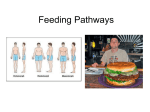
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Electrophysiology wikipedia , lookup
Single-unit recording wikipedia , lookup
Neurotransmitter wikipedia , lookup
Activity-dependent plasticity wikipedia , lookup
Aging brain wikipedia , lookup
Signal transduction wikipedia , lookup
Axon guidance wikipedia , lookup
Environmental enrichment wikipedia , lookup
Haemodynamic response wikipedia , lookup
Central pattern generator wikipedia , lookup
Synaptogenesis wikipedia , lookup
Development of the nervous system wikipedia , lookup
Selfish brain theory wikipedia , lookup
Metastability in the brain wikipedia , lookup
Nervous system network models wikipedia , lookup
Multielectrode array wikipedia , lookup
Premovement neuronal activity wikipedia , lookup
Molecular neuroscience wikipedia , lookup
Sexually dimorphic nucleus wikipedia , lookup
Pre-Bötzinger complex wikipedia , lookup
Synaptic gating wikipedia , lookup
Stimulus (physiology) wikipedia , lookup
Feature detection (nervous system) wikipedia , lookup
Optogenetics wikipedia , lookup
Neuroanatomy wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Endocannabinoid system wikipedia , lookup
Channelrhodopsin wikipedia , lookup
THE SUBFORNICAL ORGAN AND AREA POSTREMA MEDIATE THE CENTRAL EFFECTS OF CIRCULATING LEPTIN by Pauline M Smith A thesis submitted to the Graduate Program in Physiology in the Department of Biomedical and Molecular Sciences In conformity with the requirements for the degree of Philosophy Queen’s University Kingston, Ontario, Canada (October, 2012) Copyright © Pauline Smith, 2012 Abstract Leptin is an adipokine that acts centrally to regulate feeding behaviour, energy expenditure and autonomic function via activation of its receptor (ObRb) in nuclei in the central nervous system (CNS). This thesis investigates the involvement of two sensory circumventricular organs (CVOs), the subfornical organ (SFO) and area postrema (AP), in mediating the central effects of leptin using a variety of experimental approaches. We first show that acute electrical stimulation of the SFO elicits feeding in satiated rats, supporting a role for this specialized CNS structure in the control of food intake. We then demonstrate, using RT-PCR, the presence of ObRb mRNA in SFO and, using whole cell current clamp electrophysiology, reveal that leptin influences the excitability of individual SFO neurons, causing both excitatory and inhibitory responses. Furthermore, we find that leptin activates the same SFO neurons activated by amylin. Given the association between obesity and hypertension and the well-established role of the SFO in cardiovascular regulation, we show that leptin microinjection into the SFO decreases blood pressure in young rats, effects that are abolished in leptin-resistant, diet induced obese rats, suggesting that leptin-insensitivity in the SFO of obese, leptinresistant, individuals may contribute to obesity-related hypertension. Our studies also show that the medullary AP expresses ObRb and that leptin influences the excitability of AP neurons. Furthermore, we show that leptin and amylin act on the same subpopulation of neurons in the AP. Finally, our preliminary AP/SFO lesion studies reveal that animals with these lesions exhibit a profound decrease in body weight and food intake and no longer exhibit decreases in body weight in response to peripheral leptin administration. ii In summary, the data presented in this thesis suggest the SFO and AP to be important in body weight homeostasis and in mediating the central effects of leptin. In addition, these areas appear to be important in the integration of multiple signals derived from peripheral sources. Furthermore, the fact that the SFO appears to be involved in leptin effects on both energy balance and cardiovascular regulation attest to the integrative nature of the SFO in the control of diverse physiological functions. iii Co-Authorship Chapter 2: All experiments were designed, collected and analyzed by Pauline M. Smith. Gabriella Rozanski assisted with data collection in her capacity as a fourth year project student under the supervision of Pauline M. Smith. The manuscript was prepared by Pauline M. Smith with the assistance of Dr. Alastair V. Ferguson. Chapter 3: RT-PCR was performed with the assistance of Christie Hopf. Immunohistochemistry and pSTAT3 signaling experiments were performed in the laboratory of Keith Sharkey by Adam Chambers and Winnie Ho. In vivo electrophysiology was performed by Dr. Christopher Price. Design, collection, and analysis of all in vitro electrophysiology experiments were performed by Pauline M. Smith. Analysis and presentation of in vivo electrophysiology data was performed by Pauline M. Smith. The manuscript was prepared by Pauline M. Smith with the assistance of Dr. Alastair V. Ferguson and Keith Sharkey. Adam Chambers prepared immunohistochemistry and pSTAT3 methods, analysis and data presentation. Chapter 4: All experiments were designed, collected and analyzed by Pauline M. Smith. The manuscript was prepared by Pauline M. Smith with the assistance of Dr. Alastair V. Ferguson. Chapter 5: RT-PCR was performed with the assistance of Stefanie Killan. In vitro electrophysiology was performed by Kaitlin Hesketh and Christopher Arnts under the direct supervision of Pauline M Smith. In vivo electrophysiology was performed by Andrea Mimee. Analysis of the data and data presentation was performed by Pauline M. iv Smith. The manuscript was prepared by Pauline M. Smith with the assistance of Dr. Alastair V. Ferguson. Chapter 6: All experiments were designed, collected and analyzed by Pauline M. Smith. The manuscript was prepared by Pauline M. Smith with the assistance of Dr. Alastair V. Ferguson. v Acknowledgements Firstly, I need to thank Al for allowing and encouraging me to pursue my PhD in the lab after so many years as a tech. Thank you for your patience, understanding, support, and encouragement. Thank you for your continued, infectious, passion for science. Science is fun! I look forward to continuing to ‘do science’ in the Ferguson lab for as long as you will have me. To all the members of the Ferguson lab, past and present, before and during my PhD, thank you for making life in the lab interesting (to say the least). My time in the lab has certainly not been boring! Special thanks to Mark Fry and Chris Price for your support and friendship. Thanks to the ‘barn ladies’ for your friendship and fun times. You have provided me with an outlet for my frustrations and fun and supportive social environment. A very big ‘Thank You’ to my son, Christopher, who has grown up with me being in the lab and who has spent countless hours at the lab himself. Thank you for your maturity and understanding, especially when I just couldn’t be home for breakfast or dinner, and being self-sufficient enough to allow that to happen. You are a great kid and I am very proud of you. And to Richard. Thank you for your patience and understanding, especially during this final year, when things have been so busy, that I have often not been able to share, with you, the things that make our relationship so special. Thank you for hanging in there and supporting me through this process. You are a very special man and I am fortunate to have you in my life. vi Table of Contents Abstract ............................................................................................................................................ ii Co-Authorship ................................................................................................................................ iv Acknowledgements ......................................................................................................................... vi Table of Contents ............................................................................................................................ vi List of Figures .................................................................................................................................. x List of Tables .................................................................................................................................. vi List of Abbreviations ..................................................................................................................... xii Chapter 1 GENERAL INTRODUCTION ....................................................................................... 1 Hypothalamic Circuitry in the Regulation of Energy Balance................................. 3 The Discovery of Leptin................................................................................................ 4 The Arcuate Nucleus...................................................................................................... 6 The Leptin Receptor ..................................................................................................... vi Leptin Induced c-fos Activation in the Hypothalamus ........................................... 11 The Blood Brain Barrier .............................................................................................. 12 Transport Across the BBB .......................................................................................... 13 Transporting Circumventricular Organs ................................................................... 14 Transendothelial Cell Signaling ................................................................................. 15 Does the ARC have a ‘leaky’ BBB? ...................................................................... 15 Specialized ARC-ME Barrier ..................................................................................... 17 The Sensory CVOs ....................................................................................................... 19 The Subfornical Organ (SFO) .................................................................................... 20 A Role for the SFO in Regulation of Energy Balance ............................................ 22 Is the Arcuate Nucleus the Only Site of Action of Leptin in the CNS ................. 25 Caudal Brainstem and the Regulation of Energy Homeostasis.............................. 26 The Area Postrema (AP) ............................................................................................. 27 Leptin Effects on Blood Pressure ............................................................................... 29 Selective Leptin Resistance ........................................................................................ 30 STATEMENT OF THE PROBLEM ......................................................................... 31 vii Chapter 2 ACUTE ELECTRICAL STIMULATION OF THE SUBFORNICAL ORGAN INDUCES FEEDING IN SATIATED RATS ............................................................................... 33 Abstract .......................................................................................................................... 34 Introduction ..................................................................................................................... 35 Material and Methods .................................................................................................. 37 Results............................................................................................................................ 39 Discussion ..................................................................................................................... 45 Chapter 3 THE SUBFRONICAL ORGAN: AN ALTERNATIVE CNS SITE FOR CIRCULATING LEPTINTIATED RATS ………………………………………………. 50 Abstract .......................................................................................................................... 51 Introduction ..................................................................................................................... 53 Material and Methods .................................................................................................. 56 Results............................................................................................................................ 64 Discussion ..................................................................................................................... 73 Acknowledgements ...................................................................................................... 79 Chapter 4 CARDIOVASCULAR ACTIONS OF LEPTIN IN THE SUBFORNICAL ORGAN ........................................................................................................................................ 81 Abstract .......................................................................................................................... 82 Introduction ..................................................................................................................... 84 Material and Methods .................................................................................................. 87 Results............................................................................................................................ 90 Discussion ..................................................................................................................... 95 Acknowledgements .................................................................................................... 102 Chapter 5 LEPTIN INFLUENCES THE EXCITABILITY OF AREA POSTREMA NEURONS ……………………………………………………………………………………..103 Abstract ........................................................................................................................ 104 Introduction ................................................................................................................... 105 Material and Methods ................................................................................................ 107 Results.......................................................................................................................... 115 Discussion ................................................................................................................... 120 viii Acknowledgements .................................................................................................... 125 Chapter 6 LESIONS OF THE AREA POSTREMA AND SUBFORNICAL ORGAN ATTENUATE LEPTIN-INDUCED DECREASES IN BODY WEIGHT AND FOOD INTAKE ATIATED RATS ……………………………………………………………………126 Abstract ........................................................................................................................ 127 Introduction ................................................................................................................... 128 Material and Methods ................................................................................................ 130 Results.......................................................................................................................... 135 Discussion ................................................................................................................... 137 Acknowledgements .................................................................................................... 142 Chapter 7 GENERAL DISCUSSION .......................................................................................... 143 REFERENCE LIST ..................................................................................................................... 166 ix List of Figures Figure 1-1: Hypothalamic circuitry involved the control of energy balance. Figure 2-1: Animals were grouped according to anatomical location of the stimulating electrode Figure 2-2: SFO stimulation induced feeding Figure 2-3: Activity level was not different during stimulation Figure 3-1: ObRb receptor mRNA is expressed in the subfornical organ (SFO) Figure 3-2: Leptin receptor is expressed on neurons in the SFO Figure 3-3: Leptin induces pSTAT3 activation in the SFO Figure 3-4: pSTAT3 is colocalized with neuronal marker NeuN in the SFO Figure 3-5: Leptin influences the excitability of SFO neurons Figure 3-6: Leptin depolarizes amylin-sensitive SFO neurons Figure 4-1: Leptin microinjection into the SFO decreases in blood pressure Figure 4-2: Leptin microinjection into the SFO decreases BP in a dose related manner Figure 4-3: Leptin effects on blood pressure are site specific Figure 4-4: Leptin induced decreases in blood pressure are abolished in DIO rats Figure 5-1: ObRb receptor mRNA is expressed in the AP Figure 5-2: Leptin influences the excitability of AP neurons Figure 5-3: Leptin depolarizes amylin-sensitive AP neurons Figure 6-1: AP/SFO lesioned animals exhibit decreased body weight Figure 6-2: Changes in body weight following leptin administration Figure 6-3: Food and water consumption following leptin administration Figure 7-1: Proposed mechanism of leptin-angiotensin interaction at SFO neurons to regulate BP in normal weight and obese states x List of Tables Table 2-1: Summary of the effects of electrical stimulation at 100 and 200µA of the SFO on eating and drinking Table 3-1: Primer sets used in the detection of mRNA from SFO Table 5-1: Primer sets used to detect leptin mRNA from the area postema xi LIST OF ABBREVIATIONS aCSF artificial cerebral spinal fluid AgRP agouti-gene-related-peptide AMPK adenosine monophosphate kinase αMSH alpha melanocyte-stimulating hormone aCSF artificial cerebral spinal fluid ANG angiotensin AMPK adenosine monophosphate kinase AP area postrema ARC arcuate nucleus AT1 angiotensin II receptor type 1 AUC area under the curve AV3V anteroventral third ventricle BBB blood brain barrier BP blood pressure BW body weight CART cocaine-and amphetamine-related transcript cc corpus collosum CNS central nervous system CSF cerebral spinal fluid CVO circumventricular organ DMH dorsal medial hypothalamus DMN dorsomedial nucleus DMV dorsal motor nucleus of the vagus DVC dorsal vagal complex fx forix GAPDH glyceraldehyde 3-phosphate dehydrogenase GLUT-1 glucose transporter 1 GLP-1 glucagon-like peptide-1 HPD hypothalamo-pituitary disconnection xii HR heart rate HRP horseradish peroxidase icv intracerebroventricular ip intraperitoneal JAK-STAT janus activating kinase-signal transducer and activator of transcription LepR leptin receptor LH lateral hypothalamus ME median eminence MnPO median preoptic nucleus MSG monosodium glutamate NPY neuropeptide Y NTS nucleus tractus solitarius ObRa leptin receptor isoform a ObRb leptin receptor isoform b (signaling form) OVLT organum vasculosum of the lamina terminalis PI3K phosphoinositol-3-kinase POMC proopiomelanocortin PVN paraventricular Nucleus PYY peptide YY RT-PCR reverse transcriptase polymerase chain reaction SEM standard error of the mean SFO subfornical organ SON supraoptic nucleus v ventricle VMH ventral medial hypothalamus xiii Chapter 1 GENERAL INTRODUCTION 1 Obesity is a chronic metabolic condition with important public health implications associated with numerous co-morbidities including cardiovascular disease, insulin resistance, and hypertension. Globally, over 1 billion adults are overweight while approximately 500 million are obese (World Health Organization, 2011). Perhaps even more staggering, 40 million children under the age of 5 years were considered overweight in 2010 (World Health Organization, 2011). In order to maintain an ideal body weight, an organism must balance energy intake with energy expenditure. If energy intake exceeds the amount of energy an individual expends, the excess energy will be stored as adipose tissue with a prolonged imbalance leading to an individual becoming overweight and, eventually, obese. Adipose tissue, once thought of as solely a storage depot for excess triglycerides, also serves as an endocrine organ which plays a critical role in energy homeostasis, secreting adipokines that control feeding and energy metabolism. Although a certain degree of fat storage is normal, the expansion of adipose tissue that occurs in obesity alters adipokine secretion which may contribute to the development of metabolic diseases. The regulation of energy balance requires a complex interaction between signals from the periphery reflecting various aspects of metabolic status and autonomic control centers in the central nervous system (CNS) that respond to these signals to influence food intake and energy expenditure. 2 Hypothalamic Circuitry in the Regulation of Energy Balance A role for the hypothalamus in the control of feeding was proposed by Hetherington in the 1940’s when he demonstrated that hypothalamic lesions affected hunger and satiety (Hetherington & Ranson, 1940). These studies led to the development of a ‘dual center’ model for the hypothalamus in the regulation of energy homeostasis in which the lateral hypothalamus (LH) was considered the ‘feeding center’ of the brain while the ventral medial portion of the hypothalamus (VMH) was the ‘satiety center’(Stellar, 1954). Not too many years later, a role for humoral signals in the regulation of food intake was suggested by parabiotic (cross circulation) studies in which lesion of the VMH (satiety center) in one animal of the parabiotic union caused weight gain and obesity in that animal, while the unlesioned animal of the pair demonstrated a decrease in body fat (Hervey, 1959). Similarly, electrical stimulation of the LH (feeding center) in one member of the parabiotic rats produced increased feeding and fat pad mass in that animal, while fat pad mass was decreased in the non-stimulated animal(Hervey, 1959). Thus, the development of the ‘lipostatic’ theory of body weight maintenance, largely driven by data from these parabiotic studies (Kennedy, 1953;Hervey, 1959), proposed that, in addition to the roles of the feeding (LH) and satiety center (VMH), a fat-secreted “factor” that reported the status of body energy stores to the brain also was involved in regulating feeding behavior and body fat mass. Further support for the presence of a peripheral lipostatic factor controlling food intake was derived from later parabiotic studies 3 demonstrating that overfeeding one member of the pair caused the other member of the pair to reduce its food intake (Nishizawa & Bray, 1980;Harris & Martin, 1984). The Discovery of Leptin The notion that neuronal circuits in the hypothalamus and information regarding energy homeostasis provided by circulating factors play an important role in body weight homeostasis was firmly established by the discovery of leptin in 1994 (Halaas et al., 1995a). Parabiotic studies demonstrated that a circulating factor present in the wild type littermate of the ob/ob (obese) mouse (the spontaneous mutant with inherited obesity which was observed over 60 years on the C57BL/6J background at Jackson Laboratories (Ingalls et al., 1950) normalized body weight in the obese mouse (Halaas et al., 1995a). This circulating factor, coined leptin from the Greek word ‘leptos’ meaning ‘thin’, was determined to be the protein absent in the ob/ob mouse (Halaas et al., 1995a). The absence of this circulating peptide was shown to be responsible for markedly obese phenotype observed in this mouse, which is normalized by both systemic and central leptin administration (Halaas et al., 1995b;Pelleymounter et al., 1995;Campfield et al., 1995). Daily intraperitoneal (ip) injections of leptin were shown to significantly reduce food intake and body weight, preferentially reducing body fat while sparing lean tissue, in both ob/ob and wildtype mice (Halaas et al., 1995b;Halaas et al., 1997). Additional studies demonstrating that intracerebroventricular (icv) injections of leptin elicited dose dependent decreases in food intake and body weight gain suggested a central site of action for leptin (Satoh et al., 1997). Further to the observation that leptin had central 4 site(s) of action, discrete, bilateral microinjection of leptin directly into arcuate nucleus (ARC) had the greatest impact on feeding of all hypothalamic feeding centers tested (VMH, LH and ARC) leading to the conclusion that the ARC was a primary site of the satiety effect of leptin (Satoh et al., 1997). Thus, the discovery of leptin and the therapeutic potential of such an anti-obesity compound led to an explosion in research on the regulation of feeding and our knowledge of the central regulation of energy balance has grown as a result of critical work in a number of areas. Since the initial discovery of leptin, studies have revealed that central administration of a plethora of different hypothalamic neuropeptides into the ventricular system and/or a variety of discrete nuclei within the CNS affects food intake and weight gain in animals, highlighting the complexity of neural signaling within the hypothalamus in the regulation of energy balance (for review see Blouet & Schwartz, 2010;Arora & Anubhuti, 2006). In addition, the development of genetic models in which spontaneous genetic mutations or targeted gene deletions impair hypothalamic neuropeptide function causing dramatic effects on feeding behavior has identified the potential roles of specific genes in the regulation of energy balance (for review see Balthasar, 2006;Elmquist et al., 2005;Santini et al., 2009). Finally, electrophysiological and molecular characterization of hypothalamic neurons has described specific sensory or signaling roles for identified subpopulations of hypothalamic neurons in the control of energy balance (for review see Ghamari-Langroudi, 2012). Thus, our present day understanding of body weight regulation involves well-defined hypothalamic and brainstem neuronal circuits, rather 5 than discrete hypothalamic satiety and feeding centers (for review see Williams et al., 2001;Grill, 2010;Simpson et al., 2009;Blouet & Schwartz, 2010). Within all of these models, peripheral signals that convey information regarding nutritional and metabolic status of the individual must be able to access and regulate these neuronal circuits in order to regulate both food intake and energy expenditure. In addition, within many of these models the ARC has become recognized as a critical center in this integrated circuitry. The Arcuate Nucleus A role for the ARC in the control of feeding was proposed in 1969 when Olney (Olney, 1969), investigating the effects of food additives on brain architecture, observed that newborn rats administered monosodium glutamate (MSG) peripherally developed obesity. Upon microscopic examination of brain tissue, MSG administration was shown to cause severe and selective destruction of the ARC and two circumventricular organs (CVOs), the median eminence (ME) and the subfornical organ (SFO) (Olney, 1969). These observations were supported by additional studies evaluating the extent of MSG-induced damage in the CNS (Takasaki, 1978;Olney & Sharpe, 1969;Olney et al., 1972;Arees & Mayer, 1970) which confirmed Olney’s original observations. Further evidence suggesting a critical role for the ARC in the regulation of energy status stems from studies demonstrating that peripheral administration of gold thioglucose (a treatment directed toward destruction of glucosesensitive neurons in the brain) results in necrosis to the ventromedial portion of the 6 hypothalamus (the area in which the ARC is located) as well as marked hyperphagia and consequent obesity (Debons et al., 1977;Bergen et al., 1998). In addition, discrete, bilateral chemical (colchicine) lesions of the ARC caused increased food intake and body weight gain leading, eventually, to obesity (Choi & Dallman, 1999) which focused attention to the ARC as the central hypothalamic control center in the control of energy homeostasis. The ARC plays a critical role in controlling food intake via two distinct populations of neurons. The mRNA for the orexigenic neuropeptides, agouti-generelated-peptide (AgRP) and neuropeptide Y (NPY) are co-expressed in one population of ARC neurons, while a second population of ARC neurons expresses mRNA for the anorexigenic neuropeptides, alpha melanocyte-stimulating hormones (αMSH) derived from proopiomelanocortin (POMC) precursor and cocaine- and amphetamine-related transcript (CART). Dose dependent increases in food intake are observed following central icv administration of either AgRP or NPY (Rossi et al., 1998;Levine & Morley, 1984;Clark et al., 1984), while the anorexigenic peptides, αMSH and CART, decrease food intake following ICV administration (Edwards et al., 2000;Tsujii & Bray, 1989) suggesting that these neurons are critical for modulating energy balance. Individual neurons within the ARC have also been shown to be sensitive to a number of peptides known to be involved in the control of feeding behavior and, in short, signals that activate AgRP/NPY neurons have been shown to increase feeding behavior as do signals that inhibit αMSH/CART while those that inhibit AgRP/NPY neurons or 7 activate αMSH/CART neurons inhibit feeding behavior (for review see Jobst et al., 2004;Millington, 2007;Wardlaw, 2011). Anorexigenic (αMSH and CART) and orexigenic (AgRP and NPY) neurons in the ARC project to second order neurons in the hypothalamus including the paraventricular nucleus (PVN), VMH, LH, and the dorsomedial nucleus (DMN) which then process information regarding energy and metabolic status (see Figure 1-1). These hypothalamic neurons then project to the dorsal vagal complex (DVC) which includes the nucleus tractus solitarius (NTS) and the dorsal motor nucleus of the vagus (DMV) and provide a route through which meal-related satiety signals and thus, the long term regulation of energy homeostasis, may be modulated. In addition to the role of leptin in the homeostatic control of food intake, leptin has been shown to regulate the activity of the mesolimbic dopamine pathway, thus suggesting an additional role for leptin in nonfeeding-related motivated behaviors (Hommel et al., 2006;Fulton et al., 2006;Saper et al., 2002). The Leptin Receptor The presence of receptors in specific tissues suggests that the ligands specific for those receptors act on that target tissue. The leptin receptor, encoded by the ObR gene, was isolated from choroid plexus by expression cloning and is a member of the cytokine family (Tartaglia et al., 1995). Although 6 leptin receptor isoforms have been identified (ObRa – ObRf), all of which are products of the single lepr gene (Tartaglia, 1997;Chua, Jr. et al., 1997), only the long form of the receptor, ObRb, possesses the cytoplasmic 8 Figure 1-1: Hypothalamic circuitry involved the control of energy balance. The upper sagital section (0.4mm midline) shows the relative location of ARC and anatomical projections to other hypothalamic, extrahypothalamic (SFO) and brainstem (AP, NTS) areas involved in energy homeostasis. Bottom left: Expanded sagital view of the hypothalamus outlining the major anatomical projections of the hypothalamic circuitry involved in the control of feeding behavior showing projections from the ARC to VMH, DMH, LHA, and PVN. The coronal sections to the right show the relative anatomical locations of these hypothalamic nuclei. ARC: arcuate nucleus, VMH: ventromedial hypothalamus, DMH: dorsomedial hypothalamus, LHA: lateral hypothalamic area, PVN: paraventricular nucleus, SFO: subfornical organ, NTS: nucleus tractus solitaries, AP: area postrema, 3V: third ventricle, 4V: fourth ventricle. 9 domains required for signal transduction (Bjorbaek et al., 1997;Banks et al., 2000;Kloek et al., 2002). ObRb regulates multiple intracellular signaling cascades, including the janus activating kinase-signal transducer and activator of transcription (JAK-STAT) pathway and the phosphoinositol-3 kinase (PI3K) and adenosine monophosphate kinase (AMPK) pathways and is essential for the weight reducing effect of leptin (Bjorbaek et al., 1997;Bjorbaek & Kahn, 2004;Buettner et al., 2006;Bates et al., 2003;Cui et al., 2004). The functions of the shorter isoforms of leptin receptors are not fully understood. Upon the initial discovery of the multiple leptin receptor isoforms, ObRa and ObRc were suggested to be responsible for the transport of leptin across the blood brain barrier (BBB) due to their abundant expression in the choroid plexus (Liu et al., 1997), a role which, to date, has not been demonstrated. In contrast, OBRe, also known as soluble leptin receptor due to its lack of the transmembrane and cytoplasmic regions, has been shown to bind to leptin and, thus, serves to antagonize ObRb-mediated signaling (Yang et al., 2004). Circulating leptin exerts its effects on feeding and energy expenditure by binding to ObRb in the CNS (Grill & Kaplan, 2002;Burguera et al., 2000). In the CNS, ObRb is prevalent in the areas of the hypothalamus associated with the regulation of feeding including the ARC, LH, VMH, and DMN (Mercer et al., 1996;Shioda et al., 1998;Schwartz et al., 1996;Huang et al., 1996), further implicating the ARC as at least one of the hypothalamic sites at which leptin acts to exert its satiety effects. 