered essential for meeting the therapeutic needs of patients who are seriously ill, and there has not previously been any basis for suspecting that they might quietly delete normal neurons from the developing brain. Anesthetic Drugs Obstetric and pediatric patients sometimes have to undergo complex surgical procedures that require prolonged anesthesia. In essence, the nervous system must be put to sleep, sometimes for many hours, by drugs that suppress neuronal activity. All drugs that have proven useful for this purpose are either NMDA antagonists or GABAmimetics, and the most effective anesthetic protocols are based on the use of multiple agents from each of these two categories. Thus, the most effective anesthetic drug protocols act by the same combination of mechanisms as alcohol and, therefore, would presumably cause alcohol-like effects in the developing brain. Consistent with this expectation, it was shown in a recent study (5) that exposure of infant rats for a period of 6 hours to a combination of anesthetic drugs commonly used in pediatric anesthesia, produced an alcohol-like pattern of neuroapoptosis throughout many brain regions, and subsequent learning deficits that were demonstrable as the animals grew to adulthood. Antiepileptic Drugs The only known method for controlling epileptic seizure activity is to administer drugs that suppress neuronal activity. Most antiepileptic drugs (AEDs) are either GABAmimetics or sodium channel blockers. In a recent study (3), it was shown that AEDs in either of these categories trigger neuroapoptosis in the developing rat brain, and that blood levels of AEDs required to induce neuroapoptosis in the infant rat brain are in the same range as those required for effective anti-epileptic therapy. In addition, it was found that combinations of AEDs, at doses in the therapeutic range, trigger more severe neuroapoptosis than individual AEDs. Threshold Conditions Much of the developmental neuroapoptosis research performed thus far has addressed the question whether various drugs, when administered in relatively high doses, can cause large numbers of neurons to commit suicide. More recently we have begun addressing the more subtle question - what is the minimal dose or duration of exposure required for a drug to cause a small but statistically significant number of neurons to commit suicide. In a study addressing this question for alcohol we found (9) that transient blood alcohol elevations hovering in the 80 mg/dl range for approximately 60 minutes is a sufficient condition for triggering apoptotic neurodegeneration at a significantly higher rate than occurs naturally in saline-treated control animals. In a human context, a blood alcohol elevation of this magnitude would be produced by drinking about 2 cocktails. We have conducted a similar study pertaining to ketamine, which is both a drug of abuse and a drug used frequently in pediatric medicine to provide sedation or for induction of anesthesia. In this study, we found (10) that a single dose of ketamine that is sedating for an infant mouse, but does not render the animal immobile or unconscious, triggers a significant increase in neuroapoptosis in several regions of the developing brain. Extrapolating from Rodents to Humans Evidence documenting that drugs commonly used in obstetric or pediatric medicine can cause extensive apoptotic neurodegeneration in the developing rodent brain raises the important question whether a similar neurotoxic phenomenon occurs in the developing human brain following exposure to such drugs. This is a very difficult question to answer conclusively, because if a similar phenomenon did occur in humans the manifestations, except in extreme cases, would be subtle and would only become evident on a delayed basis. To document that brain injury has occurred, one must rely largely upon epidemiological evidence because, unlike the experimental animal situation, one cannot histologically examine the human brain immediately after drug exposure to obtain unequivocal evidence that an increased number of neurons are committing suicide. At the present time we do not have adequate epidemiological evidence addressing this question and we consider it unwise to speculate based on the animal data presently available. These data pertain only to rats and mice, and it is axiomatic that rodent data provide an imprecise basis at best, and irrelevant basis at worst, for evaluating human risk. An important reason for not prematurely extrapolating from rodents to humans in this particular case is that rodents and humans have a very different developmental time scale. Disruption of synaptogenesis is the proposed mechanism by which anesthetic drugs trigger neuroapoptosis. In rodents, synaptogenesis is completed within a period of weeks whereas in humans it is completed within a period of several years. Neurons are programmed to commit suicide if their synaptic mission is thwarted to some critical degree. For the rodent neuron, perhaps 2 hrs of disruption exeeds the critical time limit, but for the human neuron maybe the critical time limit is much longer, just as the life span and period of synaptogenesis is much longer. This is a credible hypothesis, and fortunately it is a testable hypothesis, because there are other species, including non-human primate species, that have substantially longer periods of synaptogenesis than rodents. Obtaining an answer to this question is an important goal of currently ongoing research. Every Cloud has a Silver Lining We think it is likely that anesthetic drugs put neurons to sleep by one mechanism, and put neurons to death by another. The mechanism by which neurons are put to death involves a chain of intracellular biochemical steps which ultimately culminate in the activation of a suicide signal. If we can identify each of the biochemical steps, it may be possible to develop blocking agents that intercede at one or more critical step and halt the chain reaction before the