10 Leptin Induced c-fos Activation in the Hypothalamus The expression of intermediate early genes, such as c-fos, by neurons in the brain is often used as an indicator of neuronal activation (direct or indirect) within that area. Systemic administration of leptin has been shown to induce c-fos protein immunoreactivity in the ARC as well as other areas in the hypothalamus involved in the energy homeostasis including the VMN, DMN, and PVN (Elmquist et al., 1997;van Dijk et al., 1996). In accordance with our understanding of critical roles for POMC neurons in the ARC in the regulation of food intake, leptin has been reported to induce c-fos expression (i.e. activation) in one subpopulation of ARC POMC neurons (Williams et al., 2010). The direct effects of leptin on ARC neurons have also been examined using electrophysiological techniques and the majority of glucose-sensitive ARC neurons were inhibited by perfusion with leptin (Shiraishi et al., 1999). In addition, leptin has also been shown to modulate different populations of hypothalamic cells in different ways. Leptin has been shown to primarily inhibit orexin-sensitive (Rauch et al., 2000) and ghrelin-sensitive NPY/AgRP neurons (Tung et al., 2001) while rapidly activating POMC neurons (Cowley et al., 2001;Williams et al., 2010), observations that are, for the most part, consistent with the effect of leptin in modulating these two populations of neurons and thus influencing feeding. The evidence presented above, clearly supports the conclusion that the actions of leptin within the ARC underlie the anorexigenic effects of this adipokine. However, there is an increasing body of evidence that suggests the ARC is securely protected from the 11 contents of the peripheral circulation by the BBB raising the intriguing possibility that circulating leptin may not be able to directly access neurons in this region of the brain. The Blood Brain Barrier The anatomical features of the BBB are well understood in that it consists, not only of endothelial cells of cerebral micro vessels joined together by tight junctions (in strict contrast to the fenestrations between endothelial cells in most capillaries), but also astrocytes, whose end feet wrap around the brain side of the endothelial cell membrane (Abbott et al., 2006;Peruzzo et al., 2000;Rodriguez et al., 2010). The tight junctions of the endothelial cells are more complex in the CNS than in the periphery, in that there are networks of strands formed by intramembranous particles joining adjacent cells (Wolburg et al., 2003;Krisch & Leonhardt, 1978). Tanycytes are a unique cell type of the mature brain lining and are a key component of the BBB and cerebrospinal fluid (CSF) brain barrier. Alpha tanycytes bridge the lumen of the third ventricle while β tanycytes form a cuff which effectively separates neurosecretory terminals from portal capillaries establishing a barrier between ARC and ME (Rethelyi, 1984;Rodriguez et al., 2005;Rodriguez et al., 1979;Rodriguez et al., 2005). The continuity of the BBB ensures that all transport occurs across the cell membrane thus limiting movement across the barrier to substances that are either lipophilic (and thus readily diffusible across the lipid bilayer), or substances that are transported across this barrier by alternative mechanisms. This compartmentalization effectively precludes autonomic control centers within the CNS from directly monitoring the varying levels of many important peripheral indicators 12 of physiological status, including leptin. This physical barrier, which prevents access to the CNS of these peripheral signals, also allows these same hormones to be produced centrally and utilized as neurotransmitters within the CNS. Therefore, although leptin unquestionably has been shown to have direct actions on distinct populations of neurons in the ARC all anatomical evidence securely positions the ARC behind the BBB. As the ARC is protected by the BBB, the question arises as to how does leptin, a large 16Kda, lipophobic, protein produced by white adipose tissue in the periphery, gain access to the neural elements in the ARC? Transport Across the BBB It has been demonstrated that specific transporters exist for a number of signaling molecules (i.e. insulin) and these transporters have been shown to facilitate transport of signaling molecules from blood to brain and/or from brain to blood (Kastin et al., 1999;Banks & Kastin, 1992). Studies using 125 I-leptin with isolated human brain capillaries in an in vitro model of the human BBB have shown evidence for a leptin receptor mediated, saturable, specific, temperature-dependent binding and endocytosis of leptin at the human BBB (Golden et al., 1997). Studies using shown that peripherally administered 125 125 I-leptin in mice have I-leptin enters the brain (as assessed by radioactivity of brain extract) and that uptake occurs in the choroid plexus, ME and ARC (as determined by autoradiography). Peak influx was shown to occur 20 minutes after intravenous leptin injection (Banks et al., 1996). However, it is not clear from this study how much 125 I-leptin was given peripherally or whether other areas of the brain showed 13 labeling. Thus, the physiological relevance of these observations is difficult to assess directly and thus remains to be established. In addition, the molecular identity of the transporter has yet to be identified more than 15 years after its initial description. Transporting Circumventricular Organs An alternative explanation, other than the presence of a saturable transporter system, exists to explain these findings. The choroid plexus and ME are CVOs, specialized CNS structures that lack the normal BBB. More specifically, the choroid plexus and ME are classified as transporting CVOs in that the choroidal cells of the choroid plexus and tanycytes of the ME have machinery to transfer substances from blood to CSF and vice versa (Knigge et al., 1976;Weindl & Joynt, 1973;Gross & Weindl, 1987). While the ARC possesses a normal BBB and a barrier to adjacent hypothalamic tissue, this nucleus is in the privileged position in that it has been shown to have direct access to the CSF present in the infundibular recess (Rodriguez et al., 2005;Rodriguez et al., 2010). Injection of tracer molecules or vital stains into the ventricular system have been shown to enter the ARC and remain confined to the ARC (Rodriguez et al., 2005;Rodriguez et al., 2010), providing a potential alternative to the saturable transporter system explanation of localization Accumulation of 125 125 125 I-leptin of in the ARC. I-leptin could be due to transport into the ARC from the CSF after I-leptin was transported from the blood to the CSF via the choroid plexus and ME (which were also labeled). This alternative explanation may also explain the time (20 minutes) of peak 125I-leptin influx into the ARC. 14 Transendothelial cell signaling Transendothelial cell signaling represents another potential mechanism by which transport of leptin across the BBB might occur. Recent work has suggested that transendothelial cell signaling occurs when molecules act on the luminal side of the cerebral vascular endothelial cell and induce the release of a second, different, signaling molecule (i.e. nitric oxide) on the other side of the barrier (Paton et al., 2007;Janigro et al., 1994). Does the ARC have a ‘leaky’ BBB? It has also been suggested that the ARC is not protected behind the BBB and, in fact, has a modified or leaky BBB. Although these myths have permeated the literature, there is no direct anatomical evidence to support this conclusion. The idea that the ARC has a modified or leaky BBB is derived from MSG lesion studies showing that peripheral MSG administration to newborn rats leads to severe and selective destruction of the ARC and CVOs (Olney, 1969;Olney, 1971;Kerkerian & Pelletier, 1986). Further analysis of this phenomenon, however, reveals that maximal damage occurs when MSG is delivered during the first post natal week (Olney, 1969;Olney, 1971;Perez et al., 1979;Peruzzo et al., 2000). MSG administration during the fourth postnatal week, on the other hand, leaves the ARC intact (Peruzzo et al., 2000). Although the mechanisms underlying the spatial and temporal selectivity of MSG lesions are not known, it has been suggested that development of ME tanycytes (Hu et al., 1998) and, in particular, the postnatal development of glucose transporter 1 (GLUT-1) in these cells is important in BBB 15 development. GLUT-1 immunoreactivity is present in endothelial cells of the BBB and, as such, is used as a BBB marker (Pardridge et al., 1990). GLUT-1 immunoreactivity is barely detectable around the ARC during the first postnatal week suggesting that the BBB is not fully developed at this time and thus explains the susceptibility of this region to MSG lesion. The BBB-ARC barrier is fully developed by the forth postnatal week, and, not surprisingly, peripheral MSG administration during this time period does not lead to ARC damage. These results show that the susceptibility of ARC to MSG administration parallels the development of the BBB in the ARC and that ARC has a fully functional BBB by the fourth postnatal week (Peruzzo et al., 2000). Others have suggested that the ARC vasculature is modified or ‘leaky’ such that ARC neurons can have access to the circulation. The ARC has, in a number of cases been referred to, and adopted as, an additional CVO despite the fact that there is no anatomical evidence suggesting that the ARC lacks the normal BBB (Krisch et al., 1978;Petrov et al., 1994;Peruzzo et al., 2000;Rodriguez et al., 2010;Rodriguez, 1976). Initial studies utilizing horseradish peroxidase (HRP) to identify regions of the brain that lack the normal BBB, showed HRP uptake in CVOs and in the ARC (Broadwell & Brightman, 1976), and these studies are often cited as evidence that the ARC lacks a normal BBB. However, the temporal aspects of the labeling of the CVOs and ARC were quite different. These studies showed that, following systemic HRP, the primary areas labeled by this molecule were the traditional CVOs while, eight hours after HRP administration, other regions of the brain showed HRP labeling, including the ARC 16 (Broadwell & Brightman, 1976). Broadwell and Brightman (Broadwell & Brightman, 1976) concluded that this labeling of HRP in the ARC was a direct result of retrograde axonal transport to ARC neurons that project to the ME. Recent studies using more sensitive fluorescence tracers have confirmed this conclusion (Cheunsuang et al., 2006;Cheunsuang & Morris, 2005). Following systemic administration of hydroxystilbamidine (a fluorogold equivalent), only ARC astrocytes show labeling (Cheunsuang & Morris, 2005). A very recent study evaluating IgG retention in the ARC, suggested the ARC to have a permeable BBB, however there is no mention of the timeframe between IgG administration and euthanasia/tissue extraction (Ciofi, 2011). If euthanasia was not immediate, the small amount of IgG accumulation seen in the ARC could be a function of IgG transport from the CSF to the ARC (following transport from the blood to CSF via the choroid (which is densely labeled) or the ME), as described earlier. Collectively, these observations do not provide any definitive evidence that ARC neurons are in a preferential position to directly access circulating substances. Specialized ARC-ME Barrier It has also been suggested that a special anatomical arrangement exists ARC and ME in which the ARC is privy to contents of the ME. However, the existence of an intact, functional ME-ARC barrier is confirmed from a number of anatomical findings. β1 tanycytes have been demonstrated to project processes into the lateral external region of ME forming a cuff which separates neurosecretory terminals from portal capillaries 17 (Rodriguez et al., 1979;Rodriguez et al., 2005). Electron microscopy and freeze-etching studies have revealed adherent and tight junctions between β1 processes and between tanycytes and neurosecretory axons (Rodriguez et al., 2010;Krisch & Leonhardt, 1978). In addition, the basal processes of the β1 tanycytes which extend along the ME-ARC border are joined by zonula and macula adherens which firmly establish a barrier between ARC and ME (Rethelyi, 1984;Rodriguez et al., 2005;Rodriguez et al., 2010). Further evidence in support of the functional existence of a complete ME-ARC barrier is provided by the use of anatomical tracers or vital stains. Tracer molecules (HRP) or vital stain (trypan blue or Evan’s blue) injected intravenously escape from portal vessels to perivascular space and intercellular space of the ME and remain confined to this region and do not penetrate the ARC (Rodriguez et al., 2010). Injection of these same tracer molecules and stains into the ventricular system has been shown to enter the ARC and remain confined to the ARC (Rodriguez et al., 2005;Rodriguez et al., 2010). Although no transport would be expected into the adjacent ME due to the MEARC barrier, no transport of these molecules into adjacent hypothalamic nuclei (i.e. VMN) has been observed. In addition, direct injection of HRP (Rethelyi, 1984) or trypan blue (Rodriguez et al., 2010) into the ARC does not spread to ME or adjacent hypothalamic structures (i.e. VMN) (Rodriguez et al., 2010). Labeling of the intercellular space stopped at lateral borders of the ME, the location of β1 tanycytes and their basal processes, further supporting the notion that β1 tanycytes are involved in the barrier mechanism between ME and ARC (Rodriguez et al., 2010). These interesting 18 observations not only confirm the presence of a complete and functional ME-ARC barrier but also suggest that there is sub compartmentalization of the ARC from the ME and the rest of the hypothalamus. The fact that the internal milieu of the ARC is open to the CSF at the infundibular recess and that labeling of wall of the infundibular recess is seen after ARC injection or trypan blue (Rodriguez et al., 2010) suggests bidirectional movement is possible across the ARC-CSF barrier in both directions. The apparent sub compartmentalization of the ARC from the rest of the hypothalamus and the bidirectional movement across the ARC-CSF barrier along with the fact the flow of CSF through this region is slowed due to the lack of multiciliated ependymal cells within the infundibular recess (Rodriguez et al., 2010) would facilitate the arrival of CSF borne signals to the ARC. Indeed, leptin mRNA has been demonstrated in brain of a variety of species providing a source for CSF derived leptin (Wilkinson et al., 2007). Thus, all available anatomical evidence suggests that the ARC of the adult possesses an intact BBB. In addition, the existence of functional, physiologically relevant, leptin transport via a saturable transport system remains theoretical. How, then, can the actions of leptin at the ARC be explained? Is there another potential interpretation of the same data? The Sensory CVOs Within the CNS there does exist a route through which circulating peptides may deliver information to the CNS without the need to cross the BBB. Specialized regions of the brain known as the circumventricular organs (CVOs) have a specialized cerebral 19 vasculature where fenestrations are found between endothelial cells (similar to the rest of the non-brain systemic vasculature) such that even large, lipophobic, substances (peptides and proteins) can cross from blood to neural tissue without having to cross the cell membrane (Gross, 1992). The fenestrated capillaries of the sensory CVOs are distinct from the rest of the CNS in that they lack the typical tight junctions between adjacent endothelial cells (Petrov et al., 1994;Rodriguez et al., 2010). The CVOs also possess an extensive and complex vascular supply as compared to other areas of the brain (Gross, 1991) presumably designed to maximize the time and area for exposure of blood borne substances to the cellular components of the CVOs (Gross, 1991). In addition to the lack of the normal BBB and the dense vascular supply, the sensory CVOs have been demonstrated to contain exceptionally dense aggregations of a variety of different receptors for peripheral signals, observations which clearly suggest the ability of neurons in these CVOs to sense circulating concentrations of these signaling molecules. Thus, these specialized CNS structures are uniquely positioned to monitor the constituents of peripheral circulation and communicate this information, via well-documented afferent projections, to autonomic control centers in the hypothalamus and medulla and thus represent potential CNS windows for autonomic feedback. The Subfornical Organ (SFO) One such sensory CVO is the subfornical organ (SFO), located in the forebrain on the midline wall of the third ventricle dorsal to the anterior commissure at the dorsal area of the lamina terminalis. The majority of anatomical data suggests that SFO neurons have 20 relatively compact dendritic trees and do not receive extensive neural inputs (Dellman & Simpson, 1979), supporting the suggested principle role of this region: receiving afferent information from peripheral circulation. The SFO then communicates this information, using well documented efferent neural projections to areas in the brain, including numerous hypothalamic autonomic control centers. SFO neurons send efferent projections to all of the important hypothalamic autonomic nuclei including the PVN, supraoptic nucleus of the hypothalamus (SON) (Miselis et al., 1979;Lind et al., 1982), median preoptic nucleus (MnPO) (Lind et al., 1982), ARC (Gruber et al., 1987), and LH (Miselis, 1981). Although the majority of input to the SFO is sensory in nature, afferent inputs to SFO are received from the LH, MnPO, lateral parabrachial nucleus, midbrain raphe, the nucleus reunions of the thalamus, and NTS (Lind, 1986;Lind et al., 1982;Zardetto-Smith & Gray, 1987). Classically, the SFO has been viewed primarily as an angiotensin sensor, with roles in body fluid homeostasis (Simpson & Routtenberg, 1975;Thrasher et al., 1982). A high density of binding sites for angiotensin II (ANG) have been demonstrated in the SFO and the SFO has been shown to be the primary CNS location mediating ANG induced dipsogenesis (Simpson & Routenberg, 1973;Mangiapane & Simpson, 1980b). Microinjection of ANG into the SFO causes drinking in satiated rats (Simpson et al., 1978;Simpson & Routenberg, 1973), while lesioning of the SFO abolishes drinking normally observed in response to icv and systemic ANG administration (Thrasher et al., 21 1982;Simpson et al., 1978). Direct electrical activation of SFO has also been shown to cause drinking in satiated rats (Smith et al., 1995). In addition to its role in fluid balance, the SFO has been shown to play a significant role in cardiovascular regulation. Studies showing that both electrical (Ishibashi & Nicolaidis, 1981;Mangiapane & Brody, 1983;Ferguson & Renaud, 1984;Smith et al., 1997) and chemical (Gutman et al., 1988b;Wall et al., 1992;Washburn et al., 1999;Mangiapane & Simpson, 1983) stimulation of this circumventricular structure causes increases in blood pressure (BP), effects that are abolished by SFO lesion (Mangiapane & Simpson, 1980a) or transection of SFO efferents (Lind et al., 1983), clearly provide support for a role for the SFO in cardiovascular regulation. The SFO has been shown to influence cardiovascular function through projections to the hypothalamus and other autonomic control centers (Lind et al., 1982;Ferguson & Renaud, 1984). A Role for the SFO in Regulation of Energy Balance Although the cause of the profound obesity seen as a consequence of MSG administration has centered on the absence of the ARC, the SFO was also damaged by these excitotoxic lesions (Takasaki, 1978;Olney & Sharpe, 1969;Olney et al., 1972;Arees & Mayer., 1970). The fact that site specific ARC lesions alone still result in increases in food intake leading to obesity (Choi & Dallman, 1999), suggest that SFO lesion is not critical for the development of this phenotype. However, it would be interesting to discern whether SFO lesion alone would lead to hyperphagia and obesity and whether discrete bilateral ARC lesions lead to the same degree of obesity, in the same time frame, 22 as when both the ARC and SFO are lesioned. Interestingly, we have shown that lesions of the SFO and the area postrema (AP), a medullary sensory CVO chronically decrease food intake and body weight in rats (Baraboi et al., 2010b;Baraboi et al., 2010a) over a 30 day period. In addition, animals with these CVO lesions no longer exhibited a decrease in food intake normally observed in response to peripheral administration of peptide YY (PYY), a peptide released by the distal gut in proportion to energy intake, supporting a role for the CVOs in mediating the central effects of this peptide (Baraboi et al., 2010a). Furthermore, exendin-4 (glucagon-like peptide-1 (GLP-1) agonist) -induced expression of c-fos mRNA in limbic structures, the hypothalamus (ARC, PVN, SON) and hindbrain was attenuated in rats with lesions of the AP and SFO (Baraboi et al., 2010b) supporting an important role for these CVOs in the activation of CNS structures involved in homeostatic control. A potential role for the SFO in energy homeostasis is also suggested by its neural projections to hypothalamic areas with well documented roles in energy homeostasis and by the distribution of a number of different receptors in the SFO for peripheral signals reflecting the animals’ energy status. Anatomical data show that SFO neurons send dense efferent projections to important hypothalamic metabolic control centers including the ARC (Gruber et al., 1987), LH (Miselis, 1981), and PVN (Miselis et al., 1979;Lind et al., 1982) nuclei. Electrophysiological studies have demonstrated functional projections from the SFO to the PVN and LH (Tanaka et al., 1986a;Tanaka et al., 1986b) while glutamate stimulation of ARC neurons alters the firing rate of SFO neurons (Rosas-Arellano et al., 23 1996). A suggested involvement for the SFO in the regulation of energy homeostasis is also derived from studies demonstrating the localization of receptor or receptor mRNA in SFO for the gut peptides ghrelin (Pulman et al., 2006), amylin (Sexton et al., 1994;Christopoulos et al., 1995;Riediger et al., 1999b;Hindmarch et al., 2008), and PYY (Kishi et al., 2005) and the adipokines adiponectin (Alim et al., 2010;Hindmarch et al., 2008) and leptin (LepR) (Hindmarch et al., 2008). ObR-like immunoreactivity has also been demonstrated in the SFO (Meister & Hakansson, 2001), however, in neither this study nor the gene array study (Hindmarch et al., 2008) were the specific leptin receptor subtypes determined. Interestingly, when we examined the number of genes that underwent 2 fold or greater changes as a consequence of water and food deprivation in or gene array studies, we found that food deprivation changed almost 30 x more genes than did water deprivation. These results further suggest an important role for the SFO in the control of energy balance. A functional role for the SFO in sensing circulating signals involved in the regulation of energy status has been suggested by electrophysiological studies demonstrating that the anorexigenic satiety signals, amylin, (Pulman et al., 2006;Riediger et al., 1999a) and adiponectin (Alim et al., 2010), the orexigenic satiety signal, ghrelin (Pulman et al., 2006) as well as glucose (Medeiros et al., 2012), influence the excitability of separate populations of SFO neurons. Together, these studies provide anatomical and functional evidence for routes through which large, lipophobic, peptidergic peripheral 24 signals which do not cross the BBB could gain access to the CNS via the SFO to influence metabolic control centers in the hypothalamus. Is the ARC the Only Site Behind the BBB for Leptin Actions? The ARC has been regarded as the primary site in the brain in which signals reflecting energy status are received and integrated and then responsible for the engagement of effector circuits required to maintain energy balance and, as such, has been the major area of focus regarding the actions of leptin in the CNS. However, numerous populations of ObRb expressing neurons exist in regions of the CNS, other than the ARC, including other regions in the hypothalamus, midbrain, and brainstem that play important role in energy balance (Elmquist et al., 1997;Elmquist et al., 1998a;Elmquist et al., 1998b;Leshan et al., 2006;Grill et al., 2002). Interestingly, support for the notion that the ARC is not solely responsible for mediating the central effects of leptin comes from a recent study from Qi et. al (Qi et al., 2010) which suggests that ARC involvement in the central action of leptin may not be as critical/imperative as previously thought. Hypothalamo-pituitary disconnection (HPD), a surgical preparation which involves the complete destruction of the ARC (Clarke et al., 1983), was used determine whether systemic leptin administration would induce c-Fos activity in the hypothalamus. Leptin influenced Fos labeling in the DMN, VMN and PVN in control and in HPD animals suggesting that the ARC is not required for neuronal activation of the ‘second order’ neurons. The authors suggest that leptin can act directly on these nuclei rather than indirectly via initial activation of the ARC. Since these 25 hypothalamic nuclei are securely protected behind the BBB these areas do not have access to the constituents of peripheral circulation. Consistent with the idea that the SFO is a central site of action for leptin, is that activation in these regions of the hypothalamus is secondary to the initial activation of SFO by leptin, with subsequent activation of these hypothalamic nuclei via afferent projections from SFO. Caudal Brainstem and the Regulation of Energy Homeostasis Other research has shown that the neural control of energy expenditure is actually distributed among several brain sites. The importance of brainstem neuronal circuits in the regulation of feeding has also been demonstrated. The caudal brainstem (CBS) is well known to control the motor aspects of feeding (ie chewing swallowing etc,). However, it is now apparent that the CBS is a site of integration via signals relayed from gastrointestinal and gustatory efferents (Norgren, 1978) and circulating signals. Decerebrate animals (animals with surgical transection of forebrain projections to the brainstem) are still able to consume food placed in the oral cavity and modulate the amount of food they consume in response to a variety of gut-derived and hormonal cues. These decerebrate animals gained less weight than controls but they did exhibit reduced energy expenditure and decreased serum leptin and insulin in response to fasting in the same manner as control animals (Harris et al., 2006) further demonstrating that the isolated caudal brainstem is sufficient to mediate many aspects of the energetic response to starvation. 26 Studies have identified POMC neurons within the commissural region of NTS in the caudal brainstem (Mountjoy et al., 1994;Kishi et al., 2003;Cone, 2005). Administration of MC4R agonists and antagonists into the fourth ventricle or directly into the caudal brainstem has been shown to influence food consumption (Grill et al., 1998; Williams et al., 2000; Zheng et al., 2005) while POMC overexpression in the NTS causes a persistant hypophagia leading to a sustained weight loss (Li et al., 2007), providing further support for a role for the caudal brainstem in energy homeostasis. The Area Postrema (AP) The AP is a sensory CVO located in the CBS at the dorsal surface of the medulla on the wall of the fourth ventricle. As is characteristic of the CVOs, the absence of tight junctions between the endothelial cells of the vasculature allow systemic dyes to be taken up by the AP, however, a border zone of astrocytes and tanycytes, joined by tight junctions, form a diffusion barrier between the ventral surface of the AP and immediately adjacent to the NTS (Wang et al., 2008;McKinley et al., 2003) which prevents the movement of dye to surrounding tissue (Wislocki & Leduc, 1952). The AP is best known for its role as the chemoreceptor trigger zone in the control of the emetic reflex (Borison & Brizzee, 1951;Carpenter et al., 1983;Miller & Leslie, 1994) but also has well-documented roles in cardiovascular regulation, fluid balance, and energy homeostasis (see Cottrell & Ferguson, 2004;Price et al., 2008 for review). The AP sends efferent projections to hypothalamic and medullary autonomic nuclei, including the NTS, lateral parabrachial nucleus, dorsal motor nucleus of the vagus, nucleus ambiguous, SON, and PVN (van der & Koda, 1983;Shapiro & Miselis, 1985a;Gross et al., 27 1990;Iovino et al., 1988; for review see Fry et al., 2007) while receiving afferent input from the NTS, DMH, PVN, lateral parabrachial nucleus and the vagus nerve (van der & Koda, 1983;Shapiro & Miselis, 1985a;Gross et al., 1990;Rogers & Hermann, 1983;Kalia & Sullivan, 1982). A role for the AP in the regulation of energy balance is well established. Not only does the AP contain receptors for numerous peripheral signals involved in feeding and metabolism (Hindmarch et al., 2008; for review see Price et al., 2008), electrophysiological studies have shown that a number of circulating signals reflecting metabolic status, such as amylin, ghrelin, cholecystokinin and adiponectin, influence the excitability of AP neurons (Riediger et al., 2002;Sun & Ferguson, 1997;Fry et al., 2006;Fry & Ferguson, 2009). Although no studies have directly examined the responsiveness of the AP to leptin, the demonstration of ObR-like immunoreactivity (Meister & Hakansson, 2001) and mRNA expression (Grill et al., 2002) in the AP suggest potential actions for leptin in this sensory CVO. In addition, recent gene array studies from our own laboratory have revealed the presence of the leptin receptor mRNA in the AP (Hindmarch et al., 2008). The specific leptin receptor subtypes were not, however, determined in any of the above studies. Measurements of phosphorylated-signal transducer and activator of transcription 3 (pSTAT3) immunoreactivity, a direct downstream marker of leptin receptor activation, has also been observed in the AP (Ellacott & Cone, 2006;Huo et al., 2007). At the physiological level, leptin injection into the 4th ventricle has been shown to suppress appetite (Skibicka & Grill, 2009) while knockdown of LepR in the mNTS and AP causes hyperphagia as well as increased body weight and adiposity (Hayes et al., 2010) 28 providing further evidence for a role of the AP in mediating the central effects of leptin. In addition, a recent study demonstrated that the AP plays a pivotal role in mediating a synergistic weight loss effect of amylin and leptin in leptin resistant diet-induced obese rats (Roth et al., 2008). Thus, the AP may provide a route in which circulating leptin may act to influence downstream autonomic nuclei that regulate feeding behaviour. Leptin Effects on Blood Pressure Not surprisingly, since the initial discovery of the weight reducing effect of leptin, this adipokine has also been suggested to play a role in blood pressure (BP) regulation. A positive correlation has been demonstrated between leptin levels and BP in obese individuals (Al-Hazimi & Syiamic, 2004;Itoh et al., 2002) suggesting that leptin may play a primary role in obesity-related hypertension. The fact that animals which are genetically leptin deficient (Mark et al., 1999) or lacking a functional leptin receptor (Chan & Johnson, 1997) do not demonstrate increases in BP despite profound obesity suggests that receptor mediated actions of leptin may be responsible for the regulation of systemic hemodynamics. Although acute systemic leptin administration has been shown to be without cardiovascular effect, chronic systemic administration causes increases in BP (Shek et al., 1998;da Silva et al., 2004). In addition, acute, central (icv) leptin administration has been shown to elevate BP in anesthetized (Dunbar et al., 1997;Rahmouni & Morgan, 2007;Lu et al., 1998) and conscious rats (Casto et al., 1998) as does chronic icv administration (Dubinion et al., 2011). Direct administration of leptin into specific cell groups within the hypothalamus expressing the leptin receptor in which neurons have been shown to be influenced by leptin, including the ARC (Rahmouni & 29 Morgan, 2007), VMH (Montanaro et al., 2005;Marsh et al., 2003) and dorsal medial hypothalamus (DMH) (Marsh et al., 2003) has been shown to cause increases in BP in anesthetized rats. Selective Leptin Resistance Ob/ob and db/db mice develop profound obesity with increased appetite and decreased energy expenditure due to a mutation in the gene that encodes leptin (ob/ob) (Campfield et al., 1995;Zhang et al., 1994) or the gene that encodes the leptin receptor (db/db) (Tartaglia et al., 1995). Although there are human correlates with this same mutation, most causes of human obesity are multifactorial and are not caused by solely by specific mutations in the leptin gene or leptin receptor gene. Most human obesity is associated with hyperleptinemia (Considine et al., 1996) and as such, obesity is not a consequence of an inability to produce leptin but rather an impairment in the ability of the obese individual to respond appropriately to the elevated circulating leptin levels. Thus, the concept of selective leptin resistance has been used to describe the phenomenon by which obese individuals are resistant to the weight reducing effects of leptin while still responsive to the sympathoexcitatory effects contributing to obesity related hypertension (Mark et al., 2002;Correia et al., 2002). 30 STATEMENT OF THE PROBLEM This thesis is composed of 5 manuscripts which examine the role of two circumventricular structures, the subfornical organ (SFO) and the area postrema (AP) in mediating the central effects of leptin, a circulating adipokine, known to act at hypothalamic and brainstem nuclei to control feeding behaviour and energy expenditure. In vitro molecular and electrophysiological studies are utilized to determine the presence of the signaling form of the leptin receptor, ObRb, in the AP and SFO and the functional consequence of ObRb activation, respectively. In vivo stimulation, microinjection, and lesion studies are employed to determine the physiological relevance of theses CVOs in autonomic functions including food intake (SFO stimulation and AP/SFO lesion studies) and blood pressure regulation (SFO microinjection studies). 1. Chapter 2 (manuscript 1) examines the effect of electrical stimulation of the SFO on feeding behaviour in conscious satiated rats. 2. Chapter 3 (manuscript 2) investigates the presence of the signaling form of the leptin receptor (ObRb) in the SFO and evaluates the effect of leptin application on membrane excitability of SFO neurons. The effect of amylin on the excitability of leptin-sensitive SFO neurons is also examined. 3. Chapter 4 (manuscript 3) evaluates the cardiovascular consequences of microinjecting leptin directly into the SFO of anesthetized rats. This study also determines whether these cardiovascular effects to leptin remain in leptin resistant, diet induced obese (DIO) rats. 31 4. Chapter 5 (manuscript 4) investigates the presence of the signaling form of the leptin receptor (ObRb) in the AP and evaluates the effect of leptin application on membrane excitability of AP neurons. The effect of amylin on the excitability of leptin-sensitive AP neurons is also examined. 5. Chapter 6 (manuscript 5) explores whether rats with lesions of the SFO and AP remain responsive to the weight reducing effect of peripheral leptin administration. 32 Chapter 2: ACUTE ELECTRICAL STIMULATION OF THE SUBFORNICAL ORGAN INDUCES FEEDING IN SATIATED RATS 33 ABSTRACT The SFO, a circumventricular organ (CVO) that lacks the normal blood-brain barrier, is an important site in central autonomic regulation. A role for the SFO in sensing circulating satiety signals has been suggested by electrophysiological studies demonstrating that the anorexigenic satiety signals, leptin and amylin, as well as the orexigenic satiety signal, ghrelin, influence the excitability of separate populations of SFO neurons. The present study examined whether acute, short duration, electrical stimulation of the SFO influenced feeding in satiated rats. Electrical stimulation (200µA) of satiated animals with subfornical organ (SFO) electrode placement (n = 6) elicited feeding in all animals tested with a mean latency to eat of 8.0 ± 4.0 min after termination of SFO stimulation (mean food consumption: 0.6 ± 0.12g /100g bw). These same rats undergoing a sham stimulation did not eat (mean food consumption: 0.0 ± 0.0g, n=6) nor did animals receiving stimulation with non-SFO electrode placements (mean food consumption: 0.0 ± 0.0g, n=6). SFO stimulation at this intensity elicited drinking in 5/6 animals with a mean latency to drink of 15.2 ± 2.6 min. Feeding effects were specific to higher stimulation intensities as lower intensity stimulation (100µA, n=6) elicited drinking (mean latency to drink: 6.2 ± 2.6 min) but did not cause any animal to eat. The results of the present study show that acute, short duration, SFO stimulation induces feeding in satiated rats, lending support for a role for the SFO as an integrator of circulating peptides that control feeding. 34 INTRODUCTION The subfornical organ (SFO), located on the midline wall of the third ventricle, belongs to a group of specialized central nervous system (CNS) structures known as the sensory circumventricular organs (CVOs). CVOs are characterized by the lack of the normal blood brain barrier (BBB), a dense vascular supply, and the presence of a wide variety of peptidergic receptors. Classically, the SFO has been viewed primarily as an angiotensin sensor, with roles in body fluid homeostasis (Simpson & Routtenberg, 1975;Thrasher et al., 1982) and cardiovascular regulation (Mangiapane & Simpson, 1980b;Ferguson & Renaud, 1984). Electrical activation of SFO has been shown to cause drinking in satiated rats (Smith et al., 1995) and increase blood pressure in anesthetized rats (Smith et al., 1997). The SFO has been shown to influence cardiovascular function through projections to the hypothalamus and other autonomic control centers (Lind et al., 1982;Ferguson & Renaud, 1984). SFO neurons send efferent projections to important hypothalamic autonomic nuclei including the paraventricular nucleus (PVN) and supraoptic nucleus of the hypothalamus (SON) (Miselis et al., 1979;Lind et al., 1982), the median preoptic nucleus (MnPO) (Lind et al., 1982), the arcuate nucleus (ARC) (Gruber et al., 1987) and lateral hypothalamic (LH) nuclei (Miselis, 1981). Although the majority of input to the SFO is sensory in nature, afferent inputs to SFO are received from the LH, MnPO, lateral parabrachial nucleus, midbrain raphe, the nucleus reunions of the thalamus, and nucleus tractus solitarius (Lind, 1986;Lind et al., 1982;Zardetto-Smith & Gray, 1987). 35 In addition to its roles in body fluid homeostasis and cardiovascular regulation, more recently, the SFO has been suggested to be involved in the regulation of energy homeostasis. The SFO has also been shown to contain receptors or receptor mRNA for a variety of peripheral signals involved in energy homeostasis including the satiation signal, amylin (Paxinos et al., 2004;Sexton et al., 1994), ghrelin (Pulman et al., 2006), a peptide that triggers meal initiation, and the adiposity signals, adiponectin (Hindmarch et al., 2008), and leptin (Hindmarch et al., 2008;Smith et al., 2009). A functional role for the SFO in sensing circulating signals involved in energy homeostasis has been suggested by electrophysiological studies from our laboratory demonstrating that the anorexigenic satiety signals, leptin (Smith et al., 2009) and amylin (Smith et al., 2009;Pulman et al., 2006;Riediger et al., 1999a), as well as the orexigenic satiety signal, ghrelin (Pulman et al., 2006), influence the excitability of separate populations of SFO neurons and is further supported by the anatomical data showing that SFO neurons send dense efferent projections to important hypothalamic metabolic control centers including the ARC (Gruber et al., 1987), LH (Miselis, 1981), and PVN (Miselis et al., 1979;Lind et al., 1982). In addition, electrophysiological studies have demonstrated functional projections from the SFO to the PVN and LH (Tanaka et al., 1986a;Tanaka et al., 1986b) and that glutamate stimulation of ARC neurons alters the firing rate of SFO neurons (RosasArellano et al., 1996). Together, these anatomical and functional data provide evidence for routes through which large, lipophobic, peptidergic peripheral signals which do not 36 cross the BBB could gain access to the CNS via the SFO to influence metabolic control centers in the hypothalamus. The present study was undertaken to determine whether electrical activation of SFO neurons influences feeding in satiated rats. MATERIALS AND METHODS Animals were maintained on a 12:12 light:dark cycle and provided food and water ad libitum for the duration of the experiment. Sodium pentobarbital anesthetized (65mg/kg, ip) male Sprague Dawley rats (175-200g) were placed in a stereotaxic frame, an incision in the skin of the skull was made, and a small burr hole drilled such that a concentric bipolar stimulating electrode (SNE100 Rhodes Medical Instruments, tip exposure 250µm) could be advanced into the region of the SFO (Bregma, -.07mm, midline, 4.5mm ventral to surface) according to the coordinates of Paxinos and Watson (Paxinos & Watson, 1982). The electrode was secured to the skull using jeweller’s screws and dental cement. The animal was allowed a minimum 7 day recovery period. Food and water intake as well as body weight was recorded daily. On the days of experimentation the animal was placed in an observation cage designed for monitoring feeding and drinking 30 min after ‘lights on’ and allowed a minimum 30 min habituation period. Immediately following the 30 min habituation period, a pre-stimulation control period was begun during which the eating and drinking behavior was monitored and the activity level of the animal was assessed every min using a modified Ellinwood and Balster’s Behavioral Arousal Scale (Ellinwood, Jr. & Balster, 1974), a nine point scale 37 where 1 indicates the animal is asleep, 5 is characterized by hyperactivity (rapid, jerky changes in position (no animal received a score this high) as previously described (Smith et al., 1995). Once the animal was considered quiescent (mean activity was less than 3, a score represented by some in-place activities such as grooming) the 5 min stimulation period was begun during which the animal received either a 5 min electrical stimulation (10Hz, 1ms pulse duration, 100μA or 200μA, center pole as cathode) or a 5 min sham stimulation (electrode connected but no current passed) during which eating and drinking were monitored and activity level was assessed every 30 sec. After termination of electrical or sham stimulation (post stimulation period), the animal remained in the observation cage for 20 min (4, 5 min post stimulation periods) during which eating, drinking and activity level was evaluated every min. Approaches to, and time spent at, the food hopper was recorded and was latency to eat and drink. The amount of food consumed was measured at the end of the 20 min post stimulation period. Animals received both electrical and sham stimulation protocols, separated by at least 72 hours and performed in a randomized order. Following these protocols, animals were overdosed with sodium pentobarbital and perfused through the left ventricle of the heart with saline followed by 10% formalin and the brain was removed. The following day, 100µm coronal sections were cut through the region of SFO, mounted and cresyl violet stained. An observer, unaware of the experimental outcome, verified the anatomical location of the stimulating electrode at the light microscopic level. 38 Animals were grouped according to the anatomical location of the stimulation electrode (SFO or non-SFO) and stimulation intensity (100 or 200µA). These groups were further divided according to stimulation protocol (electrical or sham stimulation). Latency to eat and/or drink was measured. Food intake was measured at the end of the last post stimulation period (20 min after termination of stimulation). Mean activity scores were calculated for each animal in each group for the 5 min pre-stimulation time period, the 5 min stimulation period, and each of the four, 5 min post-stimulation time periods. Mean activity scores were then calculated for each group. An ANOVA was used to determine the effect of stimulation and sham stimulation in SFO or non-SFO on these parameters. A Tukey post hoc analysis was used to determine where these differences occurred. RESULTS A total of 24 rats were used in the current study of which 10 were placed in the SFO group and 8 were placed in the non-SFO group according to the anatomical location of the stimulating electrode. Animals were placed in the SFO group when the tip of the stimulating electrode was located within 400µm of the center of SFO without penetrating the ventricle while animals with electrode placements outside this region were placed in the non-SFO group (see Figure 1). The remaining 6 animals had either extensive damage, rendering exact anatomical location of stimulation site impossible, or penetrated the ventricle and thus were excluded from further analysis. Animals within these 39 Figure 2-1: Animals were grouped according to anatomical location of the stimulating electrode Schematic diagrams on the left show the anatomical locations of the stimulating electrode. Electrode placements in the SFO group are indicated by circles while non-SFO electrode placements are indicated by the squares. Open symbols indicate stimulation intensities of 200µA while black closed symbols indicate stimulation intensities of 100µA. Closed grey circles indicate animals stimulated at both intensities. The photomicrograph on the right shows an example of a stimulating electrode site within SFO. Bregma level is indicated at the bottom of each schematic. Scale bars = 500µm. Abbreviations: SFO: subfornical organ, v: ventricle, fx: fornix, cc: corpus callosum 40 anatomical groups (SFO or non-SFO) were further divided according to the stimulation intensity (100 or 200µA) delivered. SFO stimulation induced feeding in satiated rats Electrical stimulation of the SFO at the higher intensity (200µA, 10Hz, 1ms) induced feeding in all satiated rats tested (6/6). Animals did not eat during the 30 min habituation period or the pre-stimulation period. Feeding usually occurred within 4 min of termination of SFO stimulation (5/6 animals) with a mean latency to eat of 8.0 ± 4.0 min after termination of SFO stimulation. Mean food consumption in SFO stimulated animals was 0.6 ± 0.12g /100g bw (n= 6, p < .01). These same SFO stimulated animals did not eat during sham stimulation (electrode connected but no current passed, mean food consumption = 0.0 ± 0g /100g bw, n=6, see Figure 2-2, Table 2-1). Feeding effects were specific to stimulation sites within the anatomical boundaries of SFO as animals with stimulation locations outside of the SFO did not eat in response to stimulation (mean food consumption = 0.0 ± 0g /100g bw, n=6, see Figure 2-2, Table 2-1). SFO stimulation at this intensity also elicited drinking in 5/6 animals (mean water consumption = 1.4 ± 0.4ml, n=6) with a mean latency to drink of 15.2 ± 2.6 min (see Table 2-1). These effects were also specific to SFO stimulation as these same animals undergoing sham stimulation did not drink nor did animals with stimulating electrodes outside the anatomical boundaries of SFO (non-SFO, n=6). Feeding effects were specific to higher intensity stimulation (200µA) as lower intensity SFO stimulation (100µA, n=6) did not cause feeding in any of the animals tested (0/6), although these animals did appear to have a 41 Table 2-1: Summary of the effects of electrical stimulation at 100 and 200µA of the SFO on eating and drinking 42 Figure 2-2: SFO stimulation induced feeding Electrical stimulation (200µA, 10Hz, 1ms) of the SFO induced feeding in satiated rats. Bar graph shows food intake as a result of SFO stimulation (SFO Stim, n=6), sham stimulation (SFO Sham, electrode connected but no current passed, n=6) and stimulation of locations outside of the SFO (non-SFO Stim, n=6). ** p < .01. 43 greater interest in food as determined by the number of times the animals approached the food hopper and time spent at the food hopper after termination of stimulation. SFO stimulated animals approached the hopper 7.5 ± 2.1 times for 42.8 ± 20.1sec during the first post stimulation period while sham stimulated animals did not approach the food hopper at all nor did non-SFO stimulated animals. This lower intensity stimulation did however elicit drinking (mean latency to drink: 6.2 ± 2.6 min) in 5 of 6 SFO stimulated animals, as previous reported (Smith et al., 1995). Activity level was not different during stimulation Activity levels were measured prior to (Pre), during (Stim), and after (Post) stimulation protocols. Activity levels during the pre-stimulation period (Pre) and during electrical stimulation (Stim) were not different between the SFO group (SFO) and nonSFO group (non-SFO), nor were activity levels between electrically stimulated (Stim) and sham (Sham) stimulated animals (see Figure 2-3). There was, however, an increase in activity in SFO stimulated animals throughout the entire post-stimulation period (p<.005, p<.01). This increase in activity was likely related to the feeding and subsequent grooming activities of these animals. DISCUSSION The results of the present study show, for the first time, that electrical stimulation of the SFO elicits feeding and confirm our previous reports of drinking (Smith et al., 1995) in satiated rats. 44 Figure 2-3: Activity level was not different during stimulation Activity levels were similar between SFO groups (●, n=6) and Non-SFO groups (, n=6) during the pre-stimulation period (Pre) and during electrical stimulation (Stim) and in animals undergoing sham stimulation (broken lines) of SFO or non-SFO. There was an increase in activity in SFO stimulated animals the entire post-stimulation period (Post 14). This increase in activity was related to the feeding and subsequent grooming activities of these animals. Each data point represents mean ± SEM. Error bars are within the confines of the data point if not visible. *** p<.005, ** p<.01, one way ANOVA 45 Rats were tested at the beginning of the light cycle, a period during which these animals are typically asleep and thus do not ingest either food or water. It is during the dark cycle that rats consume the majority of their daily food and water. Although small bouts of eating and drinking are known to occur during the light cycle, these bouts typically occur several hours after the beginning of the light cycle and in the last hour of the light cycle in anticipation of lights off. In this study, electrical stimulation (200µA) of the SFO elicited feeding in rats that were not previously food or water deprived. Not only did the rats eat, but they ate up to 10% of their total daily food intake in less than 30 min at a time when they are typically not eating. These effects were specific to SFO stimulation as animals with stimulating electrode placement outside the anatomical boundaries of SFO did not eat in response to stimulation. The effect of electrical stimulation on feeding was intensity dependent as stimulation at lower intensities (100µA) did not induce feeding. Although only qualitatively assessed, low intensity SFO stimulation appeared to increase the animals’ interest in food (as determined by the amount of time spent at the food hopper), however, these animals did not eat. In contrast to feeding responses, drinking occurred in animals at both stimulation intensities (100 and 200µA) as previously reported (Smith et al., 1995). Some animals receiving low intensity SFO stimulation drank while undergoing electrical stimulation whereas no animals receiving higher intensity SFO stimulation drank during the stimulation period. In fact, latency to drink in the high intensity stimulation group was 46 significantly longer than in those animals that received low intensity stimulation. Interestingly, although all animals ate in response to higher intensity stimulation, not all animals drank and, in most cases (5/6), eating preceded drinking in animals that exhibited both ingestive behaviours. Activity levels were monitored throughout the experiment to determine whether effects seen may be attributable to a general change in activity levels (arousal) rather than a specific effect on feeding. The fact that animals in all groups (SFO and non-SFO, electrically and sham stimulated) had similar activity levels prior to and during stimulation, suggests that changes in activity did not underlie changes in feeding or drinking. The only differences seen in activity levels were during the post stimulation period in SFO stimulated rats, presumably attributable to eating and drinking and associated grooming etc. behaviours that would be expected to accompany eating. One interesting question that arises from these observations is why is a higher intensity stimulus required to cause feeding while lower intensity stimulation is sufficient to elicit drinking? The fact that stimulation intensities required to induce feeding were greater than those required to elicit drinking may be related to the fact that angiotensin elicits drinking by homogenous excitatory effects on approximately 60% of SFO neurons (Li & Ferguson, 1993;Gutman et al., 1988a;Schmid & Simon, 1992). Thus angiotensin elicits drinking by homogenous excitatory effects on SFO neurons. This is in contrast to the effect of anorexigenic and orexigenic circulating satiety factors which have heterogeneous effects on the excitability of SFO neurons. 47 Studies from our own laboratory have demonstrated that separate subpopulations of SFO neurons are activated by the anorexigenic peptide, amylin, or the orexigenic peptide, ghrelin (Pulman et al., 2006). Leptin has also been shown to influence the excitability of the majority of SFO neurons, however, these effects were shown to be heterogeneous as a population of SFO neurons were inhibited by leptin while a second group of cells were excited by leptin administration. Interestingly, cells excited by the anorexigenic peptide, amylin, were also excited by leptin (Smith et al., 2009). During the light cycle, SFO neurons inhibiting food intake are likely maximally activated and, thus, in order to override this inhibitory drive to stimulate feeding, higher stimulation intensities are required to activate a sufficient proportion of SFO neurons that stimulate feeding. The results of the present study further support a role for the SFO in the regulation of energy homeostasis. Electrical stimulation in vivo mimics the effect that circulating satiety signals have on neurons in the SFO. Previous in vitro studies have demonstrated that individual circulating satiety factors influence the activity of dissociated SFO neurons (Smith et al., 2009;Pulman et al., 2006) and neurons obtained in slices (Smith et al., 2009). In contrast to electrophysiological studies, electrical stimulation does not specifically target one particular neuronal subtype based on the receptors present, but rather activates all neurons (orexigeninc and anorexigenic) in the region of the tip of the stimulating electrode. The fact that such activation elicits feeding in satiated rats attests to the integrative action of the SFO, further supporting 1) the notion of the SFO as a regulatory target for peripheral molecules reflecting an individual’s 48 energy status, and 2) a role for the SFO in influencing autonomic function through its neural projections to hypothalamic nuclei involved in energy homeostasis. 49 Chapter 3: THE SUBFORNICAL ORGAN: A CNS SITE FOR ACTIONS OF CIRCULATING LEPTIN 50 ABSTRACT Adipose tissue plays a critical role in energy homeostasis, secreting adipokines that control feeding, thermogenesis and neuroendocrine function. Leptin is the prototypic adipokine that acts centrally to signal long term energy balance. Whilst hypothalamic and brainstem nuclei are well-established sites of action of leptin, we tested the hypothesis that leptin signaling occurs in the subfornical organ (SFO). The SFO is a circumventricular organ (CVO) which lacks the normal blood brain barrier, is an important site in central autonomic regulation, and has been suggested to have a role in modulating peripheral signals indicating energy status. We report here the presence of mRNA for the signaling form of the leptin receptor in SFO, and leptin receptor localization by immunohistochemistry within this CVO. Central administration of leptin resulted in phosphorlylation of STAT3 in neurons of SFO. Whole cell current clamp recordings from dissociated SFO neurons demonstrated that leptin (10nM) influenced the excitability of 64% (46/72) of SFO neurons. Leptin was found to depolarize the majority of responsive neurons with a mean change in membrane potential of 7.3 ± 0.6 mV (39% of all SFO neurons), while the remaining cells which responded to leptin hyperpolarized (-6.9 ± 0.7 mV, 25% of all SFO neurons). Leptin was found to influence the same population of SFO neurons influenced by amylin as 3 of 4 cells tested for the effects of bath application of both amylin and leptin depolarized to both peptides These observations identify the SFO as a possible central nervous system location, with direct 51 access to the peripheral circulation, at which leptin may act to influence hypothalamic control of energy homeostasis. 52 INTRODUCTION Adipose tissue plays a critical role in energy homeostasis, secreting adipokines that control feeding, thermogenesis, immunity, and neuroendocrine function. Leptin, a 16 KDa peptide, is the prototypic adipokine that has been shown to be an important afferent signal regulating body weight. A missense mutation in the ob gene (the gene that encodes leptin) in the ob/ob mouse, results in a markedly obese phenotype which is normalized by both systemic and central leptin administration (Halaas et al., 1995b;Pelleymounter et al., 1995;Campfield et al., 1995). Leptin administration also has been shown to dosedependently decrease body weight, preferentially reducing body fat while sparing lean tissue, in both ob/ob and wild type mice (Halaas et al., 1995b;Halaas et al., 1997). Plasma leptin levels reflect both energy stores and acute energy balance. Circulating leptin decreases food intake and increases energy expenditure through activation of receptors in hypothalamic and brainstem neurons (Grill & Kaplan, 2002) . The leptin receptor, encoded by the Ob-R gene, was isolated from choroid plexus by expression cloning and is a member of the cytokine family (Tartaglia et al., 1995). Although 5 leptin receptor isoforms have been identified (Ob-Ra – Ob-Re), only the long form of the receptor, Ob-Rb, possesses the cytoplasmic domains required for signal transduction (Bjorbaek et al., 1997;Banks et al., 2000;Kloek et al., 2002). Ob-Rb regulates multiple intracellular signaling cascades, including the janus activating kinasesignal transducer and activator of transcription (JAK-STAT) pathway and the phosphoinositol-3 kinase and adenosine monophosphate kinase pathways and is essential for the weight reducing effect of leptin (Bjorbaek et al., 1997;Bjorbaek & Kahn, 53 2004;Buettner et al., 2006). In particular, Ob-Rb is found in hypothalamic nuclei involved in feeding behaviour including the arcuate nucleus (ARC), paraventricular nucleus (PVN), dorsomedial nucleus (DMH), and the lateral hypothalamic area (LHA) (Mercer et al., 1996) and it is clear that leptin signaling in these structures plays a pivotal role in regulating energy balance. The presence of the blood brain barrier (BBB) leads to the obvious question as to how this peripheral peptide gains access to the central sites. While peptide transporter systems (Banks et al., 1996) and transendothelial signaling (Paton et al., 2007) represent mechanisms through which peripheral signals may reach hypothalamic neurons behind the BBB, an alternative explanation also deserves consideration. The sensory circumventricular organs (CVOs) are a group of central nervous system structures which lack the normal BBB. These specialized regions have been shown to contain a dense vasculature, fenestrated epithelium and the presence of a large variety of peptidergic receptors. Thus, the CVOs are uniquely suited to detect the presence (or absence) of circulating signals and relay this information via well documented efferent pathways to hypothalamic autonomic nuclei (see Fry et al., 2007 for review). A role for the sensory CVO’s in mediating the weight reducing effects of leptin is supported by a recent study demonstrating that the area postrema (AP), a hindbrain CVO, plays a pivotal role in mediating a synergistic weight loss effect of amylin and leptin in leptin resistant diet-induced obesity rats (Roth et al., 2008). 