suicide signal is activated. Therefore, it may be possible to retain the therapeutic benefit of putting neurons to sleep (anesthesia), while selectively blocking the mechanism by which anesthetic drugs accidentally activate a suicide signal that puts neurons to death. Current research is aimed at identifying the steps that will have to be blocked in order to prevent the suicide signal from being activated. Developmental Neuroapoptosis and the Origins of Neuropsychiatric Disorders We consider it likely that the developmental neuroapoptosis phenomenon we are studying may contribute to a wide range of neuropsychiatric diosorders. We have observed for both alcohol and related apoptogenic drugs that, depending on whether exposure occurs during the early, mid or late phase of synaptogenesis, these agents trigger different patterns of neuronal deletion, and it follows that each pattern of neuronal loss has the potential to give rise to its own unique constellation of neurobehavioral disturbances. In a recent study by Ann Streissguth’s research group (7, 8) it was found that 72% of FAS patients, after having experienced hyperactivity/attention deficit and/or learning disorders in childhood, required psychiatric care for adult-onset disturbances, including a 44% incidence of major depression and 40% incidence of psychosis. Assuming that developmental neuroapoptosis was the mechanism by which alcohol damaged the brains of these FAS patients, these finding document that this mechanism can give rise to a wide spectrum of neuropsychiatric disorders. It is currently believed that major psychiatric disorders have a genetic predisposition that may or may not be expressed as a clinical illness, depending on the influence of relevant environmental factors. The search for relevant environmental factors is an ongoing challenge being pursued in epidemiological studies by psychiatric researchers such as Ezra Susser and colleagues. We believe that developmental neuroapoptosis is an interesting phenomenon that may be able to shed new light on this problem. This is a genetically driven phenomenon that normally does not cause unwanted deletion of neurons from the brain, but if certain environmental factors intercede, it can result in the accidental deletion of large numbers of neurons from many different brain regions. Animal studies are useful for identifying environmental factors that can trigger developmental neuroapoptosis, but the eventual proof that such factors contribute to human neuropsychiatric disorders must be established through research focused on human subjects. Environmental factors identified thus far (and there may be many more waiting to be identified) seem to trigger neuroapoptosis by interfering with the glutamate and/or GABA neurotransmitter systems. Glutamate and GABA are ubiquitous neurotransmitter/neurotrophic systems that have vitally important, but incompletely understood, roles in brain development. It is possible that many instances will be discovered whereby either an aberrant genetic or environmental factor interferes with these glutamate or GABA functions in the synaptogenesis stage of development. According to the evidence now unfolding, if such interference occurs, it will silently drive neurons in large numbers to commit suicide and cause a child to be born with neurobehavioral disturbances of occult origin that may manifest either in childhood or adulthood, or both (8). Ultimate Significance We consider it likely that the significance of the developmental neuroapoptosis phenomenon we are studying will eventually transcend the context of fetal alcohol neurotoxicity and drug abuse or iatrogenic brain damage. We view this as a "final common pathway" type of mechanism which, regardless how it is activated, has considerable potential to disrupt brain development and give rise to a wide variety of neuropsychiatric disturbances. References 1. 2. Ikonomidou, C., Bosch, F., Miksa, M., Bittigau, P., Vockler, J., Dikranian, K., Stefovska, V., Turski, L. and Olney, J.W., Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain, Science, 283 (1999) 70-74. Ikonomidou, C., Bittigau, P., Ishimaru, M.J., Wozniak, D.F., Koch, C., Genz, K., Price, M.T., Stefovska, V., Horster, F., Tenkova, T., Dikranian, K. and Olney, J.W., Ethanol-induced apoptotic neurodegeneration and fetal alcohol syndrome, Science, 287 (2000) 1056-1060. 3. Bittigau, P., Sifringer, M., Genz, K., Reith, E., Pospischil, D., Govindarajalu, S., Dzietko, M., Pesditschek, S., Mai, I., Dikranian, K., Olney, J.W. and Ikonomidou, C., Antiepileptic drugs and apoptotic neurodegeneration in the developing brain, Proc Natl Acad Sci U S A, 99 (2002) 1508994. 4. Olney, J.W., Tenkova, T., Dikranian, K., Qin, Y.Q., Labruyere, J. and Ikonomidou, C., Ethanolinduced apoptotic neurodegeneration in the developing C57BL/6 mouse brain, Dev Brain Res, 133 (2002) 115-26. 5. Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW and Wozniak DF. Early exposure to common anesthetic agents causes widespread neurodegeneration in the developing rat brain and persistent learning deficits. The Journal of Neuroscience, 23:876-882, 2003. 6. Jones KL, Smith DW. The fetal alcohol syndrome. Teratology. 1975;12:1-10 7. Streissguth AP, O’Malley K. Neuropsychiatric implications and long-term consequences of fetal alcohol spectrum disorders. Semin. Clin. Neuropsychol. 2000;5:177-190 8. Famy C, Streissguth AP and Unis AS. Mental illness in adults with fetal alcohol syndrome or fetal alcohol effects. Am J Psych, 1998; 155:552-554. 9. Olney, J.W., Tenkova, T., Ikonomidou, C. and Young, C., Threshold conditions for triggering alcohol-induced apoptotic neurodegeneration in infant mouse brain., Society for Neuroscience Abstracts, 28 (2002) 120.1. 10. Young, C., Tenkova, T., Wang, H.H., Qin, Y.Q., Labuyere, J., Jevtovic-Todorovic, V. and Olney, J.W., A Single Sedating Dose of Ketamine Causes Neuronal Apoptosis in Developing Mouse Brain, Society for Neuroscience Abstracts (2003) in press.