54 A potential role for the SFO, a forebrain CVO, in energy homeostasis is suggested by its neural projections to hypothalamic areas with well documented roles in energy homeostasis and the distribution of a number of different receptors for peripheral signals reflecting the animals’ energy status. Electrophysiological studies have demonstrated that SFO neurons project to the PVN and LHA (Tanaka et al., 1986a;Tanaka et al., 1986b) and that glutamate stimulation of ARC neurons alters the firing rate of SFO neurons (Rosas-Arellano et al., 1996). In addition, audioradiographic studies have demonstrated reciprocal SFO connections with the LHA (Miselis, 1981). A suggested involvement for the SFO in the regulation of energy homeostasis is also derived from studies demonstrating receptors in SFO for the gut peptides ghrelin (Pulman et al., 2006), amylin (Sexton et al., 1994;Christopoulos et al., 1995), and PYY (Kishi et al., 2005). A role for the SFO in modulating circulating signals reflecting energy status has been suggested by studies in which we demonstrated that ghrelin and amylin, circulating signals with opposite effects on feeding behaviour, influence the excitability of different subpopulations of SFO neurons (Pulman et al., 2006). Ob-R like immunoreactivity has been demonstrated in the sensory CVOs (SFO, AP, organum vasculosum of the lamina terminalis) (Meister & Hakansson, 2001), however, the specific leptin receptor subtypes were not determined. In addition, recent gene array studies from our own laboratory have revealed the presence of the leptin receptor mRNA in the SFO (Hindmarch et al., 2008). The present study was undertaken to test the hypothesis that leptin signaling occurs in the subfornical organ (SFO). We 55 determined if the signaling form of the leptin receptor (Ob-Rb) is found in SFO, and examined the functional effects of activation of this receptor on SFO neurons. METHODS For all experiments, animals were maintained on a 12/12 light-dark cycle and provided with food and water ad libitum prior to experimentation, except where indicated. All animal protocols were in accordance with Canadian Council for Animal Care (CCAC) guidelines and were approved by the Queen’s University and the University of Calgary Animal Care Committees. Leptin receptor localization in SFO using reverse-transcription-polymerase chain reaction (RT-PCR) Male Sprague Dawley rats (100-150g, Charles River, Quebec, Canada) were decapitated and the brain was quickly removed and placed in oxygenated ice-cold artificial cerebrospinal fluid (aCSF) containing (in mM) 124 NaCl (124), 2 KCl (2), 1.25 KH2PO4 (1.25), 2.0 CaCl2 (2.0), 1.3 MgSO4 (1.3), 20 NaHCO3 (20) and glucose (10). The SFO was visually identified at the dorsal surface of the third ventricle using a dissecting microscope and gently micro-dissected away from the immediately adjacent hippocampal commissure. Total RNA was extracted from SFO from two rats using Ambion RNAqueous kit and then DNase treated (Fermentas) by adding a mixture of 1 µl 10x buffer with MgCl2, 7 µl diethylpyrocarbonate (DEPC) treated-H2O and 1 µl 56 deoxyribonuclease to the total RNA and incubating the solution at 37°C for 30 min. After incubation, 1 µl of 25mM of EDTA was added to the solution and incubated at 65°C for 10 min. Oligo-dt based cDNA was synthesized using Superscript(TM) III reverse transcriptase kit (Invitrogen, Carlsbad, California, USA) to make a final reaction volume of 10 µl. Two microlitres of SFO cDNA was added to a PCR reaction containing: 25 µl of 2x QIAGEN Multiplex PCR Master Mix, 5 µl of Q solution, 2 µl of each primer set, and 16 µl DEPEC treated-H2O to a final volume of 50 µl. To prevent the formation of misprimed products and primer dimers, the HotStarTaq DNA polymerase (contained in the QIAGEN Multiplex PCR Master Mix) was activated by a 15min, 95ºC incubation step. The reaction tube was then cycled 45 times through a protocol of 94ºC for 60s, 60ºC for 60s, 72ºC for 60s and finally 72ºC for 10 min. Primers (Integrated DNA Technologies) directed toward an extracellular domain common to all leptin receptor isoforms were used to detect the presence of all leptin receptor isoform (LepR) mRNA (see Table 3-1). In order to determine whether the signaling form of the receptor (Ob-Rb) mRNA was present in SFO, one previously verified primer set (Burdyga et al., 2002) and one that we designed, both of which were specific for the intracellular signaling domains on Ob-Rb were used (see Table 3-1). Sets of primers were also used to detect Ob-Ra mRNA (see Table 3-1), as well as GAPDH (+ control). All of the aforementioned primers were also used in hypothalamic tissue, prepared in the same manner as the SFO, which served as a positive tissue control for the presence of LepR, Ob-Rb,and Ob-Ra mRNA. 57 An RT(–) reaction, in which the reverse transcriptase enzyme was omitted from the RTPCR reaction, was used as a negative control. PCR products were run and visualized on electrophoresis gel containing 2% agarose and ethidium bromide. Immunohistochemistry Frozen brain sections 35 µm thick were cut using a Leica CM3050 S cryostat (Richmond Hill, Ontario, Canada). For double labeling, sections were rinsed with 0.1% TritonX100 in PBS, placed in 10% goat serum 1 h, and incubated in primary antisera directed to all forms of the leptin receptor (LepR) (1:50; cat. No. SC 1834; Santa Cruz Biotechnologies, Santa Cruz, CA, the neuronal marker NeuN (1:100; cat. No. MAB 377; Chemicon, Billerica, MA), or the astrocyte marker glial fibrillary acidic protein (1:250; cat. No. BT-575 Biomedical Technologies, Inc., Stoughton, MA) for 24 h. Specificity for the leptin receptor antibody was confirmed by pre-incubating with the peptide (SC 1834P), the antibody it was raised against, which completely abolished the labeling. Donkey anti goat FITC (1:100, Jackson ImmunoResearch, West grove, PA), goat anti-mouse FITC (1:50, Jackson ImmunoResearch) and donkey anti-mouse CY3 (1:100, Biocan Scientific, Mississauga, ON, Canada) were used as secondary antisera. Staining was visualized using a Zeiss Axioplan fluorescence microscope and photographed with a digital camera, or, an Olympus Fluoview FV300 confocal microscope using krypton-argon and helium neon lasers. Differential visualization of the fluorphores FITC (excitation 490 nm and emission 520 nm) and CY3 (excitation 552 nm and emission 565 nm) was accomplished with the use of specific filter combinations. Samples were scanned sequentially and images were obtained under identical 58 Table 3-1: Primer sets used in the detection of mRNA from SFO 59 exposure conditions (pinhole aperture, laser strength, scan speed, Kalman averaging 2X). Confocal images are digital composites of Z-stacks scans 1 μm thick as detailed in the figure legends. Micrographs were generated with Fluoview Software and CorelDraw. Leptin induced pSTAT3 signaling Under ketamine /xylazine (85:15) anesthesia male Sprague-Dawley rats (250-250 g) were placed in a stereotaxic frame. The skull was exposed and a 25-gauge ga stainless steel guide cannula was positioned just above a lateral ventricle according to the coordinates of Paxinos and Watson (bregma; posterior, -1; lateral, 1; ventral, 3.2) (Paxinos & Watson, 1982) using a dental drill. The cannulae were fixed in place with dental acrylic and anchored to the skull with 2 screws. After 7 days of recovery and an overnight fast, rats were gently restrained and treated with either leptin (cat. no. L5037; Sigma-Aldrich, Oakville, ON, Canada) (5 µg; icv.; n=3 ) or vehicle (phosphate buffered saline (PBS, pH 7.4) (5 µl; n=3 ) by gravity flow using a 27-gauge needle connected to 20 cm of polyurethane tubing . Rats were returned to the home cage and anesthetized 30 min later (sodium pentobarbital, 65mg/kg; Somnotol, MTC Pharmaceuticals, Cambridge, ON, Canada). They were then intracardially perfused with saline followed by ice cold fixative (1l/kg 4% paraformaldehyde pH 7.4). The brain from each animal was post fixed overnight at 4° C , washed three times in PBS, and transferred to a 30% sucrose + PBS solution for an additional 24 h. For pSTAT3 immunohistochemistry, sections (prepared as above) were rinsed in PBS and then incubated in 1% NaOH and 1% H2O2 for 20 min, 0.3% glycine for 10 min, and 0.6% sodium dodecyl sulfate for 10 min. There were 3 X 10 min rinses in PBS between each treatment. Sections were blocked in 10% goat serum 60 and placed in anti pSTAT3 (tyr705) antibody (1:1000, cat. no. 9131s; Cell Signaling Technology, Danvers, MA) on an orbital shaker at 4°C for 24 h. Sections were rinsed and incubated in a secondary donkey anti-rabbit CY3 (1:100, Biocan Scientific, Mississauga, ON, Canada) antibody for 1 h before being mounted onto glass slides and coverslipped with bicarbonate-buffered glycerol. Cannula placement and evidence that the lateral ventricle had been punctured was confirmed during processing for immunohistochemistry in all six rats. Electrophysiology Cell Dissociation and Short Term Primary Culture: SFO tissue was acutely dissected from Male Sprague-Dawley rats (100-150g, Charles River, QC, Canada) as described above. The SFO was then incubated in 5 ml of Hibernate media (Brain Bits, Springfield, IL) containing 2mg/ml papain (Worthington Biochemical Corporation, Lakewood NJ) at 30ºC for 30 minutes. Following incubation, cells were washed, gently triturated in Hibernate media supplemented with B27 (GIBCO, Invitrogen, Burlington, ON, Canada) to liberate single cells, and centrifuged at 400g for 4 minutes at 4ºC. The supernatant was removed and the pellet resuspended in B27-supplemented Neurobasal A media (GIBCO, Invitrogen) containing 100U/ml penicillin/streptomycin and 0.5mM Lglutamine (GIBCO, Invitrogen). Cells were plated on 35-mm uncoated glass bottom culture dishes (MatTek Ashland, MA) and placed in a CO2 incubator (5% O2, 95% CO2 at 37 ºC). Additional B27-supplemented Neurobasal A was added to the culture dishes 1 hour after plating (to allow adequate time required for cells to adhere to the bottom of the 61 culture dish). Cells were maintained in the incubator until the time of experimentation (14 days). SFO Slice Preparation and Current Clamp Electrophysiology: Male SpragueDawley rats (Charles River) aged postnatal days 23-27 ( 50-100 g) were quickly decapitated. The brain was removed and placed into ice-cold slicing solution (bubbled with 95% O2 and 5% CO2), consisting of (in mM): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, and 75 sucrose. A tissue block containing the SFO was obtained and 300µm coronal slices cut using a vibratome. Slices were then incubated in a water bath at 32°C for at least 1 h before recordings commenced in oxygenated artificial cerebrospinal fluid (aCSF) composed of (in mM): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2 1.25 NaH2PO4, and 10 glucose. Slices were placed in a chamber that was continuously perfused at 2 ml/min with 28-32°C aCSF. Neurons were visualized using an infrared differential interference contrast system on an upright microscope (Nikon, Japan). Current-Clamp Electrophysiology: Whole-cell current-clamp recordings from dissociated SFO neurons were acquired using an Axopatch 700B patch-clamp amplifier (Molecular Devices, Palo Alto, CA). Stimulation and recording parameters were controlled by Spike2 (version 5) and Signal (version3) software (Cambridge Electronics Design, Cambridge, UK). Data were filtered at 1 kHz, acquired at 5 kHz, and digitized using a Cambridge Electronics Design Micro1401 interface. Capacitive transients and series resistance errors were minimized before recording. For all recordings, the external 62 recording solution contained the following (in mM): NaCl (140), KCl (5), MgCl2 (1), CaCl2 (2), HEPES (10), and glucose (10), pH 7.3 with NaOH. Patch electrodes were made from borosilicate glass (World Precision Instruments, Sarasota, FL) on a Flaming Brown micropipette puller (P87, Sutter Instrument Co. Novato CA). Electrodes were then fire polished and had resistances of 2.5–5 MΩ when filled with internal recording solution which contained (in mM): K-gluconate (130), KCl (10), MgCl2 (1), CaCl2 (2), HEPES (10), EGTA (10), Na2ATP (4), and GTP (0.1). All chemicals were purchased from Sigma (Oakville, ON, Canada). Once whole cell configuration was achieved, cells were perfused via a gravity fed perfusion system with external recording solution at a rate of 2ml/min. Cells were defined as neurons by the presence of ≥50 mV action potentials. Following a minimum 5-min stable baseline recording period (control), 10 nM leptin (rat, recombinant, Phoenix Pharmaceuticals, Belmont, CA; reconstituted in external recording solution) was bath applied (leptin) followed by a wash with external recording solution (wash). Responsiveness of SFO neurons was determined by comparing membrane potential of neurons before and after leptin perfusion. SFO neurons were considered responsive if the mean membrane potential demonstrated a shift of at least 3mV (over a 100s period) during the 30s following the initiation of bath perfusion with leptin compared with the 100s before application and demonstrated a partial recovery to baseline values following removal of the peptide from the bath (wash). Alternatively, in the few cells exhibiting regular high-frequency spontaneous action potentials (>4 Hz), a cell was considered 63 responsive if it exhibited a change of at least 1 Hz (in a 100 s period) during the 300 sec following the initiation of bath perfusion with leptin. To determine whether leptin acts on amylin-sensitive SFO neurons, the effect of bath administration of amylin (10nM) and leptin (10nM) was evaluated on the same SFO neurons. Statistics Mean change (means ± SEM) in membrane potential before (control) and during leptin administration (leptin) were calculated, and differences tested using a paired Student t-test. All statistical analyses were performed using GraphPad Prism (version 5.0, San Diego, CA). RESULTS Leptin receptor (Ob-Rb) expression in SFO Using primer sets directed toward an extracellular domain common to all leptin receptor isoforms, RT-PCR reactions performed on cDNA obtained from mRNA isolated from acutely microdissected SFO, revealed the presence of leptin receptor (LepR) mRNA (see Figure 3-1). To determine whether the signaling form of the leptin receptor, Ob-Rb, was present in SFO, two different primer sets, each directed towards unique intracellular signaling domains on the Ob-Rb receptor were used, and the data presented in Figure 3- 1 demonstrate the presence of Ob-Rb mRNA (Ob-Rb1, Ob-Rb2) in SFO. Ob-Ra mRNA was also present in SFO as illustrated in Fig 3-1. RT-PCR reactions were also performed on cDNA from acutely microdissected hypothalamus, an area of the brain previously 64 shown to express Ob-Rb (Elmquist et al., 1998b), which served as a positive tissue control (see Figure 3-1). LepR, Ob-Rb and Ob-Ra mRNA were found in all samples of hypothalamus, where appropriate sized products were localized. GAPDH served as a positive control for the PCR reactions. No labeling was found in the negative controls. The validity of all PCR products was confirmed by sequencing. The presence of the leptin receptor on SFO neurons was confirmed using immunohistochemistry with antibodies raised to epitope between amino acids 850 and 900, thereby recognizing all isoforms of the leptin receptor. Using this antibody, clear immunoreactivity was observed in the SFO. In addition, the predicted immunoreactivity in the arcuate nuleus and lack of immunoreactivity in the frontal cortex, a region known not to express leptin receptors, further supports the specificity of this antibody (see Figure 3-2). Double-labeling showed strong immunoreactivity for the Ob-Rb on neurons labeled with the neuronal nuclear marker NeuN, while Ob-Rb immunoreactivity also appeared to be co-expressed on some glial processes that double-labeled with an antibody directed against glial fibrillary acididic protein, albeit to a much lesser degree than with the neuronal marker NeuN (data not shown). Leptin receptor signaling in SFO We next examined whether central administration (lateral ventricle) of leptin induced leptin receptor signaling in the SFO by using STAT3 immunoreactivity (Hubschle et al., 2001;Hosoi et al., 2002;Inoue et al., 2006). These experiments 65 SFO MW GAPDH Ob-Rb1 Ob-Rb2 Ob-Ra LepR 500 300 200 100 Hypothalamus MW GAPDH Ob-Rb1 Ob-Rb2 Ob-Ra LepR 500 300 200 100 NTC MW GAPDH Ob-Rb1 Ob-Rb2 Ob-Ra LepR 500 300 200 100 Figure 3-1: ObRb receptor mRNA is expressed in the subfornical organ (SFO) Agarose gels showing RT-PCR analysis of SFO cDNA for leptin receptor expression (top). Ob-Rb receptor mRNA (Ob-Rb1, Ob-Rb2) as well as Ob-Ra receptor mRNA (ObRa), and leptin receptor (LepR) mRNA (expression common to all leptin receptor isoforms) were also expressed in SFO. The hypothalamus (middle), a positive control tissue, also shows Ob-Rb receptor (Ob-Rb1, Ob-Rb2), Ob-Ra (Ob-Ra) and LepR expression. A primer set specific for GAPDH served as a positive control in both SFO and hypothalamic tissue. PCR products for GAPDH and all leptin receptors (Ob-Rb1, Ob-Rb2, Ob-Rba and LepR) are not observed in the NTC (no template control, RT-) lane in which template has been omitted from the cDNA synthesis reaction. Product size (base pairs) is shown in the leftmost lane of each panel. 66 demonstrated that leptin induced pSTAT3 immunoreactivity in the SFO and hypothalamus of leptin-treated rats (see Figure 3-3, B and D). To verify that pSTAT3 activation was a consequence of leptin administration and not due to inflammatory effects of the cannulation and microinjection procedures, the effects of PBS (vehicle) injections into the lateral ventricle were also examined, and, as can been seen in Figure3-3, these injections did not induce pSTAT3 immunoreactivity in the SFO or hypothalamus (see Figure 3-3, A and D). The pattern of staining in the hypothalamus was consistent with what has been reported by others (Levin et al., 2004) with extensive activation in the ventral medial and arcuate nuclei. Neuronal localization of pSTAT3 immunoreactivity in SFO was confirmed using the neuronal-specific marker, NeuN as illustrated in Figure 34. Electrophysiology Dissociated SFO Neurons: Whole cell current clamp techniques were used to evaluate the direct effects of leptin receptor activation on dissociated SFO neurons. The process of dissociation leaves us with single SFO neurons in synaptic isolation (no visibledendritic contacts), which are ideally suited for the assessment of direct effects of exogenously applied leptin (10nM) on the excitability of SFO. Cells were classified as neurons if they exhibited spontaneous action potentials or if application of a short (100ms) depolarizing current pulse evoked action potentials. SFO neurons were required to demonstrate action potentials (spontaneous or evoked) of at least 50 mV and stable resting membrane potentials in order to be considered ‘healthy’ and tested for the effects 67 Figure 3-2: Leptin receptor is expressed on neurons in the SFO Immunofluorescence micrographs of leptin receptor immunoreactivity in the SFO (A and B), arcuate nucleus (C), and frontal cortex (D). Leptin receptor immunoreactivity was detected in the SFO and, as expected, in the arcuate nucleus (Bregma -2.8mm), which served as a positive control. No labeling was observed in the frontal cortex at the same level as the subfornical organ (Bregma -0.8mm). Scale bars: 100µm 68 Figure 3-3: Leptin induces pSTAT3 activation in the SFO Immunofluorescence micrographs of pSTAT3 immunoreactivity in the SFO (A and B) and hypothalamus (C and D) of a vehicle (A and C) and leptin treated (B and D) rat. pSTAT3 immunoreactivity was not detected in animals injected intracerebroventricularly with saline. Note that pSTAT3 immunoreactivity was observed in the ventromedial and arcuate nuclei in the leptin-treated animal (Bregma -3.4mm). Scale bars: 100µm. 69 of leptin. In accordance with these criteria, current clamp recordings were obtained from 72 SFO neurons. Of these neurons, the majority (85%, 61/72 cells) were spontaneously active. The remaining cells were quiescent, but fired action potentials in response to a brief depolarizing current pulse. The mean resting membrane potential was -48.8 ± 1.2mV for all cells obtained. There was no difference in resting membrane potential between those cells which were spontaneously active (mean resting membrane potential = -48.9 ± 1.7mV) and quiescent cells (mean resting membrane potential = -47.7 ± 3.0mV, p = .72). Bath application of leptin (10nM) influenced 64% (46/72) of SFO neurons tested. The majority of responsive neurons (28/46) exhibited a depolarization in response to leptin administration. All depolarizing responses began within 100 seconds of leptin application, with many cells beginning their depolarizing response within 30 seconds of bath perfusion with leptin. The depolarizing responses seen in response to leptin administration were of a long duration, lasting several minutes upon termination of leptin application. In fact, only half of the depolarizing effects were completely reversible, showing a recovery to baseline membrane potentials during the period of the recording (see Figure 3-5, top trace). The mean change in membrane potential of these depolarizing cells was 7.3 ± 0.6 mV (n=28) and was typically accompanied by an expected increase in action potential firing frequency. The remaining affected cells (18/46) demonstrated hyperpolarizing effects in response to leptin administration. Similar to the depolarizing 70 Figure 3-4: pSTAT3 is colocalized with neuronal marker NeuN in the SFO Confocal immunofluorescence micrographs of pSTAT3 (A and D) and NeuN (B and E) immunoreactivity in the SFO of a vehicle (A-C) and leptin treated (D-F) rat. Micrographs of the SFO of a vehicle treated rat illustrate no pSTAT3 immunoreactivity (A) in neurons whose nuclei are labeled with NeuN (B). C is an overlay of the two images (26, 1-µm optical sections). Micrographs of the SFO of a leptin-treated rat illustrate pSTAT3 immunoreactivity (D) in neurons whose nuclei are labeled with NeuN (E). F is an overlay of the two images (13, 1-µm optical sections). Arrows indicate where pSTAT3 and NeuN colocalize. Note that in the SFO NeuN is relatively weakly immunoreactive compared to other regions of the brain. Scale bars: 50µm. 71 responses, these effects began within 100 seconds of peptide administration, lasted several minutes, and were reversible in 50% of neurons (see Figure 3-5, bottom trace). The mean change in membrane potential of hyperpolarizing SFO neurons was -6.9 ± 0.8 mV (n=18). In spontaneously active neurons, these hyperpolarizing effects were accompanied with a decrease in action potential firing frequency. To ensure leptin responsiveness of SFO neurons was not the result of transformation of these cells following dissociation, the effect of leptin on SFO neurons in slice preparations were also evaluated. Whole cell current-clamp recordings from 8 SFO neurons in SFO slices showed similar responsiveness to bath application of 10 nM leptin with 50% of cells depolarized (mean change in membrane potential 4.8 ± 0.6 mV, n=4, see Figure 3-5), one cell hyperpolarized (-4.9mV), and the remaining three cells tested being unaffected by peptide application. To determine whether leptin influenced the same population of SFO neurons influenced by amylin which has been shown to only depolarizes SFO neurons (38), 11 cells were tested for the effects of bath administration of both leptin and amylin. Three of 4 cells that depolarized to leptin also depolarized to amylin (see Figure 3-6). Cells that hyperpolarized in response to leptin (n=3) were not influenced by bath application of amylin, while the remaining 4 cells were not influenced by administration of either peptide. 72 Figure 3-5: Leptin influences the excitability of SFO neurons A: Currentclamp recording from a spontaneously active dissociated SFO neuron showing that bath application of 10nM leptin s caused a depolarization accompanied by an increase in action potential frequency. B:.current-clamp recording from a different dissociated SFO neuron that exhibited a hyperpolarizing response and a decrease in action potential frequency in response to bath application of 10nM leptin. In both neurons, a return to baseline membrane potential and frequency after removal of leptin from the bath was seen. C: current Clamp recording obtained from a spontaneously active SFO in slice preparation illustrating a depolarizing response to 10nM bath application of leptin. D: bar graph summarizing the effects of leptin on membrane potential of dissociated SFO neurons (black bars) and SFO neurons obtained from a slice preparation (gray bars) illustrating similar effects on membrane excitability. Numbers in brackets indicate number of cells tested. Time and duration of leptin administration is indicated by the gray bar above each current clamp recording. 73 10-8 Leptin 10-8 Amylin 20 mV 30 sec Figure 3-6: Leptin depolarizes amylin-sensitive SFO neurons Current clamp recording obtained from the same SFO neuron illustrating depolarizing responses to leptin (top trace) and amylin (bottom trace). Time and duration of leptin (top trace, solid bar) and amylin (bottom trace, hatched bar) administration is indicated by the bar above each current clamp recording. 74 DISCUSSION In this study we demonstrate the presence of the signaling form of the leptin receptor in the SFO and the responsiveness of SFO neurons to leptin. Using RT-PCR technology we have, for the first time, demonstrated the presence of the signaling form of the leptin receptor, Ob-Rb mRNA as well as the presence of Ob-Ra mRNA in acutely dissected whole SFO. The same primers also detected Ob-Rb mRNA and Ob-Ra mRNA in acutely dissected hypothalamus, an area known to contain both receptor types (see Jequier, 2002 for review). These findings confirm and extend recent findings of gene array studies from our laboratory demonstrating the presence of the leptin receptor mRNA in the SFO (Hindmarch et al., 2008). In the present study, we sought to identify whether mRNA was translated into a functional receptor using 2 approaches: 1) immunohistochemistry for the receptor and 2) leptin-induced activation of STAT3, revealed as nuclear labeling of pSTAT3 immunoreactivity. We observed immunoreactivity in the SFO by using an antibody raised to epitope between amino acids 850 and 900 (thus identifying all leptin receptor isoforms), confirming the presence of the leptin receptor in SFO. The neuronal identity of some of these cells was confirmed using the neuronal marker NeuN. Leptin receptor immunoreactivity was also co-expressed on a small population of glial processes in the SFO, suggesting potential roles for leptin in influencing both neurons and glial cells. These findings were not completely unexpected, as previous studies have shown that OB-Rb is expressed in both neurons (Grill & Kaplan, 2002;Mercer et al., 1998;Burguera et al., 2000) and primary glial cell cultures (Hosoi et al., 2000). We are 75 aware that the specificity of widely used antibodies directed toward the leptin receptor has been a point of controversy in the field of obesity research (Montanaro et al., 2005). While some labeling using antibodies is “real”, there are some sites in the brain where immunoreactivity is observed in the absence of the mRNA identified by in situ hybridization. In view of this issue, we chose not to rely exclusively on this approach to identify functional leptin receptor expression in SFO. In our studies, we also examined the localization of pSTAT3, a well accepted marker of Ob-Rb receptor activation as an additional indicator of the presence of functional Ob-Rb in SFO. Central and peripheral leptin administration has been shown to increase STAT3 phosphorylation in hypothalamic and brainstem nuclei involved in the regulation of feeding (Hosoi et al., 2002), and that pSTAT3 activation is necessary for the inhibitory effect of leptin on food intake and body weight (Piper et al., 2008). In the present study we have shown leptin induced pSTAT3 expression in the SFO (as well as in the hypothalamus as previously described) while little or no expression was observed in vehicle-treated animals indicating that pSTAT activation was specific to leptin treatment and not due to inflammatory processes that may have arisen as a consequence of cannula placement/microinjection into the ventricle. Having shown that leptin activates transcriptional events in SFO neurons, we wanted to explore whether there were short-term signaling consequences of leptin receptor activation in this organ. The results of our current clamp experiments clearly demonstrate that leptin influences the excitability of the majority (64%) of dissociated 76 SFO neurons tested. The effects of leptin on dissociated neurons are the result of direct actions of leptin, as the process of dissociation isolates single SFO neurons rendering them devoid of any synaptic input. Our findings that similar effects were seen in slice preparations confirms that such responsiveness is not a consequence of transformation of these cells following dissociation. The presence of subpopulations of neurons in the SFO may explain the finding of both depolarizing and hyperpolarizing responses to leptin administration. Previous studies, from our laboratory and others, have demonstrated heterogeneous responses of SFO neurons (depolarizing and hyperpolarizing responses) to a variety of peptidergic substances (Cottrell et al., 2004). Although not addressed in the present study, perhaps the heterogeneity in excitability of SFO neurons in response to leptin administration reflects different subpopulations of SFO neurons with specific, yet different hypothalamic projection sites, which together contribute to the coordinated effects of leptin on food intake. Evidence in support of a potential role for the SFO in the control of energy balance is emerging from a number of different observations. Neurons in the SFO have been shown to be responsive to both the appetite-suppressing hormone, amylin (Riediger et al., 1999b;Barth et al., 2004) and the appetite-stimulating hormone, ghrelin (Pulman et al., 2006) with this recent study from our own lab showing that different populations of SFO neurons are depolarized by either amylin or ghrelin, circulating signals which have opposite effects on food intake, with no SFO cells responding to both peptides. The findings of the present study, demonstrating depolarizing and hyperpolarizing effects of 77 leptin on SFO neurons, also support the existence of subpopulations of SFO neurons and would suggest that neurons that depolarize to leptin may be those that are similarly influenced by the anorexigenic peptide, amylin. To test this hypothesis, we evaluated the effects of bath application of both amylin and leptin on individual SFO neurons and have shown that leptin depolarizes amylin-responsive SFO neurons. Together these findings suggest that leptin influences separate subpopulations of neurons to influence the hypothalamic regulation of feeding, enhancing its ability to inhibit food intake; however, currently it is not known whether leptin signaling in the SFO affects energy balance. Changes in circulating leptin concentrations clearly influence feeding behaviour and the activity of neurons in regulatory centers in the hypothalamus including the ARC. The actions of leptin on neurons in the ARC is well documented, with depolarizing and hyperpolarizing effects observed on specific neuronal populations (Cowley et al., 2001). Whole animal studies have demonstrated Fos activation in ARC neurons following systemic leptin administration, while the excitability of ARC neurons is, unquestionably, influenced by direct leptin application in hypothalamic slices (Cowley et al., 2003). However, an issue that must be taken into consideration is how leptin gains access to central feeding circuits protected behind the BBB. It has been suggested that leptin directly accesses the ARC through a leaky BBB; however, anatomical studies have clearly demonstrated that the ARC has an intact BBB as it does not contain type III fenestrated capillaries (Shaver et al., 1992). Although a saturable blood-to-brain transport system for leptin has been demonstrated (Banks et al., 1996), it is not clear what 78 concentrations of transported substances are delivered to their target normally protected by the BBB. Perspectives and Significance The SFO, a sensory CVO, possesses fenestrated capillaries permitting circulating leptin direct access to neurons within this central nervous system structure. The lack of the normal BBB and, therefore, access to peripheral signals as well mean that the SFO is well suited for a role in monitoring circulating signals indicating energy status. In addition, the well-documented projections from the SFO to hypothalamic nuclei with established roles in feeding behaviour, including the ARC, lateral hypothalamus, and paraventricular nucleus (Lind et al., 1982;Rosas-Arellano et al., 1996), suggests potential roles for SFO efferents in the regulation of energy balance.. The present study identifies the SFO as a possible central nervous system location with direct access to the peripheral circulation, at which leptin may act to influence hypothalamic metabolic control centers. Our data show that the signaling form of the leptin receptor is present in the SFO and the responsiveness of SFO neurons to leptin; however, the physiological relevance of these observations remains to be fully explored. ACKNOWLEDGEMENTS: This work was supported by a Canadian Institutes for Health Research Team Grant on the Neurobiology of Obesity (to A. V. Ferguson and K. A. Sharkey), a Heart and Stroke Foundation of Canada Studentship (to P. M. Smith), an Alberta Heritage Foundation for Medical Research (AHFMR) Studentship (to A. P. Chambers), and an AHFMR Scientist award (to K. A. Sharkey). K. A. Sharkey is the 79 Crohn’s and Colitis Foundation of Canada Chair in Inflammatory Bowel disease Research at the University of Calgary. 80 Chapter 4: CARDIOVASCULAR ACTIONS OF LEPTIN IN THE SUBFORNICAL ORGAN ARE ABOLISHED BY DIET INDUCED OBESITY 81 ABSTRACT The subfornical organ (SFO), a sensory circumventricular organ lacking the normal blood brain barrier with well documented roles in cardiovascular regulation, has recently been identified as a potential site at which the adipokine, leptin, may act to influence central autonomic pathways. Systemic and central leptin administration has been shown to increase blood pressure and it has been suggested that selective leptin resistance contributes to obesity related hypertension. Given the relationship between obesity and hypertension, the present study was undertaken to investigate the cardiovascular consequences of direct administration of leptin into the SFO of young lean rats and in the diet induced obesity (DIO) rat model which has been shown to be leptin resistant. Leptin administration (500 fmol) directly into the SFO of young rats resulted in rapid decreases in blood pressure (BP) (mean area under the curve (AUC) = -677.8 ± 167.1 mmHg*sec, n=9), without an effect on HR (mean AUC = -21.2±13.4 beats, n=9), effects which were dose related as microinjection of 5 pmol leptin into the SFO had a larger effect on BP (mean AUC = -972.3±280.1 mmHg*sec, n=4). These BP effects were also shown to be site specific as leptin microinjection into non-SFO regions or into the ventricle was without effect on BP (non SFO: mean AUC = -22.4 ± 55.3 mmHg*sec, n=4; ventricle: mean AUC = 194.0 ± 173.0 mmHg*sec, n=6). In contrast, microinjection of leptin into leptin resistant DIO rats was without effect on BP (mean AUC = 205.2±75.1 mmHg*sec, n=4). These observations suggest that the SFO may be an important relay center through which leptin, in normal weight, leptin responsive rats, acts to maintain BP within normal physiological limits through descending autonomic 82 pathways involved in cardiovascular control and that in obese, leptin-resistant, rats leptin no longer influences SFO neurons resulting in elevated BP, thus contributing to obesity related hypertension. 83 INTRODUCTION Obesity is a chronic metabolic condition with important public health implications associated with numerous co-morbidities including cardiovascular disease, insulin resistance, and hypertension. Adipose tissue plays a critical role in energy homeostasis, secreting adipokines that control feeding and energy metabolism. Leptin is one such adipokine that acts centrally to signal long term energy balance. Plasma leptin levels reflect both energy stores and acute energy balance and circulating leptin has been shown to decrease food intake and increase energy expenditure through activation of receptors in hypothalamic and brain stem neurons (Grill & Kaplan, 2002). Consistent with its effects on energy expenditure, leptin increases sympathetic nerve activity to brown adipose tissue, kidney, adrenal gland, and hindlimb (Dunbar et al., 1997;Haynes et al., 1997). Not surprisingly, since the initial discovery of the weight reducing effect of leptin, this adipokine has also been suggested to play a role in blood pressure (BP) regulation. A positive correlation has been demonstrated between leptin levels and BP in obese individuals (Al-Hazimi & Syiamic, 2004;Itoh et al., 2002) suggesting that leptin may play a primary role in obesity-related hypertension. The fact that animals which are genetically leptin deficient (Mark et al., 1999) or lacking a functional leptin receptor (Chan & Johnson, 1997) do not demonstrate increases in BP despite profound obesity, suggests that receptor mediated actions of leptin may be responsible for the regulation of systemic hemodynamics. Although acute systemic leptin administration has been shown to be without cardiovascular effect, chronic systemic administration causes increases in BP (Shek et al., 1998;da Silva et 84 al., 2004). Similarly, acute central (intracerbroventricular, icv) leptin administration has also been shown to elevate BP in anesthetized (Dunbar et al., 1997;Rahmouni & Morgan, 2007;Lu et al., 1998) and conscious rats (Casto et al., 1998) as does chronic icv administration (Dubinion et al., 2011). Direct administration of leptin into specific cell groups within the hypothalamus possessing the leptin receptor and shown, electrophysiologically, to be influenced by leptin including the arcuate nucleus (ARC) (Rahmouni & Morgan, 2007), the ventromedial hypothalamus (VMH) (Montanaro et al., 2005;Marsh et al., 2003) and the dorsomedial hypothalamus (DMH) (Marsh et al., 2003) has been shown to cause increases in BP in anesthetized rats. The presence of the blood-brain barrier (BBB) leads to the obvious question as to how leptin, a 16KDa peripherally derived protein, gains access to central sites to influence BP. While peptide transporter systems (Banks et al., 1996) and transendothelial signaling (Paton et al., 2007) represent mechanisms through which leptin may reach hypothalamic neurons behind the BBB, the subfornical organ (SFO), a sensory circumventricular organ lacking the normal BBB with well documented roles in cardiovascular regulation has recently been implicated as a CNS structure involved in energy homeostasis (see Fry et al., 2007;Hoyda et al., 2009 for review). The SFO communicates with hypothalamic autonomic control centers through efferent projections to autonomic nuclei involved in energy homeostasis and/or cardiovascular regulation including the ARC, paraventricular nucleus, supraoptic nucleus, organum vasculosum of the lamina terminalis, and lateral hypothalamus (Miselis, 1982;Lind et al., 1982;Lind, 85 1986;Gruber et al., 1987). The SFO has been shown to play a significant role in cardiovascular regulation by studies showing that both electrical (Ishibashi & Nicolaidis, 1981;Mangiapane & Brody, 1983;Ferguson & Renaud, 1984) and chemical (Gutman et al., 1988b;Wall et al., 1992;Washburn et al., 1999;Mangiapane & Simpson, 1983) stimulation of this circumventricular structure causes increases in BP, effects that are abolished by SFO lesion (Mangiapane & Simpson, 1980a) or transection of SFO efferents (Lind et al., 1983). Acute electrical stimulation of the SFO has been shown to elicit feeding in satiated rats (Smith et al., 2010) suggesting a role for this specialized CNS structure in energy homeostasis. Further support for a role of the SFO in energy homeostasis is derived from studies which demonstrate the localization of receptors and receptor mRNA for a variety of peripherally derived metabolic signals including adiponectin, amylin, ghrelin (GHSR receptor) and leptin in the SFO (Alim et al., 2010;Pulman et al., 2006;Riediger et al., 1999b;Smith et al., 2009;Hindmarch et al., 2008). In addition, each of these peptides has been shown to influence the excitability of individual SFO neurons (Alim et al., 2010;Pulman et al., 2006;Riediger et al., 1999b;Smith et al., 2009), suggesting a functional role for actions of these circulating peptides at the SFO. Leptin has been shown to cause both depolarizing and hyperpolarizing effects on different SFO neurons (Smith et al., 2009). Interestingly, leptin has been shown to activate the same SFO neurons activated by amylin (Smith et al., 2009), supporting a role for the SFO in 86 integrating peripherally derived signals regulating food intake. Together these findings suggest a functional role for leptin at the SFO. Given the well documented role of the SFO in cardiovascular regulation and accumulating evidence suggesting SFO involvement in mediating the central effects of leptin, the present study was undertaken to first investigate the cardiovascular consequences of direct administration of leptin into the SFO of young normal weight rats, and subsequently to determine if these effects were modified in leptin resistant diet induced obese (DIO) rats. MATERIALS AND METHODS All procedures were conducted in accordance with the Canadian Council on Animal Care regulations and approved by Queen’s University Animal Care Committee. Urethane-anesthetized (1.4 g/kg) male Sprague Dawley rats (150–350 g) were placed on a feedback-controlled heating blanket for the duration of the experiment to maintain body temperature at 37°C. Animals were fitted with an indwelling femoral arterial catheter for the measurement of blood pressure (BP) and heart rate (HR). The animal was then placed in a stereotaxic frame and a midline incision was made through the skin of the skull. A small burr hole was drilled in the skull such that a microinjection cannula (150 μm tip diameter; Rhodes Medical Instruments) could be advanced into the region of SFO according to the coordinates of Paxinos and Watson (Paxinos & Watson, 1982). After a minimum 2 minute stable baseline recording was obtained leptin (0.5 μl of 10-5 (5 pmol) or 10−6 M (500fmol)) or artificial cerebral spinal fluid (0.5 μl aCSF, vehicle control) was 87 microinjected into the region by a pressure driven 10 μl Hamilton micro-syringe over 10 sec and the effects on BP and HR assessed. At the conclusion of the experiment, animals were overdosed with anesthetic and perfused with 0.9% saline, followed by 10% formalin, through the left ventricle of the heart. The brain was removed and placed in formalin for at least 24 hours. Using a vibratome, 100μm coronal sections were cut through the region of SFO, mounted, and cresyl violet stained. The anatomical location of the microinjection site was verified at the light microscope level by an observer unaware of the experimental protocol or the data obtained. Data Analysis Animals were assigned to one of 3 anatomical groups (SFO, non-SFO or ventricle) according to the location of the microinjection site. Animals with injection sites that were not wholly confined within any of these regions were excluded from further analysis. Animals with confirmed microinjection sites in the SFO were further divided according to the concentration of leptin microinjected (5pmol or 500fmol) or whether aCSF (vehicle control) was microinjected into the region. Normalized BP and HR data (mean baseline BP and HR data were calculated for 60 sec before injection and subtracted from all data points before and after injection) were obtained for each animal 60 s before the time of microinjection (control period) until 200 sec after microinjection. Area under the curve (AUC, area between baseline and each BP and HR response) was calculated for each animal for each leptin concentration and aCSF for the 200 sec time period immediately following the injection. Mean AUC for BP and HR responses for 88 each group were then calculated. A one way analysis of variance (one way ANOVA) was used to determine whether BP and HR responses observed in response to substance administered (5pmol or 500fmol leptin or aCSF) into the SFO differed and to determine whether responses to 5pmol leptin were different based on anatomical location (SFO, non-SFO and ventricle) of leptin microinjection. Diet Induced Obesity In order to determine whether leptin effects seen in lean, young animals would be present in obese animals, a second series of experiments was undertaken in a diet induced model of obesity (DIO). Upon arrival, male Sprague–Dawley rats (125-150 g) were housed in pairs in a temperature controlled room on a 12 h light–dark cycle and exposed to a high fat diet (Research Diets, New Brunswick, NJ #D124521, composition 45% kcal% fat, 35% kcal% carbohydrate, and 20% kcal% protein) with water ad libitum. Weight gain was measured on a weekly basis and, after 10 weeks of ad libitum feeding on a high fat diet, animals were divided into diet induced obese (DIO) or diet resistant (DR) based on those who gained the greatest and least weight, respectively. Animals that were greater than 700 grams were placed in the DIO group while animals that weighed less than 600 grams were considered DR. The DIO phenotype was validated in accordance with the DIO model of others (Levin & Dunn-Meynell, 2002;Hyland et al., 2007;Li et al., 2011) in our own colony. Rats classified as DIO based on weight gain after 10 weeks on the medium high fat diet had a mean body weight of 718.6 ± 6.8g (n=16). DR rats weighed the same as age-matched chow fed controls (DR mean body 89 weight = 570.7 ± 6.3g, n=25; chow fed mean body weight= 570.9 ± 16.5g, n=8; one way ANOVA p<.0001, chow vs DR, p> .05, n.s. Newman-Keuls post hoc analysis) and weighed significantly less than the rats classified as DIO (DIO vs DR: p< .05 NewmanKeuls post hoc analysis). Chow fed animals were not used further in the present study. Leptin microinjection (5pmol) and data analysis was performed in DIO and DR rats as described above. A one way analysis of variance (one way ANOVA) was used to determine whether BP and HR responses observed in response to leptin were different between young, DR and DIO rats. RESULTS Leptin Microinjection into young lean rats A total of 37 animals were used in this study, 18 of which had microinjection locations entirely within the SFO (SFO group), while 4 had microinjection locations wholly outside of the SFO (non-SFO) and in a further 6 animals, leptin injection was made into the ventricle (ventricle). The remaining animals (n=9) were included for calculation of anesthetized baseline BP but were excluded from further analysis of microinjection effects as sites could not be reliably classified into any of the above 3 groups. Microinjection of 500 fmol leptin into the SFO of young rats resulted in rapid decreases in BP (mean area under the curve (AUC) = -677.8 ± 167.1 mmHg*sec, n=9), without an effect on HR (mean AUC = -21.2±13.4 beats, n=9) (see Figure 4-1, Figure 42). Leptin microinjection into the SFO decreased BP in a dose related manner as 90 Figure 4-1: Leptin microinjection into the SFO decreases in blood pressure Blood pressure (BP, upper panel) and heart rate (HR, lower panel) recordings from a single animal showing the cardiovascular response elicited by 500 fmol (0.5µl) bolus leptin microinjection into the SFO. Time of injection is indicated by the arrow. The anatomical location of the injection site is shown on the photomicrograph inset. 91 microinjection of 5 pmol leptin into the SFO had a larger effect on BP (mean AUC = 972.3±280.1 mmHg*sec, n=4), again without affecting HR (mean AUC = 19.7±5.7 beats, n=4) (see Figure 4-2). These effects were due to the administration of leptin as microinjection of aCSF (vehicle) was without effect on BP (-64.2 ± 166.6 mmHg*sec, n=5) or HR (mean AUC = 0.78 ± 3.3 beats, n=5) (see Figure 4-2). In addition, BP effects were shown to be site specific as leptin microinjection (500 fmol) into non-SFO regions or into the ventricle was without effect on BP (non SFO: mean AUC = -22.4 ± 55.3 mmHg*sec, n=4; ventricle: mean AUC = 194.0 ± 173.0 mmHg*sec, n=6) (see Figure 43) or HR (non SFO: mean AUC = -6.5 ± 16.2 beats, n=4; ventricle: mean AUC = -20.4 beats ± 2.6, n=6). BP was also not elevated up to 2 hours after icv leptin administration (data not shown). DIO Phenotype A total of 40 rats were fed the medium high fat diet in this study, of which 20% (n=8) were classified as DIO, and 42.5% (n=16) were placed in the DR group. DIO and DR rats displayed a significant difference in weight at the 10 week time period (DIO 720.0 ± 6.8 g, n = 8 versus DR 570.2 ± 5.7 g, n = 17, p< 0.001, Student’s t test). The remaining animals had body weights that fell between those of DR and DIO rats (633.0 ± 4.6 g, n = 16) and were removed from further study as animals with these intermediate body weights (601 – 699g) could not be reliably classified as DIO or DR. 92 Figure 4-2: Leptin microinjection into the SFO decreases BP in a dose related manner Normalized mean BP (upper left) and HR (lower left) traces in response to a bolus (0.5µl) microinjection of 5 pmol (▲) or 500 fmol ( ) leptin, or vehicle (●). Time of injection is indicated by the arrow. Bar graphs to the right summarize the mean BP (upper) and HR (lower) area under the curve (AUC). * p<.05, Neuman-Keuls post hoc analysis. 93 Figure 4-3: Leptin effects on blood pressure are site specific This bar graph shows the mean area under the curve (AUC) following administration of 500fmol leptin into SFO (green bar), non-SFO (grey bar) or ventricle (brown bar). * p<.05, ** p<.01 Neuman-Keuls post hoc analysis. 94 Leptin Microinjection in DIO and DR rats Baseline BPs of anesthetized young lean rats (84.4 ± 2.7 mmHg, n = 37) DIO (73.2 ± 3.6 mmHg, n=7) DR (77.2 ± 4.3 mmHg, n=10) rats were not significantly different (p=.13, one way ANOVA). Leptin microinjection (5pmol) into the SFO of DR rats resulted in similar decreases in BP (AUC = -832.9±159.6 mmHg*sec, n=10) as the young rats. In contrast, microinjection of leptin into the SFO of DIO rats was without effect on BP (mean AUC = 205.2±75.1 mmHg*sec, n=4; p<.01) as illustrated in Figure 4-4. DISCUSSION The results of the present study demonstrate that leptin administration into the SFO of young rats (<300g) caused rapid, site specific decreases in BP without influencing HR. Although the lack of effect on HR is congruent with the effects of central leptin administration on HR (Lu et al., 1998;Rahmouni & Morgan, 2007;Casto et al., 1998), the hypotensive effects observed following microinjection of leptin into the SFO were unexpected in light of previous studies demonstrating increases in BP observed following leptin microinjection directly into hypothalamic feeding centers (Montanaro et al., 2005;Marsh et al., 2003;Rahmouni & Morgan, 2007) or following icv leptin administration (Dunbar et al., 1997;Rahmouni & Morgan, 2007;Casto et al., 1998;da Silva et al., 2004;Lu et al., 1998). In addition, the well-established association between obesity, high circulating leptin levels, and hypertension had suggested to us that leptin microinjection into SFO would result in increased BP. Interestingly, the decreases in BP 95 Figure 4-4: Leptin induced decreases in blood pressure are abolished in DIO rats Normalized mean BP trace in response to a bolus (0.5µl) microinjection of 5 pmol leptin into the SFO of young (▲), diet resistant (DR ■), and diet induced obesity (DIO, ●) rats. Time of injection is indicated by the arrow. Inset bar graph summarizes the mean BP area under the curve (AUC). ** p<.01, Neuman-Keuls post hoc analysis. 96 observed following leptin injection into SFO in the current study were elicited in response to less leptin (8 ng) than studies that found hypertensive responses following leptin microinjection into various hypothalamic nuclei (100–500ng) (Marsh et al., 2003;Rahmouni & Morgan, 2007) or into the ventricle (5-10µg) (Lu et al., 1998;Rahmouni & Morgan, 2007;Dunbar et al., 1997). In addition, decreases in BP elicited by leptin microinjection in SFO occurred almost immediately (< 1 minute), while increases in BP following leptin injection into the ARC were not observed until 2 hours after injection despite the higher leptin dose (Rahmouni & Morgan, 2007). Although microinjection of larger amounts of leptin (100ng) into the VMH elicited increases in BP within a similar time frame to our studies in SFO (<100sec) (Montanaro et al., 2005) when the dose of leptin was reduced (16ng) the latency to the peak BP increase was much longer, increasing to 15-20 minutes (Marsh et al., 2003). While our observation that acute icv leptin is without effect on BP is in contrast to previous studies showing icv leptin increases BP in anesthetized rats, this difference is most likely a consequence of the fact that we administered much less leptin (80ng) that was used in these previous studies (5 -10µg) which elicited increases in BP (Lu et al., 1998;Rahmouni & Morgan, 2007;Dunbar et al., 1997). Previous studies have also been consistent in demonstrating that the hypertensive effects of icv administration of leptin are long lasting (>1hr), although the latency to these long-duration BP responses appears to be somewhat variable (5 min – 2 hours) (Dunbar et al., 1997;Rahmouni & Morgan, 2007;Casto et al., 1998;da Silva et al., 2004;Lu et al., 1998). In light of these previous 97 observations, we measured cardiovascular effects for up to 2 hours following icv leptin administration but did not observe any changes in BP or HR at any point during this time period. These results suggest that the hypotensive effects of leptin administration into SFO were due to actions at the SFO and not as a consequence of leakage into the ventricle and that the dose of leptin used was not sufficient to induce cardiovascular changes when administered icv. A positive correlation has been demonstrated between leptin levels and BP in obese individuals (Al-Hazimi & Syiamic, 2004;Itoh et al., 2002) suggesting that leptin may play a primary role in obesity-related hypertension. It has been demonstrated in both genetic and diet-induced models of obesity that, while these animals are resistant to the weight reducing effects of both systemic and peripheral leptin, they remain sensitive to leptin effects on renal sympathetic output (Correia et al., 2002;Rahmouni et al., 2005;Rahmouni et al., 2002), an action which has been suggested contribute to hypertension (for review see Mark et al., 2002;Correia & Haynes, 2004). Our findings, in the present study, showing that leptin actions in the SFO to influence BP are abolished in DIO animals supports the concept of selective leptin resistance. In addition, the fact that depressor effects are abolished in the ‘leptin-resistant’ DIO model may serve to exacerbate the hypertension present in obese individuals. Although genetic defects causing leptin deficiency and leptin receptor defects have been shown to cause profound obesity, the most common cause of obesity has been linked to overeating or the consumption of a high fat diet (Unger et al., 2010) both of 98 which lead to a marked increase in plasma leptin levels (Friedman & Halaas, 1998;Wang et al., 2001). In order to investigate whether decreases in BP would be maintained in a diet induced model of obesity (in which leptin resistance occurs) leptin microinjection was performed into the SFO of DIO animals. In contrast to genetic models of obesity, DIO develops as a consequence of consuming a high fat diet and not all animals that display a preference for high fat diets develop obesity (Levin et al., 1989). What causes some animals to become obese while others do not (diet-resistant; DR) is unknown (Levin et al., 1989). Interestingly, the DIO phenotype is only partially explained by increased food intake (Levin et al., 1989). It has been demonstrated that rodents fed a high fat diet develop a resistance to peripheral leptin followed by central leptin resistance if continued on the high fat diet (El-Haschimi et al., 2000;Lin et al., 2000;Wang et al., 2001;Boyle et al., 2011) and that this leptin resistance may be one of the factors underlying DIO phenotype. Although not specifically measured in the present study, numerous studies have shown markedly elevated serum leptin levels in the DIO rat model (Lu et al., 1998;Zhang et al., 2010;Tumer et al., 2007;Dubinion et al., 2011;Elmarakby & Imig, 2010;Levin & Dunn-Meynell, 2002) despite profound obesity. In conscious animals, BP has been shown to be elevated in rats fed HFD as compared to those fed a normal diet (Boustany et al., 2004;Dubinion et al., 2011;Zhang et al., 2010;Tumer et al., 2007). Our findings that baseline BP was not different in any of the groups under urethane anesthetic concurs with a previous study which showed that 99 there was no difference in BP between HFD fed rats and lean controls under the same anesthetic regimen (Lu et al., 1998). The finding in the present study that leptin induced depressor responses are abolished in DIO rats while maintained in age matched DR rats suggests that a decreased sensitivity to leptin underlies the lack of depressor responses, in concordance with the observation of central leptin resistance demonstrated in previous studies (for review see Mark et al., 2002;Correia & Haynes, 2004). Interestingly, it has also been shown rats that become obese following 8 weeks on a high fat diet no longer demonstrate pressor responses to icv leptin administration (Lu et al., 1998), supporting the conclusion that leptin sensitivity may be crucial for mediating the effect of icv leptin on BP. Although we have shown pSTAT activation in the SFO in normal (non-obese) rats (Smith et al., 2009), follow-up studies evaluating whether pSTAT is activated in SFO following leptin administration in DIO animals would lend credence to the suggestion of leptin resistance in the SFO in this model. The results of the present study suggest that leptin may play a positive role in cardiovascular regulation where under normal (non-obese) conditions when the individual is leptin-responsive, leptin acts at the SFO to maintain BP within normal physiological limits. In leptin resistant, obese individuals, leptin is no longer able to act at the SFO to maintain BP and, without this braking effect, BP begins to rise. Thus, this leptin insensitivity in the SFO would serve to further exacerbate the effect of central selective leptin resistance in the development of obesity related hypertension. 100 Such a hypothesis fits nicely with the well-documented roles of SFO in cardiovascular regulation and body fluid homeostasis. Within this context, ANG has been shown, electrophysiologically, to elicit solely excitatory responses on approximately 70% of SFO neurons both in vivo (Gutman et al., 1988a;Tanaka et al., 1987) and in vitro (Li & Ferguson, 1993;Okuya et al., 1987;Schmid & Simon, 1992). It has been previously demonstrated that approximately 65% of SFO neurons are responsive to leptin, half of which are excited by leptin and half inhibited (Smith et al., 2009). Such observations support integrative functions in this CNS structure and further suggest that ANG and leptin influence some of the same SFO neurons. Given that ANG and leptin exert opposite effects on BP through actions at the SFO, perhaps a subpopulation of SFO neurons activated by ANG are also inhibited by leptin. Thus, under normal conditions, when individuals are capable of responding to leptin, the net effect of the action of ANG (which alone would increase BP) and leptin (which alone would decrease BP) at the SFO is a maintenance of BP within normal physiological limits. In obesity, when an individual is leptin resistant and no longer able to respond appropriately to leptin (despite elevated leptin levels) these same neurons in the SFO, once responsive to ANG and leptin, respond only to ANG causing an elevation in BP. CONCLUSIONS Our data demonstrate that exogenous leptin administration into the SFO results in significant decreases in BP in young and DR rats while leptin microinjection into leptinresistant DIO rats is without effect. These observations suggest that the SFO may be an 101 important relay center through which leptin, in normal weight, leptin responsive individuals, acts to maintain BP within normal physiological limits through descending autonomic pathways involved in cardiovascular control. In contrast, in obese, leptinresistant, individuals leptin is no longer able to influence SFO neurons thus resulting in elevated BP, contributing to obesity related hypertension. ACKNOWLEDGEMENTS: This work is funded by the Canadian Institutes for Health Research. PMS is supported by a CIHR Banting and Best PhD fellowship. 102 Chapter 5: LEPTIN INFLUENCES THE EXCITABILITY OF AREA POSTREMA NEURONS 103 ABSTRACT Leptin is an adipocyte-derived peptide hormone encoded by the ob gene that circulates at concentrations proportional to adipose tissue mass and communicates levels of fat stores to hypothalamic and brainstem nuclei to influence food intake and energy expenditure, thus playing an important role in long-term regulation of energy balance. The area postrema (AP) is a sensory circumventricular organ (CVO) which lacks the normal blood brain barrier, with important roles in central autonomic regulation. This medullary structure has been shown to express the leptin receptor and has been suggested to have a role in modulating peripheral signals indicating energy status. Using RT-PCR we have shown the presence of mRNA for the signaling form of the leptin receptor, ObRb, in AP and whole cell current clamp recordings from dissociated AP neurons demonstrated that leptin (10nM) influenced the excitability of 51% (42/82) of AP neurons. The majority of responsive neurons (62%) exhibited a depolarization (mean change in membrane potential = 5.3 ± 0.7 mV, n=26) in response to leptin administration The remaining affected cells (16/42) demonstrated hyperpolarizing effects (mean change in membrane potential = -5.96 ± 0.95 mV, n=16) in response to leptin administration. Furthermore, amylin was found to influence the same population of AP neurons. Of the 7 cells that depolarized in response to bath administration of leptin, 5 also depolarized in response to bath perfusion of amylin. These findings suggest that the AP, a sensory CVO with direct access to peripheral circulation, is a site at which leptin may act to influence CNS control of energy homeostasis. 104 INTRODUCTION Leptin is an adipocyte-derived peptide hormone encoded by the ob gene that circulates at concentrations proportional to adipose tissue mass and communicates levels of fat stores to the central nervous system (CNS), thus playing an important role in longterm regulation of energy balance. Circulating leptin exerts its effects on feeding and energy expenditure by binding to leptin receptors in the CNS (Grill & Kaplan, 2002;Burguera et al., 2000). Although 6 leptin receptor isoforms have been identified (Tartaglia et al., 1995), only the long form of the receptor, ObRb, possesses the cytoplasmic domains required for signal transduction (Bjorbaek et al., 1997;Banks et al., 2000;Kloek et al., 2002) and this form is essential for the weight reducing effect of leptin (Bjorbaek et al., 1997;Bjorbaek & Kahn, 2004;Buettner et al., 2006;Bates et al., 2003;Cui et al., 2004) The arcuate nucleus of the basal hypothalamus has been a principle focus of research directed towards elucidating leptin actions in the CNS (Satoh et al., 1997) (for review see Cone et al., 2001). However, more recently, the role of other hypothalamic, forebrain, and brainstem regions have been shown to be critical in mediating the central effects of leptin (Grill & Hayes, 2009;Qi et al., 2010;Hommel et al., 2006;Huo et al., 2007;Myers, Jr. et al., 2009;Dhillon et al., 2006;Hayes et al., 2010). Whether at the arcuate or in these other CNS regions where leptin receptors are found, in order for circulating leptin to act centrally to influence body weight homeostasis, this adipokine must cross the blood brain barrier (BBB) to reach receptors in the hypothalamic and brainstem nuclei. The BBB, a regulatory interface between the 105 brain and periphery, restricts the central access of circulating molecules (Banks & Kastin, 1990) and leads to the obvious question as to how leptin gains access to central sites to influence body weight regulation. Although a saturable leptin transport system (Banks et al., 1996) and transendothelial signaling (Paton et al., 2007) represent potential mechanisms through which peripheral signals may reach neurons protected by the BBB, recent evidence (Smith et al., 2009) suggests that the sensory circumventricular organs (CVOs), a group of specialized CNS structures which lack the normal BBB, may play a role. The area postrema (AP), located on the wall of the fourth ventricle, is a sensory CVO well known for its role in the emetic reflex (Miller & Leslie, 1994). The AP also has well-documented roles in immune function, cardiovascular regulation and energy homeostasis (see Cottrell & Ferguson, 2004;Price et al., 2008 for review). The AP sends extensive efferent projections to hypothalamic and medullary autonomic nuclei (Shapiro & Miselis, 1985a;Gross et al., 1990; for review see Fry et al., 2007) and has receptors for numerous peripheral signals involved in feeding and metabolism (Hindmarch et al., 2008; for review see Price et al., 2008). Although few studies have examined the responsiveness of the AP to leptin, the demonstration of ObR-like immunoreactivity (Meister & Hakansson, 2001), and mRNA expression (Grill et al., 2002) in the AP suggest potential actions for leptin in this sensory CVO. In addition, recent gene array studies from our own laboratory have revealed the presence of the leptin receptor mRNA in the AP (Hindmarch et al., 2008). The specific leptin receptor subtypes were not, however, determined in any of the above studies. 106 Measurements of phosphorylated-signal transducer and activator of transcription 3 (pSTAT3) immunoreactivity, a direct downstream marker of leptin receptor activation, has also been observed in the AP (Ellacott & Cone, 2006;Huo et al., 2007). At the physiological level, leptin injection into the 4th ventricle has been shown to suppress appetite (Skibicka & Grill, 2009) while knockdown of LepR in the mNTS and AP causes hyperphagia as well as increased body weight and adiposity (Hayes et al., 2010) providing further evidence for a role of the AP in mediating the central effects of leptin. In addition, a recent study demonstrated that the AP plays a pivotal role in mediating a synergistic weight loss effect of amylin and leptin in leptin resistant diet-induced obese rats (Roth et al., 2008). Thus, the AP may provide a route in which circulating leptin may act to influence downstream autonomic nuclei that regulate feeding behavior. The present study was undertaken to determine whether the signaling form of the leptin receptor (ObRb) is found in AP and to examine the functional effects of activation of this receptor on AP neurons. MATERIALS AND METHODS All rats were maintained on a 12/12 light-dark cycle and were provided with food and water ad libitum before decapitation. All animal protocols conformed to the Canadian Council for Animal Care (CCAC) standards and were approved by the Queen’s University Animal Care Committee. 107 Leptin Receptor Localization Using RT-PCR Unanesthetized male Sprague Dawley rats (100-150g) were decapitated and the brains were quickly removed and placed into oxygenated (95% O2/5% CO2), ice cold (14°C) artificial cerebrospinal fluid (aCSF) containing (in mM): 124 NaCl, 2 KCl, 1.25 KH2PO4, 2.0 CaCl2, 1.3 MgSO4, 20 NaHCO3 and 10 glucose. Brain slices (300 µm) were cut through the medulla using a Vibratome (Leica, Nussloch Germany) and placed in Hibernate medium (Brain Bits, Springfield, Illinois) supplemented with 1 X B27 (Invitrogen, Burlington, Ontario, Canada). Under a dissecting microscope, the AP was identified based on its distinct anatomical boundaries and its location on the border of the fourth ventricle and carefully microdissected to ensure no surrounding tissue was included. Total RNA was extracted from AP from 3 rats using an Ambion RNAqueous kit and was then DNase treated (Fermentas) by adding 1μl 10X buffer with MgCl2, and 1μl deoxyribonuclease to the total RNA, followed by incubation at 37°C for 30 min. After incubation, 1μl of 25mM ethylenediaminetetraacetic acid (EDTA) was added to the solution and incubated at 65°C for 10 min to stop the DNAse reaction. Reverse transcription of the RNA to cDNA was immediately undertaken by adding 26mM dithiothreitol, 100ng random hexamers, 10mM dNTP mix, 5mM MgCl2, 20 U RNase inhibitor and 100 U superscript II (Invitrogen, Carlsbad, CA), and incubated overnight at 37oC. 108 Five microliters of AP cDNA were added to a multiplex PCR reaction containing 25μl of 2X QIAGEN Multiplex PCR Master Mix, 5μl of Q solution, 0.4μM of each primer set, and diethyl pyrocarbonate-treated H2O to a final volume of 50μl. To prevent the formation of misprimed products and primer dimers, the HotStarTaq DNA polymerase (contained in the QIAGEN Multiplex PCR Master Mix) was activated by a 15 min, 95°C-incubation step. The reaction tube was then cycled 35 times through a protocol of 94°C for 30 sec, 60°C for 90 sec, 72°C for 90 sec, and finally, 72°C for 10 min. The product of the amplification was purified with the QIAquick PCR Purification Kit to remove excess primers, nucleotides, and other impurities. To ensure full amplification of the target sequences, the PCR product was used in a second round PCR amplification. Two microliters of the first reaction was used as a template in a second round of nested PCR using the same reagent volumes as in the multiplex reaction. The HotStarTaq DNA polymerase (contained in the QIAGEN Multiplex PCR Master Mix) was again activated by a 15 min, 95°C incubation step, and then cycled 30 times through a protocol of 94°C for 20 sec, 60°C for 30 sec, 72°C for 40 sec, and finally, 72°C for 10 min. Primers (Integrated DNA Technologies) directed toward an extracellular domain common to all leptin receptor isoforms were used to detect the presence of leptin receptor (LepR) mRNA (see Table 5-1). To determine whether the signaling form of the receptor (ObRb) mRNA was present in AP, two different primer set, one previously verified primer set (Burdyga et al., 2002) and one we designed ourselves, specific for the 109 Gene Primer Glyceraldehyde-3phosphate GAPDH dehydrogenase Leptin receptor b ObRb1 Leptin receptor b ObRb2 Leptin receptor a ObRa Leptin receptor LepR Position Sequence Sense tgccactcagaagactgtgg Antisense acctggtcctcagtgtagcc Sense gattccacaaggggttcta Antisense ctggaggattctgatgtc Sense tgaccactccagattccaca Antisense tgaacagacagtgagctggg Sense acactgttaatttcacaccagag Antisense agtcattcaaaccatagtttagg Sense tcgtcctacatcagagctca Antisense gtggagagtcaagtgaacct Product Size (bp) Table 5-1: Primer sets used to detect leptin mRNA from the area postema 110 Ascension number 297 NM_017008 510 NM_012596 501 NM_012596 237 AF304191 140 NM_012596 intracellular signaling domains on ObRb were used (See Table 5-1). Sets of primers were also used to detect Ob-Ra mRNA (see Table 5-1), as well as GAPDH (positive control). All of the aforementioned primers were also used in hypothalamic tissue, prepared in the same manner as the AP, which acted as a positive tissue control, and with sterile H2O which served as a negative, no template, control. All PCR products were run and visualized on electrophoresis gel containing 2% agarose and RedSafe nucleic acid dye. The validity of all PCR products was confirmed by sequencing (Robarts Institute, London, ON, Canada). Dissociated Cell Preparation and Cell Culture AP was isolated as described above for RT-PCR. Following isolation, the AP was incubated in Hibernate-A medium containing 2 mg/ml papain (Brainbits, Worthington, Lakewood, NJ) at 30°C for 30 min. After incubation, AP tissue was washed and triturated in B-27 supplemented Hibernate-A medium, and dissociated cells were centrifuged at 500 rpm for 8 min. The supernatant was then removed and the pellet was re-suspended in B-27 supplemented Neurobasal-A medium (Invitrogen) supplemented with 5mM glucose, 100 U/ml penicillin-streptomycin, and 0.5mM L-glutamine (Invitrogen). Dissociated cells were plated on 35mm uncoated glass-bottom culture dishes (MatTek, Ashland, MA) at a low density (∼10 cells/mm2) to ensure that synaptic contacts did not form, and then incubated at 37°C in 5% CO2 for 1–5 days. 111 AP slice preparation Unanesthetized, male Sprague-Dawley rats (Charles River, 23-27 days) were quickly decapitated and the brain removed and placed into oxygenated (95% O2/5% CO2), ice-cold (1-4°C) slicing solution consisting of (in mM): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose, and 75 sucrose. A medullary tissue block containing the AP was isolated and 300μm coronal slices were obtained using a Vibratome (Leica, Nussloch Germany). Slices were then incubated at 32°C for at least 1 h in oxygenated aCSF (external recording solution) composed of (in mM): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2 CaCl2, 2 MgCl2 1.25 NaH2PO4, and 10 glucose, pH 7.2 with NaOH. Slices were transferred to a recording chamber and continuously perfused at a rate of 1-2 ml/min with 32°C aCSF. Neurons were visualized using an infrared differential interference contrast system on an upright microscope (Nikon, Japan). Electrophysiology Whole cell current-clamp recordings from AP neurons were obtained using an Axopatch 700B (dissociated cell recording) or Multiclamp 700B (slice recordings) patchclamp amplifier (Molecular Devices, Palo Alto, CA). Stimulation and recording parameters were controlled by Spike2 (version 6) and Signal (version 3) software (Cambridge Electronics Design, Cambridge, UK). For dissociated cell recordings, data were acquired at 8 kHz, filtered at 2 kHz, and digitized using a Micro1401 interface (Cambridge Electronics Design) while for slice recordings data were acquired at 10kHz 112 and filtered at 2.4KHz. Capacitive transients and series resistance errors were minimized before recording. For dissociated cell recordings, the external recording solution contained the following (in mM): 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and 10 glucose, pH 7.2 with NaOH. Patch electrodes were made from borosilicate glass (World Precision Instruments, Sarasota, FL) on a Flaming Brown micropipette puller (model P97; Sutter Instrument, Novato, CA). Electrodes used for dissociated cell recordings were then fire polished and had resistances of 2.5–5 MΩ when filled with internal recording solution that contained (in mM) 130 K-gluconate, 10 KCl, 1 MgCl2, 2 CaCl2, 10 HEPES, 10 EGTA, and 2 NaATP. For slice recording, pipettes were filled with an intracellular solution made of (in mM): 125 potassium gluconate, 10 KCl, 2 MgCl2, 0.1 CaCl2, 5.5 EGTA, 10 HEPES, 2 NaATP (pH 7.2 with KOH) and had a resistance of 3-5 MΩ. All chemicals were purchased from Sigma (Oakville, ON, Canada). Once whole cell configuration was achieved, cells were perfused via a gravity-fed perfusion system with external recording solution at a rate of 1-2 ml/min. Cells were defined as neurons by the presence of ≥50 mV action potentials. Following a minimum 5 min. stable baseline recording period (control), 100 pM or 10 nM leptin (Phoenix Pharmaceuticals, Belmont, CA; reconstituted in external recording solution) was bath applied for 100-200 sec followed by a wash with external recording solution. Leptin concentrations were chosen based on the low and high boundaries of leptin concentrations normally measured in rat circulation (Landt et al., 1998;Landt et al., 2001). 113 We also examined the effects of leptin in amylin-sensitive AP neurons, by determining the effect of bath administration of amylin (10 nM) and leptin (10 nM) on the same dissociated AP neurons. Leptin was applied before amylin in some recordings and in other recordings the order of peptide application was reversed. The second peptide was not applied until the neuron fully recovered and stabilized at the original baseline potential, or the neuron partially recovered and stabilized at a new membrane potential. Electrophysiological Data Changes in membrane potential were calculated from the maximal difference between the average membrane potential in 100 sec segments immediately before and for 100 sec epochs after peptide application. AP neurons were considered responsive if this difference was ≥2 SDs of the mean baseline membrane potential and the cell showed recovery toward baseline. To be considered significant, a response must have initiated during the period of leptin application. The response magnitude was measured at its maximum value (mean over 100s epoch) before returning to baseline. Changes in action potential frequency were assessed by comparison of the difference between the mean action potential frequency 100 s immediately before leptin application and that after leptin application. Statistics Mean change (mean ± SEM) in membrane potential before (control) and during leptin peptide administration (leptin) were calculated, and differences were tested using a 114 paired Student's t-test. A Fisher’s exact test was used to determine if the proportion of depolarizing and hyperpolarizing responses were different at different concentrations of leptin (100pM and 10nM). All statistical analyses were performed using GraphPad Prism (version 5.0; San Diego, CA). RESULTS ObRb mRNA is expressed in the area postrema RT-PCR analysis of cDNA obtained from mRNA isolated from acutely microdissected AP using primer sets directed toward an extracellular domain common to all leptin receptor isoforms confirmed the presence of leptin receptor (LepR) mRNA in AP (see Figure 5-1). To determine whether the signaling form of the leptin receptor (ObRb) was present in AP, primer sets directed toward two different unique intracellular signaling domains on the ObRb receptor were used and revealed the presence of ObRb mRNA in AP (see Figure 5-1). Ob-Ra mRNA was also present in SFO as illustrated in Figure 5-1. LepR, ObRb1, ObRb2, and ObRa mRNA were also localized in the hypothalamus, an area of the brain previously shown to express ObRb (Elmquist et al., 1998b), which thus served as a positive control (see Figure 5-1). GAPDH served as a positive control for the PCR reactions. PCR products for GAPDH and all leptin receptors (ObRb1, ObRb2, ObRa, and LepR) were not observed in the no template control (NTC) lane in which the template has been omitted from the cDNA synthesis reaction and no 115 Figure 5-1: ObRb receptor mRNA is expressed in the AP Agarose gels showing RT-PCR analysis of AP cDNA for leptin receptor expression (left gel). Ob-Rb receptor (ObRb1, ObRb2) mRNA, as well as Ob-Ra receptor (ObRa) mRNA, and leptin receptor (LepR) mRNA (expression common to all leptin receptor isoforms) were also expressed in the AP. The hypothalamus (right gel) served as a positive control tissue and shows ObRb (ObRb1, ObRb2), ObRa (Ob-Ra), and LepR receptor expression. GAPDH served as a positive control in both AP and hypothalamic tissue. Product size (base pairs) is shown in the leftmost lane of each gel. 116 labeling was found in the negative controls (data not shown). The validity of all PCR products was confirmed by sequencing. Leptin influences the excitability of area postrema neurons Whole cell current-clamp techniques were used to evaluate the direct effects of leptin receptor activation on neuronal excitability of dissociated AP neurons. Currentclamp recordings were obtained from 103 AP neurons. Bath application of leptin (10nM) influenced the excitability of the majority (42/82) of AP neurons tested. The majority of responsive neurons (62%) exhibited a depolarization (mean change in membrane potential = 5.3 ± 0.7 mV, n=26) in response to leptin administration (see Fig. 2). All depolarizing responses began within 100 sec of leptin application. The depolarizing responses seen in response to leptin administration were of a long duration, often lasting several minutes upon termination of leptin application, and were typically accompanied by an expected increase in action potential firing frequency. The remaining affected cells (16/42) demonstrated hyperpolarizing effects (mean change in membrane potential = 5.96 ± 0.95 mV, n=16) in response to leptin administration (see Figure 5-2). Similar to the depolarizing responses, these effects began within 100 s of peptide administration, lasted several minutes, and were often accompanied with a decrease in action potential firing frequency. A 100-fold lower concentration of leptin (100pM) was bath-applied to 21 dissociated neurons to determine whether leptin responses were concentration dependent. There was no difference in the number of AP neurons affected, the distribution of 117 Figure 5-2: Leptin influences the excitability of AP neurons Left Panel (Dissociated) Current-clamp recordings from 2 different dissociated AP neurons showing that bath application of 10nM leptin caused a depolarization (upper trace) and an accompanying increase in action potential firing frequency while a different neuron (lower panel) exhibited a hyperpolarization and an accompanying decrease in action potential firing frequency in response to leptin administration. The right panel (Slice) shows current-clamp recordings from 2 different AP neurons obtained from slice preparations. The upper trace shows a depolarizing response to bath administration of 10nM leptin, while the neuron shown in the lower trace was hyperpolarized by leptin application. Time and duration of leptin administration is indicated by the gray bar above each current-clamp recording. 118 depolarizing and hyperpolarizing responses, or the magnitude of the response. Of the 21 neurons tested, 52% (11/21) neurons were influenced by bath administration of leptin with 8 neurons showing a depolarizing response (mean change in membrane potential = 4.3 ± 2.7 mV), 3 neurons exhibiting a hyperpolarization (mean change in membrane potential was -3.7 ± 1.1 mV), and the remaining cells (n=10) being unaffected by leptin administration. Although the magnitude of the effects of 100pM leptin on membrane potential were smaller than observed after administration of 10nM, these changes were not statistically significant (depolarization: p=0.6; hyperpolarization p=0.32), nor were the proportion of cells demonstrating depolarizing and hyperpolarizing responses (p = .72, Fisher’s exact test). To ensure that the effects on the neuronal excitability of AP neurons in response to leptin administration were not the result of changes in these cells as a consequence of the dissociation procedure, the effect of leptin administration on AP neurons in slice preparations was also evaluated. Whole cell current-clamp recordings from 10 AP neurons in slice preparation showed similar responsiveness to bath application of 10 nM leptin with 30% of cells exhibiting depolarizations (mean change in membrane potential 6.95 ± 1.2 mV, n = 3, see Figure 5-2), while 4 cells hyperpolarized (−4.5 ± 0.95 mV, see Figure 5-2). The remaining cells tested (n=3) were unaffected by leptin application. Leptin influences amylin sensitive area postrema neurons We next examined whether leptin influenced amylin sensitive AP neurons, using wholecell current clamp recordings from 14 dissociated AP neurons which were tested for 119 responsiveness to both leptin (10nM) and amylin (10 nM). Of the 7 cells that depolarized in response to bath administration of leptin, 5 also depolarized in response to similar bath perfusion of amylin (see Figure 5-3). None of the AP neurons that hyperpolarized in response to leptin were influenced by amylin (n=2). The remaining 5 cells were unaffected by application of either peptide. DISCUSSION In this study we have, for the first time, demonstrated the presence of the signaling form of the leptin receptor, ObRb, in the AP and have also shown direct effects of the adipocyte-derived hormone, leptin on the excitability of AP neurons. Although the hypothalamic arcuate nucleus is perceived as the principal mediator of leptin signaling, there is a growing body of evidence suggesting that leptin also acts in the caudal brainstem (Grill, 2010;Myers, Jr. et al., 2009). Leptin receptor mRNA has been shown to be expressed in these regions including the AP, NTS and dorsal motor nucleus of the vagus (Hindmarch et al., 2006;Buyse et al., 2001) and this expression is complemented by immunohistochemical experiments showing expression of the LepR receptor itself in these same areas (Grill et al., 2002). Using RT-PCR we have confirmed the presence of the leptin receptor (LepR) in the AP and have shown that the signaling form of the leptin receptor, ObRb, is present in this medullary CVO, suggesting a role for the AP in mediating the central effects of leptin. A functional role for ObRb in AP is supported by the fact that pSTAT3 activation is observed in the AP (Huo et al., 2007). 120 10nM Amylin 20mV 60 s 10nM Leptin Figure 5-3: Leptin depolarizes amylin-sensitive AP neurons. Current-clamp recording obtained from the same SFO neuron illustrating depolarizing responses to 10nM amylin (top trace) and 10nM leptin (bottom trace). Time and duration of amylin (top trace, black bar) and leptin (bottom trace, grey bar) administration is indicated by the bar above each current-clamp recording. 121 Further support for a functional role of ObRb receptor activation in AP is shown by the results of our electrophysiological recordings in the present study. Using current clamp techniques to record from AP neurons in both dissociated cell and medullary slice preparations, we showed that more than half the AP neurons were influenced by leptin, with either depolarization or hyperpolarization observed in response to this adipokine. The dissociation process leaves us with single SFO cells in synaptic isolation (no visible dendritic contacts), which are thus ideally suited for the assessment of direct effects of exogenously applied leptin on the excitability of AP neurons. Although it is possible that the dissociation procedure itself or the 1-5 day culture period influenced receptor expression, it is not likely as leptin-mediated responses on neuronal excitability were not altered during our recording period and similar responses were observed in cells obtained from our AP slice preparation. Similar findings from dissociated neurons and neurons obtained from acutely prepared brain slices have been previously demonstrated for adiponectin actions in the AP (Fry et al., 2006) and leptin actions in the SFO (Smith et al., 2009).The observation of both excitatory and inhibitory responses is consistent with previous studies suggesting the existence of neuronal subpopulations in the AP that respond differentially to specific peptides (Cai & Bishop, 1995;Fry et al., 2006;Yang & Ferguson, 2003;Ingves & Ferguson, 2010). Although not evaluated in the present study, the differential responses (depolarization vs hyperpolaization) may reflect different projection sites of subpopulations of AP neurons. Major projections of the AP include the NTS and the lateral parabrachial nucleus (Shapiro & Miselis, 1985a), both of which are 122 major sites of sensory-autonomic integration. The NTS is of particular interest due to the presence of proopiomelanocortin (POMC) neurons, which are proposed to be the primary cell type mediating the central anorexic effects of leptin (Cowley et al., 2001). The energetic and anorexic effects of leptin injection into the fourth ventricle are blocked by antagonizing hindbrain receptors for melanocortins (a product of the POMC gene), thereby demonstrating the importance of POMC signaling in leptin effects in the caudal brainstem (Skibicka & Grill, 2009). It is unknown, however, whether leptin directly activates NTS POMC neurons or binds leptin receptors on upstream neurons that regulate NTS POMC gene expression. Our demonstration of AP leptin-sensitivity, coupled with well-documented AP projections to the NTS, suggests that the AP is a plausible candidate for regulating POMC-mediated leptin signaling in the caudal brainstem. Our demonstration of both depolarizing and hyperpolarizing effects of leptin is consistent with the presence of different neuronal populations in the AP. We investigated whether the subpopulation of AP neurons that is activated by leptin is also activated by amylin, a pancreatic peptide that is co-secreted with insulin in response to food intake and increases in blood glucose (Cooper et al., 1989), reducing food intake by binding its receptors in the CNS (Morley & Flood, 1991). The AP has been shown to mediate the anorexic effects of amylin as these effects on food intake are eliminated in AP-lesioned animals (Lutz et al., 2001;Lutz et al., 1998). Electrophysiological studies have revealed that amylin depolarizes about half of AP neurons tested and about 90% of these neurons are also sensitive to glucose (Riediger et al., 2001;Riediger et al., 2002), suggesting 123 integrative actions of energy homeostasis related circulating factors at the AP. Given that leptin and amylin both inhibit food intake in response to positive energy balance, albeit that they function over different time periods (leptin is a long-term adiposity signal, whereas amylin is a short-term satiety signal), it is plausible that a subpopulation of AP neurons may mediate the anorexic effects of both these circulating factors, and thus exhibit excitatory responses to both peptides. Our observation that the majority of neurons that were depolarized by leptin were also depolarized by amylin while neurons that were hyperpolarized by leptin, or did not respond to leptin, were insensitive to amylin, is in accordance with such a hypothesis. Therefore, it is likely that amylin and leptin activate the same subpopulation of AP neurons. Our findings correspond with similar experiments in the SFO demonstrating complementary effects of leptin and amylin on neuronal excitability (Smith et al., 2009) Leptin-amylin agonism has implications for pharmacological obesity interventions as preclinical trials in rats and humans have shown synergistic weight loss with leptin-amylin co-administration, exceeding the predicted additive effect of therapy with either peptide alone (Roth et al., 2008;Trevaskis et al., 2008). The physiological effects of amylin have been shown to be almost exclusively mediated by actions in the AP (Roth et al., 2009). Our finding that amylin and leptin activate the same population of AP neurons may provide insight into mechanisms of leptin-amylin synergistic effects on weight loss. 124 In summary, the present study demonstrates the direct actions of leptin on the excitability of AP neurons and suggests that leptin action in the AP may contribute to its regulation of long-term energy balance. The AP, an ideal target for leptin due to its ability to monitor the contents of the circulation and transmit this information via wellestablished projections to autonomic nuclei in the brainstem and hypothalamus, provides a route by which the circulating adiposity signal, leptin, which cannot cross the BBB, can act to regulate energy expenditure and feeding behaviour. Furthermore, the effects of leptin and amylin on the same subpopulation of neurons in the AP may be relevant to the development of obesity interventions based on the synergistic effects of leptin and amylin on weight loss. ACKNOWLEDGEMENTS: This work was supported by a Canadian Institutes for Health Research Grant (AVF). PMS is supported by a Banting and Best PhD studentship from the Canadian Institutes for Health Research. AM is supported by a National Sciences and Engineering Research Council PGS-D scholarship and a Fonds de Recherche du Quebec scholarship. The authors would like to thank Stefanie Killen for excellent technical assistance. 125 Chapter 6: LESIONS OF THE AREA POSTREMA AND SUBFORNICAL ORGAN ATTENUATE LEPTIN-INDUCED DECREASES IN BODY WEIGHT AND FOOD INTAKE 126 ABSTRACT Leptin, an adipocyte-derived peptide hormone circulates at concentrations proportional to adipose tissue mass and communicates levels of fat stores to the central nervous system (CNS). Peripheral (intraperitoneal, ip) administration of leptin has been shown to decrease food intake and body weight via actions in hypothalamus and brainstem. Recent evidence suggests that neurons in the area postrema (AP) and subfornical organ (SFO), two sensory circumventricular organs (CVOs) which lack the normal BBB and contain the leptin receptor, may mediate the central effects of leptin via well-established efferent connections to hypothalamic and medullary metabolic control centers. The current study was undertaken to determine whether lesions of the AP and SFO would influence the ability of systemic leptin to decrease body weight and food intake. While ip leptin administration reduced 24 hour body weight gain and food intake in animals with intact AP and SFO, animals with lesions of these two CVOs did not demonstrate a reduction in body weight in response to peripheral leptin administration. Interestingly, leptin administration caused an increase in food intake and water intake in AP/SFO lesioned animals. The results of the present study clearly demonstrate that AP/SFO lesions attenuate the decrease in body weight gain normally observed following peripheral administration of leptin, supporting a critical role for the AP and SFO in mediating the central, metabolic actions of leptin. 127 INTRODUCTION Leptin, an adipocyte-derived peptide hormone encoded by the ob gene, circulates at concentrations proportional to adipose tissue mass and communicates levels of fat stores to the central nervous system (CNS), thus playing an important role in long-term regulation of energy balance. Peripheral (intraperitoneal, ip) administration of leptin has been shown to dose-dependently decrease food intake and body weight in both ob/ob obese mice as well as wild type littermates (Halaas et al., 1995b;Halaas et al., 1997), effects which occur as a result of actions in the CNS (Grill & Kaplan, 2002) (Burguera et al., 2000). Although 6 leptin receptor isoforms have been identified (Tartaglia et al., 1995), only the long form of the receptor, ObRb, possesses the cytoplasmic domains required for normal signal transduction (Bjorbaek et al., 1997;Banks et al., 2000;Kloek et al., 2002) associated with the weight reducing effect of leptin (Bjorbaek et al., 1997;Bjorbaek & Kahn, 2004;Buettner et al., 2006;Bates et al., 2003;Cui et al., 2004). The arcuate nucleus of the hypothalamus (ARC) has been a principle focus of research directed towards elucidating leptin actions in the CNS (Satoh et al., 1997) for review see Cone et al., 2001). However, more recently, the role of extrahypothalamic (forebrain and brainstem regions) have also been shown to be critical in mediating the central effects of leptin (Grill & Hayes, 2009;Qi et al., 2010;Hommel et al., 2006;Huo et al., 2007;Myers, Jr. et al., 2009;Dhillon et al., 2006;Hayes et al., 2010;Qi et al., 2010). In order for this peripherally derived adipokine to influence body weight homeostasis through direct actions in the CNS, it must cross the blood brain barrier (BBB) to reach receptors in the hypothalamic and brainstem nuclei. 128 The BBB is a regulatory interface between the brain and periphery designed to restrict access of circulating molecules to the CNS. Although a saturable leptin transport system (Banks et al., 1996) represents a potential mechanism through which peripheral signals may reach neurons protected by the BBB, recent evidence (Smith et al., 2009) suggests that the sensory circumventricular organs (CVOs), a group of specialized CNS structures which lack the normal BBB and contain exceptionally dense aggregations of a variety of different receptors for peripheral signals, may play a role. The area postrema (AP), located on the wall of the fourth ventricle, and the subfornical organ (SFO), located in the forebrain on the midline wall of the third ventricle dorsal to the anterior commissure, are sensory CVOs that have been shown to be involved in the control of energy balance (see Cottrell & Ferguson, 2004;Price et al., 2008 for review). Interestingly, we have previously shown that lesions of the AP and SFO chronically decrease body weight in rats (Baraboi et al., 2010b;Baraboi et al., 2010a) over a 30 day period. In addition, these CVOs were shown to be critical for peptide YY (PYY) elicited decreases in food intake as animals with lesions of the AP and SFO no longer exhibited the initial decrease in food intake normally observed in response to peripheral administration of PYY (Baraboi et al., 2010a). Furthermore, exendin-4 (glucagon-like peptide-1 (GLP-1) agonist) induced expression of c-fos in hypothalamic (ARC, PVN, SON) and hindbrain nuclei with important roles in energy homeostasis was attenuated in rats with lesions of the AP and SFO (Baraboi et al., 2010b) further 129 supporting an important role for the CVOs in the activation of CNS structures involved in homeostatic control. A role for the AP and SFO in mediating the central effects of leptin is highlighted by recent studies demonstrating that both the AP and the SFO express mRNA for the signaling form of the leptin receptor, ObRb, (Smith et al., 2009) and electrophysiological studies have revealed that leptin administration influences the excitability of AP and SFO neurons (Smith et al., 2009). Thus, the AP and SFO are uniquely positioned to detect circulating leptin and communicate this information to hypothalamic and medullary autonomic nuclei involved in energy homeostasis (Shapiro & Miselis, 1985a;Gross et al., 1990;Miselis et al., 1979;Lind et al., 1982;Gruber et al., 1987;Miselis, 1981). The current study was undertaken to determine whether lesions of the AP and SFO would influence decreases in body weight normally observed in response to peripheral leptin administration. MATERIAL AND METHODS Animals and Diet All procedures were performed in accordance with Canadian Council on Animal Care and Queen’s University Animal Care Committee. The animals were individually housed, exposed to a 12:12hour light:dark cycle, and were provided ad libitum standard rodent laboratory chow (Purina) and water, unless otherwise specified. 130 Electrolytic Lesions Male Sprague Dawley rats (100-125 g) were randomly assigned to 1 of 2 groups; 1) SFO + AP lesion or 2) SFO Intact + AP Intact. Animals with intact SFO and AP included animals that either 1) received sham lesions to these structures whereby the electrode was placed into AP and SFO however, no current was passed or 2) had lesion sites that did not incorporate AP (or the adjacent NTS) or SFO (or its rostral ventral stock – see below). AP lesion + SFO lesion (AP/SFO lesion) SFO lesion: Under sodium pentobarbitol anesthesia (65mg/kg, ip), rats were placed in a stereotaxic apparatus and the head was horizontally fixed. A midline incision was made in skin of the skull and a small hole was drilled such that a monopolar Parylene C-insulated tungsten electrode with a tip exposure of 100 μm (Micro-Probe) could be stereotaxically positioned into the region of the SFO according to the coordinates of Paxinos and Watson (Paxinos & Watson, 1982) (midline, 0.7 mm caudal to bregma, 4.5 mm ventral to surface). The rat was then subjected to an electrolytic lesion (Keithley Instruments 225 Current Source) by passing 0.5 mA direct current for 30 sec. The electrode was removed and the skin closed. The electrode was then positioned into the AP. AP lesion: Following SFO lesion, rats were placed in a stereotaxic apparatus in the nose-down position. A midline incision was made in the skin overlying the occipital 131 bone to the atlas and the musculature was blunt dissected such that the cisterna magna could be opened to permit access to the fourth ventricle. The AP was visualized and the electrode was positioned in the center of AP (0.3mm ventral to surface). This area was then electrolytically lesioned by passing 0.5 mA direct current for 30 sec. The musculature was closed followed by the skin. SFO Sham lesion + AP Sham lesion (AP/SFO Intact). Rats underwent the procedures outlined above to position the electrode into the region of SFO for 30 sec however no current was delivered to the site. The incision was closed and the electrode was then positioned into the AP for 30 sec but no current was delivered to the region. Postoperative Care Immediately following surgery and during postoperative days 1 and 2, rats received the analgesic, Metacam (Day 1: 2mg/kg, SQ, Day 2 and 3: 1mg/kg SQ), and the antibiotic Tribrissen (120mg/kg, SQ). Animals were weighed daily and, in addition to ad libitum access to normal rat chow and water, animals were provided with free access to liquid chocolate-flavored Ensure as a palatable, well-balanced diet to promote resumption of eating and drinking and to promote weight gain following surgery. The animals were allowed access to Ensure for at least 2 days or until they had begun to gain weight following surgery, after which they were returned to a normal laboratory chow diet and water only. Rats were allowed 2 weeks to recover before any further investigation, 132 during which time body weight, food and water intake were measured daily, 1 hour prior to the beginning of the dark cycle. All measurements and manipulations were performed by the same experimenter at the same time of day throughout the course of the experiment to minimize the likelihood that any changes observed were due to handling/manipulation by novel personnel. Experimental Protocol: Leptin Injections Leptin (rat recombinant, Phoenix Pharmaceuticals) was prepared in 15mM HCl (0.5ml/mg) and 7.5mM NaOH (0.3ml/mg) according to manufacturer’s directions and further diluted in aCSF. As such, the same HCl/NaOH/aCSF solution (without leptin) served as vehicle control. In order to ensure effects on body weight and food intake were due to the effects of leptin and not a consequence of handling, restraint, and/or injections, rats were treated with vehicle for 2 consecutive days, immediately followed, on day 3, by leptin (1mg/kg). Body Weight, Food Intake, Water Intake Body weight, food and water intake were measured daily 1 hour prior to lights off during all postoperative days. On the days of experimentation (vehicle or leptin injection), animals were weighed and food and water intake measured as had been done all previous days. Leptin or vehicle volume was calculated based on body weight obtained on the day of injection and was administered (ip) 30 minutes prior to lights out. Body weight, food and water intake were then measured 24 hours later. 133 Histological Analysis of AP and SFO Lesions As the conclusion of the experiment (2 days vehicle, followed by 1 day leptin), rats were overdosed with sodium pentobarbitol (200 mg\kg ip) and perfused through the left ventricle of the heart with 0.9% saline followed by formalin. The brain was removed and placed in formalin for at least 24 hours. Coronal sections were then cut through AP (50µm) and SFO (100µm), cresyl violet stained, and lesion sites were then validated by histological examination of brain sections by an observer unaware of the experimental protocol or the data obtained. Only animals having complete lesions of both the AP and SFO were included in the AP/SFO lesion group. AP lesions included rats with complete AP lesions and animals were excluded if there was any AP remaining or if damage to the adjacent NTS was evident. SFO lesions included only animals in which the SFO was totally destroyed or in which the rostral SFO and rostroventral stalk were sufficiently damaged to disconnect the SFO from its targets in the ventral forebrain. Neuronal SFO projections to the preoptic region and hypothalamus exit the SFO via its rostroventral stalk (Miselis, 1981). Data from rats with partial damage to the SFO without destruction of the rostroventral stalk were not included in the analyses. AP sham lesioned animals showed no damage present in AP or surrounding NTS tissue. SFO sham lesioned animals showed evidence of the electrode tracks into the hippocampal commissure but no evidence of damage to the SFO. 134 Statistical Analysis: Animals were grouped according into either the SFO/AP lesion group or SFO/AP intact group according to histological assessment of electrolytic lesions. For comparison of body weight at the beginning of injections, mean body weight for each group was calculated and compared for significance using a Student’s t-test. Percent change in 24 hr body weight and daily food per 100g body weight and water intake per 100g body weight were calculated daily for each animal and a mean calculated for each group. A paired ttest was used to evaluate whether differences prior to (Pre) and after leptin (Leptin) injections were significant. RESULTS A total of 15 animals were used in the present study in which 5 were classified as SFO/AP Lesion and 3 were placed in the AP/SFO Intact group. The remaining 7 animals were excluded from further analysis as they did not meet the criteria for either the lesion or intact group. Surgeries were performed over 2 consecutive days and vehicle and leptin injections were carried out, 2 weeks later during the same 3 day period. Despite similar recover times following from surgery, rats with SFO/AP lesions had significantly lower baseline body weights (mean body weight = 146.8 ± 1.6, n=5) at the time of the first vehicle injection when compared to SFO/AP intact animals (mean body weight = 227.1 ± 12.5, n=3; p<.001, Student’s t- test, see Figure 6-1). 135 Figure 6-1: AP/SFO Lesioned animals exhibit decreased body weight Mean body weight (BW) of AP/SFO Intact animals (red bar) and AP/SFO Lesion animals (blue bar) at two weeks following surgery. *** p<.001, Student’s t test. 136 Vehicle injection was without effect on body weight gain, food or water intake in either group (data not shown). As previously reported (Halaas et al., 1995b;Halaas et al., 1997;Bojanowska & Nowak, 2007;Nowak & Bojanowska, 2008), ip leptin administration significantly reduced 24 hour body weight gain in animals with intact AP and SFO (SFO/AP Intact) (preinjection = 3.7 ± 0.5%; leptin = 2.6 ± 0.5%, p<0.01) as illustrated in Figure 6-2. This decrease in body weight was accompanied by reduced 24 hour food intake/100g body weight (preinjection = 9.9 ± 0.4g; leptin = 8.7 ± 0.5g, p=0.06, see Figure 6-3) without influencing water consumption/100g body weight (preinjection = 15.7 ± 0.9ml; leptin = 15.5 ± 0.8ml, p= 0.56, see Figure 6-3). In contrast, animals with lesions of the AP and SFO (AP/SFO Lesion) did not demonstrate a reduction in 24 hour body weight gain in response to peripheral leptin administration (preinjection= 1.5 ± 0.5%; leptin = 2.0 ± 0.95%, p=0.67, see Figure 6-2). Interestingly, leptin administration caused an increase in food intake/100g body weight (preinjection = 4.6 ± 0.9g; leptin = 7.17 ± 1.0g, p=0.08) and water intake/100g body weight (preinjection = 14.8 ± 0.9ml; leptin = 19.8 ± 1.3ml, p= 0.01) in AP/SFO Lesion animals (see Figure 6-3). DISCUSSION The results of the present study show that AP/SFO lesions cause a profound decrease in body weight and food intake and attenuate the decrease in body weight gain normally observed following peripheral administration of leptin (Halaas et al., 1995b;Halaas et al., 1997). 137 Figure 6-2: Changes in body weight following leptin administration Mean percent (%) change in body weight (BW) in AP/SFO Intact animals (red bars) and AP/SFO Lesion animals (blue bars) prior to leptin injection (Pre; solid bars) and following leptin injection (Leptin; hatched bars) showing that leptin administration (1 mg/kg) no longer caused a decrease in % body weight gain in the 24 hours following leptin administration in AP/SFO Lesion animals. ** p<.01, paired Student’s t-test. 138 Figure 6-3: Food and water consumption following leptin administration These bar graphs show the 24 hour food intake (upper bar graphs) and water intake (lower bar graphs) in AP/SFO Intact animals (left bar graphs) and AP/SFO Lesion animals (right bar graphs) prior to leptin injection (Pre; solid bars) and following leptin injection (Leptin; hatched bars). Leptin administration caused a decrease in food intake in AP/SFO Intact animals (as expected) while in AP/SFO Lesion animals leptin administration caused an increase in both food and water intake. * p<.05, paired Student’s t-test. 139 The observations that lesions of the AP and SFO cause a significant reduction in both body weight and food intake suggest that the AP and SFO play an important role in the control of body weight. These observations are in agreement with previous studies demonstrating a persistent decrease in body weight in animals with AP and SFO lesions for up to 30 days post lesion (Baraboi et al., 2010a;Baraboi et al., 2010b). The magnitude of the decrease in body weight and food intake observed in the present study is greater than that seen in the studies by Baraboi (Baraboi et al., 2010a;Baraboi et al., 2010b). These observations may be due to the fact that, in the present study, the animals were subjected to lesions at a much earlier age (100 g vs 350g) and suggest that destruction of these CVOs at an earlier stage in development has more profound effects on the regulation of body weight and food intake than if AP/SFO lesions are performed in adult animals. The results of the present also show that AP/SFO lesions attenuate the decrease in body weight gain normally observed following peripheral administration of leptin (Halaas et al., 1995b;Halaas et al., 1997), supporting a role for the AP and SFO in mediating the central, weight reducing effect of leptin. The fact that leptin administration resulted in a decrease in body weight gain and food intake in AP/SFO intact animals, as predicted from previous studies (Halaas et al., 1995b;Halaas et al., 1997), indicates that the amount of leptin delivered in this study was sufficient to achieve previously observed effect on body weight and food consumption. A particularly interesting finding of this study is that leptin increased food intake in AP/SFO lesioned animals, an effect which is 140 opposite to that normally observed in response to peripheral leptin administration. This increase in food intake was also accompanied by an increase in water consumption. This study did not address whether the increase in food intake and water intake were dependent on the other, and if so, which preceded which, or the mechanism(s) by which AP and/or SFO lesions reverse the effect of acute peripheral leptin on food consumption. It is important to point out that these are preliminary studies and that more animals are needed to support the observations described above. In addition to increasing numbers in the AP/SFO Intact group, which will conceivably strengthen the observation that leptin administration decreases body weight gain and food intake, it will be important to assess whether AP or SFO lesion alone would be sufficient to attenuate leptin-induced effects on body weight gain observed in the present study. Previous studies showing that lesions of both AP and SFO are required to attenuate the initial decrease in food intake observed in response PYY (Baraboi et al., 2010a) and exendin-4 induced expression of cfos in the CNS (Baraboi et al., 2010b) suggests that neither AP nor SFO lesion alone may not be sufficient to blunt the peripheral actions of leptin. The fact that food intake is markedly decreased by AP/SFO lesions, may, in itself, impair the ability of leptin to decrease food intake further in these lesioned animals. Thus, additional experiments in older animals (>300g), in which lesions have been shown to chronically reduce body weight (Baraboi et al., 2010a;Baraboi et al., 2010b), but not to the magnitude observed in the current study, may be warranted to confirm that AP/SFO lesions attenuate lepin-induced decreases in body weight and food intake. 141 The AP and SFO are sensory CVOs and, as such, are uniquely positioned to monitor the constituents of the blood and, through well-documented efferent projections to autonomic nuclei in the brainstem and hypothalamus, communicate this information to centers behind the BBB responsible for controlling energy homeostasis. The preliminary data presented in the present study, demonstrating that animals with AP and SFO lesions exhibit a profound decrease in body weight and no longer demonstrate decreases in body weight in response to peripheral leptin administration, suggests that the AP and SFO play a critical role in the central control of body weight and in mediating the central effects of leptin. ACKNOWLEDGEMENTS: This work is funded by the Canadian Institutes for Health Research. PMS is supported by a CIHR Banting and Best PhD fellowship. 142 Chapter 7: GENERAL DISCUSSION 143 The discovery of leptin in 1994 (Halaas et al., 1995a) prompted a renewed interest and resurgence of research into the regulation of body weight homeostasis. A possible ‘lipostatic factor’ for the control of body weight, proposed 40 years (Kennedy, 1953) before this ground-breaking discovery, had been identified. With the demonstration that this adipose tissue derived circulating factor acted centrally to decrease food intake and increase energy expenditure, research into the central control of body weight grew exponentially. The research that followed has shaped our current understanding of body weight homeostasis in that it is now well established that peripherally derived signals (of which leptin is one) act centrally to influence hypothalamic, brainstem and reward circuits to coordinate integrative behaviours designed to maintain ideal body weight. In accordance with this understanding much attention has also focused on how pertubations in any part of this system, can lead to impaired energy homeostasis. Since the initial discovery of leptin a number of other peripheral factors released from adipose tissue (ie leptin, adiponectin, angiotensin), the pancreas (amylin, insulin), and the gastrointestinal tract (ie cholecystokinin, ghrelin, and peptide YY) have been shown to influence autonomic nuclei within the CNS to regulate body weight homeostasis (see Badman & Flier, 2005 for review). In addition, the development of genetic models in which a specific target has been deleted or over-expressed further emphasizes the importance of these signaling molecules in the regulation of body weight (for review see Balthasar, 2006;Elmquist et al., 2005;Santini et al., 2009). 144 Although all of these peptides have been shown to influence autonomic nuclei within the CNS, these nuclei are protected behind the BBB and thus do not have access to information regarding energy status provided by these circulating peptides. With the exception of insulin, which has been shown to be transported across the BBB via by a saturable transport system (Banks & Kastin, 1998), all of these peripherally derived signals are precluded from CNS access. Although mechanisms exist for the transmission of information regarding energy status carried by a few peptides produced in the periphery to the CNS have been demonstrated (ie CCK via vagal efferents to the caudal brainstem; selective insulin transporter for the movement of insulin across the BBB), how most of these signals influence neuronal elements in the CNS is unknown or speculative at best. Although a saturable transport system has been identified for leptin (Banks et al., 1996), this transport remains theortical as the physiological relevance remains to be shown as does the molecular identity of the transporter. How, then, do large, lipophobic peptides, and leptin in particular, gain access to the CNS to influence neuronal populations protected behind the BBB? The CVOs are specialized CNS structures that provide a route through which circulating peptides may deliver information to the CNS without the need to cross the BBB. The CVOs possess specialized fenestrated cerebral vasculature which allows large, lipophobic substances (peptides and proteins) to cross from blood to neural tissue without having to cross the cell membrane (Gross, 1992). These fenestrated capillaries are distinct from the rest of the CNS in that they lack the typical tight junctions between adjacent 145 endothelial cells (Petrov et al., 1994;Rodriguez et al., 2010). In addition, the CVOs possess an extensive and complex vascular supply as compared to other areas of the brain (Gross, 1991) presumably designed to maximize the time and area for exposure of blood borne substances to the cellular components of the CVOs (Gross, 1991). The demonstration of exceptionally dense aggregations of a variety of different receptors for peripheral signals clearly suggests that neurons in these CVOs have the ability to sense circulating concentrations of signaling molecules for which receptors are present. Thus, these specialized CNS structures are uniquely positioned to monitor the constituents of peripheral circulation and communicate this information, via well-documented afferent projections, to autonomic control centers in the hypothalamus and medulla and thus represent potential CNS windows for autonomic feedback. The subfornical organ (SFO) is a forebrain CVO with efferent neural projections to all of the important hypothalamic autonomic control centres (Miselis, 1982;Lind, 1986;Gruber et al., 1987) including those involved in energy homeostasis such as the ARC (Miselis, 1982;Lind, 1986;Gruber et al., 1987), PVN, and LH (Miselis, 1982;Lind et al., 1982). This anatomical connectivity provides a clear route through which information regarding energy status can be communicated from the SFO to hypothalamic autonomic control centers. Work from our laboratory and others have revealed the presence of a myriad of receptors in the SFO with our recent microarray studies, which allows a scan of the entire transcriptome, which not only confirmed the presence of previously reported receptors for a number of peripheral signals reflecting energy status 146 and/or influencing food intake including amylin (Hindmarch et al., 2008;Christopoulos et al., 1995;Ueda et al., 2001), ghelin (Hindmarch et al., 2008;Pulman et al., 2006), PYY (Hindmarch et al., 2008), adiponectin (Hindmarch et al., 2008;Alim et al., 2010), CCK (Hindmarch et al., 2008) and apelin (Hindmarch et al., 2008) but also identified novel receptor mRNA for others such as leptin and the cannabinoid receptor (Hindmarch et al., 2008). The functional relevance for the presence of these receptors has been demonstrated by whole cell current clamp electrophysiology showing that the excitability of SFO neurons is influenced by each of these peptides (Riediger et al., 1999a;Pulman et al., 2006;Alim et al., 2010, unpublished observations). Thus, given the presence of receptors for a variety of signals involved in body weight homeostasis and functional evidence of actions of these peptide at SFO neurons we asked the question ‘What effect would electrical stimulation in the SFO have on feeding?’ We had used this approach previously to evaluate the effect of electrical stimulation on drinking and have reported that acute, short term SFO stimulation elicited drinking in satiated rats (Smith et al., 1995). In this earlier experiment, electrical stimulation was used to mimic the excitatory effect of angiotensin II on SFO neurons which has been shown to be disogenic (Simpson & Routenberg, 1973;Simpson et al., 1978;Thrasher et al., 1982;Phillips et al., 1982;Badoer & McKinlay, 1997). We demonstrated that short duration (5 min) electrical stimulation of the SFO elicited robust feeding in satiated rats. Not only did the rats eat, but they ate 10% of their total daily food intake in less than 30 min. This is remarkable in that this food 147 consumption occurred during a time when the animals normally do not eat. Rats were tested at the beginning of the light cycle, a period during which rats are typically asleep and do not ingest either food or water. Small bouts of eating and drinking are known to occur during the light cycle, however, these bouts typically occur several hours after the beginning of the light cycle and in the last hour of the light cycle in anticipation of lights off. It is during the dark cycle that rats consume the majority of their daily food and water. These effects on feeding were specific to SFO stimulation as animals with stimulating electrode placement outside the anatomical boundaries of SFO did not eat in response to stimulation nor did animals with SFO electrode placement receiving sham stimulation (animals undergoing the same experimental protocol without current being passed through the electrode). The effect of electrical stimulation on feeding was also shown to be intensity dependent as stimulation at lower intensities (100µA) did not induce feeding, although this low intensity SFO stimulation appeared to increase the animals’ interest in food. This increased interest was not quantitatively measured but rather determined by the amount of time spent at the food hopper. Interestingly, in contrast to feeding responses, drinking occurred in animals at both stimulation intensities (100 and 200µA) as previously reported (Smith et al., 1995). In some animals receiving low intensity (100µA) SFO stimulation, drinking occurred during the 5 min electrical stimulation period, whereas no animals receiving higher intensity (200µA) SFO stimulation drank during the stimulation period. Furthermore, although all animals ate in 148 response to higher intensity stimulation, not all animals drank and, in most cases (5/6), eating preceded drinking in animals that exhibited both behaviours. The latency to drink in the high intensity stimulation group was significantly longer (latency to drink = 15.2 ± 2.6 min) than in those animals that received low intensity stimulation (latency to drink = 6.2 ± 2.6 min). These observations not only suggest that eating and drinking are independent behaviours, that is eating does not drive drinking or vice versa, but also attests to the complicated nature of these ingestive behaviours. The fact that stimulation intensities required to induce feeding were greater than those required to elicit drinking may be related to the fact that angiotensin elicits drinking by homogenous excitatory effects on approximately 60% of SFO neurons (Li & Ferguson, 1993;Gutman et al., 1988a;Schmid & Simon, 1992). This is in contrast to the effect of anorexigenic and orexigenic circulating satiety factors which have heterogenous effects on the excitability of SFO neurons. Studies from our own laboratory have demonstrated that separate subpopulations of SFO neurons are activated by the anorexigenic peptide, amylin, or the orexigenic peptide, ghrelin (Pulman et al., 2006). Adiponectin also has been shown to have both excitatory and inhibitory effects on the excitability of SFO neurons (Alim et al., 2010). In contrast to electrophysiological studies, electrical stimulation does not specifically target one particular neuronal subtype based on the receptors present, but rather activates all neurons (orexigeninc and anorexigenic) in the region of the tip of the stimulating electrode. Thus, a possible explanation for the intensity dependence of stimulation induced feeding is that during the 149 light cycle, when the animal is satiated and typically sleeping, SFO neurons inhibiting food intake are likely maximally activated and, in order to override this inhibitory drive to stimulate feeding, higher stimulation intensities are required to activate a sufficient proportion of SFO neurons that stimulate feeding. These stimulation studies support a role for the SFO in the regulation of energy homeostasis. The fact that such activation elicits feeding in satiated rats attests to the integrative action of the SFO, further supporting 1) the notion of the SFO as a regulatory target for peripheral molecules reflecting an individual’s energy status, and 2) a role for the SFO in influencing autonomic function through its neural projections to hypothalamic nuclei involved in energy homeostasis. Once we had determined that activation of SFO neurons elicited feeding in satiated rats and based on evidence that the leptin receptor was present in SFO (Hindmarch et al., 2008), we sought to determine whether the signaling form of the leptin receptor, ObRb, was present in SFO and, if so, to evaluate whether leptin, the prototypical adipokine, had effects at the SFO. Initially, we confirmed the presence of mRNA for the leptin receptor (LepR) reported in the microarray analysis (Hindmarch et al., 2008). The primer sets used to identify LepR are primer sets directed to an extracellular region of the leptin receptor which is shared by all leptin receptor subtypes. Since the weight reducing effects of leptin have been shown to be mediated by long form of the leptin receptor, ObRb(Bjorbaek et al., 1997;Bjorbaek & Kahn, 2004;Buettner et al., 2006;Bates et al., 2003;Cui et al., 150 2004), which processes the cytoplasmic domains required for signal transduction (Bjorbaek et al., 1997;Banks et al., 2000;Kloek et al., 2002) we used primer sets directed to unique intracellular domains of ObRb to specifically determine its presence in the SFO. Using these specific primer sets we demonstrated the presence of the ObRb in SFO using RT-PCR. These same primers also detected ObRb mRNA in acutely dissected hypothalamus, an area known to contain ObRb (see Jequier, 2002 for review). Using leptin receptor localization in SFO by immunohistochemistry and leptin-induced pSTAT3 expression, a well-accepted marker of ObRb receptor activation, in SFO, we demonstrated that leptin receptor mRNA was translated into functional protein in SFO neurons. Central and peripheral leptin administration has been shown to increase STAT3 phosphorylation in hypothalamic and brainstem nuclei involved in the regulation of feeding (Hosoi et al., 2002) and that pSTAT3 activation is necessary for the inhibitory effect of leptin on food intake and body weight (Piper et al., 2008). Having demonstrated the presence of ObRb mRNA in SFO neurons and its translation to functional protein, we then evaluated the functional consequence of leptin receptor activation in SFO neurons. Using whole cell current-clamp techniques in dissociated SFO neurons we have shown that leptin influences the excitability of the majority (64%) SFO neurons, causing both depolarizing and hyperpolarizing effects on different populations of SFO neurons, thus demonstrating functional roles for leptin actions at SFO neurons. The presence of subpopulations of neurons in the SFO may explain the finding of both depolarizing and hyperpolarizing responses to leptin 151 administration. Previous studies, from our laboratory and others, have demonstrated heterogeneous responses of SFO neurons (depolarizing and hyperpolarizing responses) to a variety of peptidergic substances (Cottrell et al., 2004;Alim et al., 2010). Although not addressed in the present study, the heterogeneity in excitability of SFO neurons in response to leptin administration may reflect different subpopulations of SFO neurons with specific, yet different hypothalamic projection sites, which together contribute to the coordinated effects of leptin on food intake. Previous studies from our laboratory have shown that SFO neurons with known projection to the PVN to demonstrate a unique electrophysiological profile dominated by a large transient potassium conductance and were shown to be osmo- and angiotensin II sensitive, suggesting specific functional roles for this anatomically defined group of SFO neurons (Anderson et al., 2001). Amylin, a pancreatic peptide that is co-secreted with insulin in response to food intake and increases in blood glucose (Cooper et al., 1989), reducing food intake by binding its receptors in the CNS (Morley & Flood, 1991), has been shown to influence 59% of SFO neurons, eliciting only excitatory effects on SFO neurons. Given that leptin and amylin both inhibit food intake in response to positive energy balance, albeit that they function over different time periods (leptin is a long-term adiposity signal, whereas amylin is a short-term satiety signal), it is plausible that a subpopulation of SFO neurons may mediate the anorexic effects of both these circulating factors, and thus exhibit excitatory responses to both peptides. Our finding that that leptin depolarizes amylin- 152 responsive SFO neurons supports a role for the SFO in integrating peripherally derived satiety signals. Having demonstrated that leptin influenced SFO neurons we then evaluated the effect(s) of leptin in the AP, a medullary sensory CVO. There is a growing body of evidence suggesting that leptin acts in the caudal brainstem, with particular attention to the NTS, to influence energy homeostasis (Grill, 2010;Myers, Jr. et al., 2009). However, the fact that the NTS is located behind the BBB means that circulating leptin does not have direct access to this region. The AP, which lies immediately adjacent to the NTS and has extensive projections to the NTS (Shapiro & Miselis, 1985b), provides a route by which leptin may access the medulla and influence NTS neurons. Evidence for leptin actions in the AP is provided by studies demonstrating leptin receptor mRNA (LepR) expression in the AP (Hindmarch et al., 2006) and immunohistochemical experiments showing expression of the LepR receptor (Grill et al., 2002;Buyse et al., 2001). Using RT-PCR, we have confirmed the presence of the leptin receptor (LepR) in the AP and have shown that the signaling form of the leptin receptor, ObRb, is present this medullary CVO, suggesting a role for the AP in mediating the central effects of leptin at the level of the causal brainstem. A functional role for ObRb in AP is supported by the fact that pSTAT3 activation is observed in the AP (Huo et al., 2007). Further support for a functional role of ObRb receptor activation in AP is shown by the results of our electrophysiological recordings demonstrating that the majority (51%) of dissociated AP neurons were influenced by leptin, with either depolarization or 153 hyperpolarization observed, responses which were also present in AP neurons recorded from a medullary slice preparation. The observation of both excitatory and inhibitory responses is consistent with previous studies suggesting the existence of neuronal subpopulations in the AP that respond differentially to specific peptides (Cai & Bishop, 1995;Fry et al., 2006;Yang & Ferguson, 2003;Ingves & Ferguson, 2010) and, although not evaluated in the present study, the differential responses (depolarization vs hyperpolaization) may reflect different projection sites of subpopulations of AP neurons. Electrophysiological studies have previously shown that amylin excites approximately half of AP neurons tested (Riediger et al., 2001;Riediger et al., 2002) and, in accordance with the hypothesis that the AP is involved in integrative actions of energy homeostasis-related circulating factors at the AP, we investigated whether the subpopulation of AP neurons that is activated by leptin would also be activated by amylin. Our observation that the majority of neurons that were depolarized by leptin were also depolarized by amylin while neurons that were hyperpolarized by leptin, or did not respond to leptin, were insensitive to amylin, is in accordance with such a hypothesis. This idea is also supported by the previous observation that about 90% of amylinsensitive AP neurons are also sensitive to glucose (Riediger et al., 2002). Our finding that amylin and leptin activate the same population of AP neurons may provide insight into mechanisms of synergistic leptin-amylin effects on weight loss if we can understand their combined actions on these neurons. 154 Our studies demonstrating that ObRb is present in two of the sensory CVOs, the SFO and AP, and that leptin influences the excitability of neurons in SFO and AP, suggest a role for these specialized CNS structures in mediating the central actions of leptin. As discussed earlier, the lack of the BBB in these regions uniquely positions the SFO and AP to detect circulating leptin and transmit this information to feeding centers in the hypothalamus and brainstem to influence food intake and energy expenditure. Leptin circulates at levels proportional to adiposity, with plasma leptin being about 20ng/ml in women and 6ng/ml in men (Swartz et al, 1996). Peripheral leptin administration has been previously shown to reduce body weight and food intake (Halaas et al., 1995b;Halaas et al., 1997). To directly assess the physiological role of the SFO and AP in mediating the decrease in body weight gain and food intake in response to peripherally administered leptin, we undertook a series of experiments in which animals were subjected to SFO and AP lesions and then we evaluated their responsiveness to ip leptin. Analysis of body weight gain, post surgery, in AP/SFO lesion animals reveal a profound decrease in body weight when compared to AP/SFO intact animals. These observations are supported by previous studies showing that AP/SFO lesions chronically decrease body weight in rats (Baraboi et al., 2010b;Baraboi et al., 2010a) over a 30 day period. Our observations that lesions of the AP and SFO cause a significant reduction in both body weight and food intake suggest that the AP and SFO play an important role in the regulation of body weight. Furthermore, our findings that AP/SFO lesions attenuate 155 the decrease in body weight gain normally observed following peripheral administration of leptin support an important role for the AP and SFO in mediating the central, weight reducing effect of leptin. The observation that the sensory CVOs are critical in mediating central weight-reducing effect of leptin, is supported by previous studies showing that AP/SFO lesions attenuate the initial component of the PYY-induced decrease in food intake (Baraboi et al., 2010a) and exendin-4 induced expression of c-fos in the CNS (Baraboi et al., 2010b). Early studies that centered on the absence of the ARC as the cause of profound obesity seen as a consequence of MSG-induced cytotoxic damage in the ARC had also shown the SFO to be damaged by these excitotoxic lesions (Takasaki, 1978;Olney & Sharpe, 1969;Olney et al., 1972;Arees & Mayer, 1970). The fact that site specific ARC lesions still result in increases in food intake leading to obesity (Choi & Dallman, 1999), suggest that SFO lesion is not critical for the development of this phenotype. However, our findings that AP/SFO lesions cause a significant reduction in body weight and attenuate leptin-induced body weight and food reduction, suggest that these structures are important in maintaining a normal body weight and in mediating the central effects of leptin, respectively. A particularly interesting finding of this study, the leptin induced increase in food intake in AP/SFO lesion animals, is in stark contrast to the decrease in food intake normally observed in response to acute peripheral leptin administration (Halaas et al., 1995b;Halaas et al., 1997). Equally interesting is our finding that water intake increased in response to peripheral leptin administration in AP/SFO lesioned rats. These leptin156 induced effects on food and water intake in AP/SFO lesioned animals certainly warrant further investigation. It will also be important to assess the relative contributions of each of the CVOs to determine whether AP or SFO lesion alone would be sufficient to attenuate leptin-induced effects on body weight gain and/or food and water intake observed in the present study. Obesity is associated with numerous co-morbidities including hypertension. Not surprisingly, in addition to its role in energy homeostasis, leptin has also been shown to be involved in BP regulation and has been suggested that leptin may play a primary role in obesity-related hypertension. The observations that a positive correlation exists between leptin levels and BP in obese individuals (Al-Hazimi & Syiamic, 2004;Itoh et al., 2002) and that chronic systemic administration causes increases in BP (Shek et al., 1998;da Silva et al., 2004) suggests a role for leptin in obesity-related hypertension. Furthermore, direct application of leptin into the ARC (Rahmouni & Morgan, 2007), VMH (Montanaro et al., 2005;Marsh et al., 2003), and DMH, (Marsh et al., 2003), hypothalamic structures involved in energy balance, has been shown to cause increases in BP, suggesting that leptin acts centrally to influence BP. Given the well documented role of the SFO in cardiovascular regulation and our data showing ObRb to be present in SFO and that leptin influences the excitability of SFO neurons(Smith et al., 2009), we investigated whether the SFO may be involved in leptin-induced cardiovascular effects of leptin by microinjecting leptin into the SFO of anesthetized rats. We showed that leptin administration into the SFO of young, normal 157 weight rats caused rapid, site specific decreases in BP without influencing HR. These findings were, initially, quite surprising to us. Given the well-established association between obesity, high circulating leptin levels, and hypertension and previous studies demonstrating increases in BP observed following icv leptin administration (Dunbar et al., 1997;Rahmouni & Morgan, 2007;Casto et al., 1998;da Silva et al., 2004;Lu et al., 1998) and as a consequence of direct microinjection of leptin directly into hypothalamic feeding centers (Montanaro et al., 2005;Marsh et al., 2003;Rahmouni & Morgan, 2007) all of which suggested that leptin microinjection into SFO would result in increased BP. The concept of selective leptin resistance which describes the phenomenon in which obese animals (both genetic and diet-induced models of obesity) are resistant to the weight reducing effects of both systemic and central leptin while remaining sensitive to leptin effects on renal sympathetic output (Correia et al., 2002;Rahmouni et al., 2005;Rahmouni et al., 2002), an action suggested contribute to hypertension (for review see Mark et al., 2002;Correia & Haynes, 2004), might explain our apparently contrary findings. Could leptin, under normal (non obese) conditions be playing a positive role in cardiovascular regulation whereby when the individual is leptin-responsive, leptin acts as a brake at the SFO to maintain BP within normal physiological limits while in obese, leptin-resistant, individuals, leptin no longer acts at the SFO to maintain BP and, without this braking effect, BP begins to rise? To test this hypothesis, we then asked the question ‘Would the decreases in BP observed in our young, normal weight animals remain in obese ‘leptin resistant’ animals?’ 158 Although genetic defects causing leptin deficiency and leptin receptor defects have been shown to cause profound obesity, the most common cause of obesity has been linked to overeating or the consumption of a high fat diet (Unger et al., 2010) which leads to a marked increase in plasma leptin levels (Friedman & Halaas, 1998;Wang et al., 2001). Diet induced obesity (DIO) develops as a consequence of consuming a high fat diet and, not dissimilar to what occurs in the human population, not all animals who display a preference for high fat diets develop obesity (Levin et al., 1989). Rodents fed a high fat diet develop a resistance to peripheral leptin followed by central leptin resistance if continued on the high fat diet (El-Haschimi et al., 2000;Lin et al., 2000;Wang et al., 2001;Boyle et al., 2011). Numerous studies have shown markedly elevated serum leptin levels in the DIO rat model (Lu et al., 1998;Zhang et al., 2010;Tumer et al., 2007;Dubinion et al., 2011;Elmarakby & Imig, 2010;Levin & Dunn-Meynell, 2002) despite profound obesity, thus demonstrating an insensitivity to the weight-reducing effects of leptin. As such, we chose the DIO rat model to investigate whether decreases in BP observed in response to leptin microinjection into the SFO would be maintained in DIO rats. Our findings that leptin-induced depressor responses are abolished in DIO rats, while maintained in age matched diet resistant (DR) rats supports the concept of selective leptin resistance in that a decreased sensitivity to leptin underlies the lack of depressor responses seen in the DIO animals. In fact, the lack of the depressor effect of leptin in the ‘leptin-resistant’ DIO model may serve to exacerbate the hypertension present in obese individuals. 159 These results suggest that leptin may play a positive role in cardiovascular regulation. In normal (non-obese) conditions when the individual is leptin-responsive, leptin acts at the SFO to maintain BP within normal physiological limits. In leptin resistant, obese individuals, leptin is no longer able to act at the SFO to maintain BP and, without this braking effect, BP begins to rise. Thus, this leptin insensitivity in the SFO would serve to further exacerbate the effect of central selective leptin resistance in the development of obesity related hypertension. Such a hypothesis fits nicely with the well-documented roles of SFO in cardiovascular regulation, body fluid homeostasis, and energy homeostasis, and the integrative functions of the SFO. We have shown that a population of SFO neurons responds to both leptin and amylin, observations which support integrative functions in this CNS structure. Within this context of a role for the SFO in the integration of multiple signals, ANG has been shown, electrophysiologically, to elicit solely excitatory responses on approximately 70% of SFO neurons both in vivo (Gutman et al., 1988a;Tanaka et al., 1987) and in vitro (Li & Ferguson, 1993;Okuya et al., 1987;Schmid & Simon, 1992). We have shown that that approximately 65% of SFO neurons are responsive to leptin, half of which are excited by leptin and half inhibited (Smith et al., 2009). Thus, some of the same neurons in the SFO must be responsive to both ANG and leptin. Given that ANG and leptin exert opposite effects on BP through actions at the SFO, perhaps a subpopulation of SFO neurons activated by ANG are also inhibited by leptin. Perhaps, under normal conditions, when individuals are capable of responding to 160 leptin, the net effect of the action of ANG (which alone would increase BP) and leptin (which alone would decrease BP) at the SFO is to maintain BP within normal physiological limits. In obesity, when an individual is leptin resistant and no longer able to respond appropriately to leptin, despite elevated leptin levels, these same neurons in the SFO, once responsive to ANG and leptin, now only respond only to ANG causing an elevation in BP (see Figure 7-1). Further support for possible integrative actions of leptin and ANG at the SFO is provided by preliminary data from our laboratory showing that single SFO neurons express mRNA for both the ObRb and AT1 receptor. Conclusion Based on the findings presented in this thesis, the current focus in much of the literature on the ARC as the primary (and sole) site of action for leptin in the CNS deserves to be revisited. Undeniably, the ARC is important in central leptin signaling and although more recent evidence points to integrated actions for leptin at multiple appetite centers in both hypothalamic (PVN, LH, VMH), forebrain, and brainstem (DMV, NTS) sites (Grill & Hayes, 2009;Qi et al., 2010;Hommel et al., 2006;Huo et al., 2007;Myers, Jr. et al., 2009;Dhillon et al., 2006;Hayes et al., 2010), all of these regions, in contrast to the SFO and AP, are protected by the BBB and thus do not have direct access to circulating leptin. The SFO and AP are sensory CVOs and, as such, are uniquely positioned to monitor the constituents of the blood and, through well-documented efferent projections to autonomic nuclei in the hypothalamus and brainstem, communicate this information to centers behind the BBB responsible for controlling energy homeostasis. 161 Figure 7-1: Proposed mechanism of leptin-angiotensin interaction at SFO neurons to regulate BP in normal weight and obese states ANG has excitatory effects on SFO neurons (lower left schematic) which leads to increases in BP. Leptin elicits both excitatory and inhibitory responses and leptininduced hyperpolarization (upper left schematic) may underlie the depressor responses observed in response to leptin microinjection into the SFO. We propose that neurons excited by ANG are the same SFO neurons inhibited by leptin (right hand column). In a normal weight, leptin-sensitive individual, the net effect of leptin (hyperpolarize) and ANG (depolarize) at the SFO is to maintain BP within normal physiological limits (upper right schematic). In the obese, leptin-resistant individuals, leptin can no longer act at SFO allowing the excitatory effect of ANG to predominate, leading to increased BP and contributing to obesity related hypertension (lower right schematic). 162 We have provided evidence for important roles for the CVOs in mediating energy homeostasis with a variety of experimental approaches. We have shown that acute electrical stimulation of the SFO elicits feeding in satiated rats, supporting a role for this specialized CNS structure in the control of food intake. In addition, our demonstration that ObRb is present in SFO and that leptin influences the excitability of individual SFO neurons suggests a functional role for leptin actions in this CVO. The fact that leptinactivates the same SFO neurons activated by amylin, supports a role for the SFO in integrating peripherally derived signals regulating food intake. Furthermore, our demonstration that exogenous leptin administration into the SFO results in significant decreases in BP in young and DR rats while leptin microinjection into leptin-resistant DIO rats is without effect, suggests the SFO to be an important site for mediating the central actions of leptin in BP regulation. These findings suggest that leptin-insensitivity in the SFO of obese, leptin-resistant, individuals may contribute to obesity-related hypertension. These studies also provide evidence for the SFO in the integration of diverse, but related, autonomic functions, a disturbance of which can lead to obesity and obesity-related hypertension. The SFO mediates leptin actions related to both energy homeostasis and cardiovascular control presumably via activation of different autonomic nuclei governing energy balance and cardiovascular function. Our studies also show that the medullary sensory CVO, the AP, expresses ObRb and the excitability of individual neurons in the AP is influenced by the adipocyte- 163 derived hormone, leptin. Furthermore, the fact that leptin and amylin act on the same subpopulation of neurons in the AP suggests integrative actions of this CVO. And finally, our preliminary studies demonstrating that animals with AP and SFO lesions exhibit a profound decrease in body weight and food intake and no longer demonstrate decreases in body weight in response to peripheral leptin administration suggest that the AP and SFO play an integral, physiological role in the regulation of body weight and in mediating the central weight reducing effects of leptin. In summary, our data suggest that the direct actions of leptin on the excitability of AP and SFO neurons contribute to the regulation of long-term energy balance, and, in the case of the SFO, cardiovascular regulation. The AP and SFO, are ideal targets for leptin due to the lack of the BBB, thus have the ability to monitor the contents of the circulation. This information can then be transmitted via well-established projections to autonomic nuclei in the brainstem and hypothalamus, providing a route by which the circulating adiposity signal, leptin, can act to regulate energy expenditure, feeding behavior, and cardiovascular regulation via actions in hypothalamic and brainstem autonomic nuclei. Furthermore, the effects of leptin and amylin on the same subpopulation of neurons in the AP and SFO, attests to the integrative functions of these CVOs in energy homeostasis. The SFO also appears to have the potential to integrate the actions of leptin in cardiovascular control and in energy balance. Thus, the sensory CVOs are important centers in the control of energy homeostasis. The fact that these specialized structures lack the normal BBB allows 164 peripherally derived signaling molecules to directly access the CNS at these regions without the need for special transporters. The action of leptin at these areas not only suggest that they are important in the regulation of leptin actions in the CNS but the responsiveness of single neurons in the AP and SFO to both leptin and amylin suggest that the SFO and AP may also be important in the integration of multiple signals derived from peripheral sources. Furthermore, the fact that the SFO appears to be involved in leptin effects on both energy balance and cardiovascular regulation attest to the integrative nature of the SFO in the control of diverse physiological parameters. 165 REFERENCES Abbott NJ, Ronnback L, & Hansson E (2006). Astrocyte-endothelial interactions at the blood-brain barrier. Nat Rev Neurosci 7, 41-53. Al-Hazimi AM & Syiamic AY (2004). Relationship between plasma angiotensinII, leptin and arterial blood pressure. Saudi Med J 25, 1193-1198. Alim I, Fry WM, & Ferguson AV (2010). Actions of adiponectin on the excitability of subfornical organ neurons are altered by food deprivation. Brain Res. Anderson JW, Smith PM, & Ferguson AV (2001). Subfornical organ neurons projecting to paraventricular nucleus: whole-cell properties. Brain Res 921, 78-85. Arees EA & Mayer J (1970). Monosodium glutamate-induced brain lesions: electron microscopic examination. Science 170, 549-550. Arora S & Anubhuti (2006). Role of neuropeptides in appetite regulation and obesity--a review. Neuropeptides 40, 375-401. Badman MK & Flier JS (2005). The gut and energy balance: visceral allies in the obesity wars. Science 307, 1909-1914. Badoer E & McKinlay D (1997). Effect of intravenous angiotensin II on Fos distribution and drinking behavior in rabbits. Am J Physiol 272, R1515-R1524. Balthasar N (2006). Genetic dissection of neuronal pathways controlling energy homeostasis. Obesity (Silver Spring) 14 Suppl 5, 222S-227S. Banks AS, Davis SM, Bates SH, & Myers MG, Jr. (2000). Activation of downstream signals by the long form of the leptin receptor. J Biol Chem 275, 14563-14572. Banks WA & Kastin AJ (1990). Peptide transport systems for opiates across the bloodbrain barrier. Am J Physiol 259, E1-E9. 166 Banks WA & Kastin AJ (1992). Bidirectional passage of peptides across the blood-brain barrier. Prog Brain Res 91, 139-148. Banks WA & Kastin AJ (1998). Differential permeability of the blood-brain barrier to two pancreatic peptides: insulin and amylin. Peptides 19, 883-889. Banks WA, Kastin AJ, Huang W, Jaspan JB, & Maness LM (1996). Leptin enters the brain by a saturable system independent of insulin. Peptides 17, 305-311. Baraboi ED, Michel C, Smith P, Thibaudeau K, Ferguson AV, & Richard D (2010a). Effects of albumin-conjugated PYY on food intake: the respective roles of the circumventricular organs and vagus nerve. Eur J Neurosci 32, 826-839. Baraboi ED, Smith P, Ferguson AV, & Richard D (2010b). Lesions of area postrema and subfornical organ alter exendin-4-induced brain activation without preventing the hypophagic effect of the GLP-1 receptor agonist. Am J Physiol Regul Integr Comp Physiol 298, R1098-R1110. Barth SW, Riediger T, Lutz TA, & Rechkemmer G (2004). Peripheral amylin activates circumventricular organs expressing calcitonin receptor a/b subtypes and receptoractivity modifying proteins in the rat. Brain Res 997, 97-102. Bates SH, Stearns WH, Dundon TA, Schubert M, Tso AW, Wang Y, Banks AS, Lavery HJ, Haq AK, Maratos-Flier E, Neel BG, Schwartz MW, & Myers MG, Jr. (2003). STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature 421, 856-859. Bergen HT, Mizuno TM, Taylor J, & Mobbs CV (1998). Hyperphagia and weight gain after gold-thioglucose: relation to hypothalamic neuropeptide Y and proopiomelanocortin. Endocrinology 139, 4483-4488. Bjorbaek C & Kahn BB (2004). Leptin signaling in the central nervous system and the periphery. Recent Prog Horm Res 59, 305-331. Bjorbaek C, Uotani S, da SB, & Flier JS (1997). Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem 272, 32686-32695. 167 Blouet C & Schwartz GJ (2010). Hypothalamic nutrient sensing in the control of energy homeostasis. Behav Brain Res 209, 1-12. Bojanowska E & Nowak A (2007). Interactions between leptin and exendin-4, a glucagon-like peptide-1 agonist, in the regulation of food intake in the rat. J Physiol Pharmacol 58, 349-360. Borison HL & Brizzee KR (1951). Morphology of emetic chemoreceptor trigger zone in cat medulla oblongata. Proc Soc Expt Biol Med 77, 38-42. Boustany CM, Bharadwaj K, Daugherty A, Brown DR, Randall DC, & Cassis LA (2004). Activation of the systemic and adipose renin-angiotensin system in rats with dietinduced obesity and hypertension. Am J Physiol Regul Integr Comp Physiol 287, R943R949. Boyle CN, Rossier MM, & Lutz TA (2011). Influence of high-fat feeding, diet-induced obesity, and hyperamylinemia on the sensitivity to acute amylin. Physiol Behav 104, 2028. Broadwell RD & Brightman MW (1976). Entry of peroxidase into neurons of the central and periferal nervous systems from extracerebral and cerebral blood. J Comp Neurol 166, 257-284. Buettner C, Pocai A, Muse ED, Etgen AM, Myers MG, Jr., & Rossetti L (2006). Critical role of STAT3 in leptin's metabolic actions. Cell Metab 4, 49-60. Burdyga G, Spiller D, Morris R, Lal S, Thompson DG, Saeed S, Dimaline R, Varro A, & Dockray GJ (2002). Expression of the leptin receptor in rat and human nodose ganglion neurones. Neuroscience 109, 339-347. Burguera B, Couce ME, Long J, Lamsam J, Laakso K, Jensen MD, Parisi JE, & Lloyd RV (2000). The long form of the leptin receptor (OB-Rb) is widely expressed in the human brain. Neuroendocrinology 71, 187-195. 168 Buyse M, Ovesjo ML, Goiot H, Guilmeau S, Peranzi G, Moizo L, Walker F, Lewin MJ, Meister B, & Bado A (2001). Expression and regulation of leptin receptor proteins in afferent and efferent neurons of the vagus nerve. Eur J Neurosci 14, 64-72. Cai YR & Bishop VS (1995). Effects of arginine vasopressin and angiotensin II on area postrema neurons in rabbit brain slice preparation. Neurosci Lett 190, 125-128. Campfield LA, Smith FJ, Guisez Y, Devos R, & Burn P (1995). Recombinant mouse OB protein: Evidence for a peripheral signal linking adiposity and central neural networks. Science 269, 546-549. Carpenter DO, Briggs DB, & Strominger N (1983). Responses of neurons of canine area postrema to neurotransmitters and peptides. Cell Mol Neurobiol 3, 113-126. Casto RM, VanNess JM, & Overton JM (1998). Effects of central leptin administration on blood pressure in normotensive rats. Neurosci Lett 246, 29-32. Chan CB & Johnson KJ (1997). Reduced sensitivity of fa/fa Zucker rats to adrenomedullin. Can J Physiol Pharmacol 75, 1138-1141. Cheunsuang O & Morris R (2005). Astrocytes in the arcuate nucleus and median eminence that take up a fluorescent dye from the circulation express leptin receptors and neuropeptide Y Y1 receptors. Glia 52, 228-233. Cheunsuang O, Stewart AL, & Morris R (2006). Differential uptake of molecules from the circulation and CSF reveals regional and cellular specialisation in CNS detection of homeostatic signals. Cell Tissue Res 325, 397-402. Choi S & Dallman MF (1999). Hypothalamic obesity: multiple routes mediated by loss of function in medial cell groups. Endocrinology 140, 4081-4088. Christopoulos G, Paxinos G, Huang XF, Beaumont K, Toga AW, & Sexton PM (1995). Comparative distribution of receptors for amylin and the related peptides calcitonin gene related peptide and calcitonin in rat and monkey brain. Can J Physiol Pharmacol 73, 1037-1041. 169 Chua SC, Jr., Koutras IK, Han L, Liu SM, Kay J, Young SJ, Chung WK, & Leibel RL (1997). Fine structure of the murine leptin receptor gene: splice site suppression is required to form two alternatively spliced transcripts. Genomics 45, 264-270. Ciofi P (2011). The arcuate nucleus as a circumventricular organ in the mouse. Neurosci Lett 487, 187-190. Clark JT, Kalra PS, Crowley WR, & Kalra SP (1984). Neuropeptide Y and human pancreatic polypeptide stimulate feeding behavior in rats. Endocrinology 115, 427-429. Clarke IJ, Cummins JT, & de Kretser DM (1983). Pituitary gland function after disconnection from direct hypothalamic influences in the sheep. Neuroendocrinology 36, 376-384. Cone RD, Cowley MA, Butler AA, Fan W, Marks DL, & Low MJ (2001). The arcuate nucleus as a conduit for diverse signals relevant to energy homeostasis. Int J Obes Relat Metab Disord 25 Suppl 5, S63-S67. Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, & . (1996). Serum immunoreactiveleptin concentrations in normal-weight and obese humans. N Engl J Med 334, 292-295. Cooper GJ, Day AJ, Willis AC, Roberts AN, Reid KB, & Leighton B (1989). Amylin and the amylin gene: structure, function and relationship to islet amyloid and to diabetes mellitus. Biochim Biophys Acta 1014, 247-258. Correia ML & Haynes WG (2004). Obesity-related hypertension: is there a role for selective leptin resistance? Curr Hypertens Rep 6, 230-235. Correia ML, Haynes WG, Rahmouni K, Morgan DA, Sivitz WI, & Mark AL (2002). The concept of selective leptin resistance: evidence from agouti yellow obese mice. Diabetes 51, 439-442. Cottrell GT & Ferguson AV (2004). Sensory circumventricular organs: central roles in integrated autonomic regulation. Regul Pept 117, 11-23. 170 Cottrell GT, Zhou QY, & Ferguson AV (2004). Prokineticin 2 modulates the excitability of subfornical organ neurons. J Neurosci. Cowley MA, Cone RD, Enriori P, Louiselle I, Williams SM, & Evans AE (2003). Electrophysiological actions of peripheral hormones on melanocortin neurons. Ann N Y Acad Sci 994, 175-186. Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Cone RD, & Low MJ (2001). Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature 411, 480-484. Cui Y, Huang L, Elefteriou F, Yang G, Shelton JM, Giles JE, Oz OK, Pourbahrami T, Lu CY, Richardson JA, Karsenty G, & Li C (2004). Essential role of STAT3 in body weight and glucose homeostasis. Mol Cell Biol 24, 258-269. da Silva AA, Kuo JJ, & Hall JE (2004). Role of hypothalamic melanocortin 3/4-receptors in mediating chronic cardiovascular, renal, and metabolic actions of leptin. Hypertension 43, 1312-1317. Debons AF, Krimsky I, Maayan ML, Fani K, & Jemenez FA (1977). Gold thioglucose obesity syndrome. Fed Proc 36, 143-147. Dellman HD & Simpson JB (1979). The subfornical organ. Int Rev Cytol 58, 333-421. Dhillon H, Zigman JM, Ye C, Lee CE, McGovern RA, Tang V, Kenny CD, Christiansen LM, White RD, Edelstein EA, Coppari R, Balthasar N, Cowley MA, Chua S Jr, Elmquist JK, & Lowell BB (2006). Leptin directly activates SF1 neurons in the VMH, and this action by leptin is required for normal body-weight homeostasis. Neuron 49, 191-203. Dubinion JH, da Silva AA, & Hall JE (2011). Chronic blood pressure and appetite responses to central leptin infusion in rats fed a high fat diet. J Hypertens 29, 758-762. Dunbar JC, Hu Y, & Lu H (1997). Intracerebroventricular leptin increases lumbar and renal sympathetic nerve activity and blood pressure in normal rats. Diabetes 46, 20402043. 171 Edwards CM, Abbott CR, Sunter D, Kim M, Dakin CL, Murphy KG, Abusnana S, Taheri S, Rossi M, & Bloom SR (2000). Cocaine- and amphetamine-regulated transcript, glucagon-like peptide-1 and corticotrophin releasing factor inhibit feeding via agoutirelated protein independent pathways in the rat. Brain Res 866, 128-134. El-Haschimi K, Pierroz DD, Hileman SM, Bjorbaek C, & Flier JS (2000). Two defects contribute to hypothalamic leptin resistance in mice with diet-induced obesity. J Clin Invest 105, 1827-1832. Ellacott KL & Cone RD (2006). The role of the central melanocortin system in the regulation of food intake and energy homeostasis: lessons from mouse models. Philos Trans R Soc Lond B Biol Sci 361, 1265-1274. Ellinwood EH, Jr. & Balster RL (1974). Rating the behavioral effects of amphetamine. Eur J Pharmacol 28, 35-41. Elmarakby AA & Imig JD (2010). Obesity is the major contributor to vascular dysfunction and inflammation in high-fat diet hypertensive rats. Clin Sci (Lond) 118, 291-301. Elmquist JK, Ahima RS, Elias CF, Flier JS, & Saper CB (1998a). Leptin activates distinct projections form the dorsomedial and ventromedial hypothalamic nuclei. Proc Natl Acad Sci USA 95, 741-746. Elmquist JK, Ahima RS, Maratos-Flier E, Flier JS, & Saper CB (1997). Leptin activates neurons in ventrobasal hypothalamus and brainstem. Endocrinology 138, 839-842. Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, & Saper CB (1998b). Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol 395, 535-547. Elmquist JK, Coppari R, Balthasar N, Ichinose M, & Lowell BB (2005). Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J Comp Neurol 493, 63-71. Ferguson AV & Renaud LP (1984). Hypothalamic paraventricular nucleus lesions decrease pressor responses to subfornical organ stimulation. Brain Res 305, 361-364. 172 Friedman JM & Halaas JL (1998). Leptin and the regulation of body weight in mammals. Nature 395, 763-770. Fry M & Ferguson AV (2009). Ghrelin modulates electrical activity of area postrema neurons. Am J Physiol Regul Integr Comp Physiol 296, R485-R492. Fry M, Hoyda TD, & Ferguson AV (2007). Making Sense of It: Roles of the Sensory Circumventricular Organs in Feeding and Regulation of Energy Homeostasis. Experimental Biology and Medicine 232, 14-26. Fry M, Smith PM, Hoyda TD, Duncan M, Ahima RS, Sharkey KA, & Ferguson AV (2006). Area Postrema Neurons Are Modulated by the Adipocyte Hormone Adiponectin. J Neurosci 26, 9695-9702. Fulton S, Pissios P, Manchon RP, Stiles L, Frank L, Pothos EN, Maratos-Flier E, & Flier JS (2006). Leptin regulation of the mesoaccumbens dopamine pathway. Neuron 51, 811822. Ghamari-Langroudi M (2012). Electrophysiological analysis of circuits controlling energy homeostasis. Mol Neurobiol 45, 258-278. Golden PL, Maccagnan TJ, & Pardridge WM (1997). Human blood-brain barrier leptin receptor. Binding and endocytosis in isolated human brain microvessels. J Clin Invest 99, 14-18. Grill HJ (2010). Leptin and the systems neuroscience of meal size control. Front Neuroendocrinol 31, 61-78. Grill HJ & Hayes MR (2009). The nucleus tractus solitarius: a portal for visceral afferent signal processing, energy status assessment and integration of their combined effects on food intake. Int J Obes (Lond) 33 Suppl 1, S11-S15. Grill HJ & Kaplan JM (2002). The neuroanatomical axis for control of energy balance. Front Neuroendocrinol 23, 2-40. 173 Grill HJ, Schwartz MW, Kaplan JM, Foxhall JS, Breininger J, & Baskin DG (2002). Evidence that the caudal brainstem is a target for the inhibitory effect of leptin on food intake. Endocrinology 143, 239-246. Gross PM (1991). Morphology and physiology of capillary systems in subregions of the subfornical organ and area postrema [Review]. Can J Physiol Pharmacol 69, 1010-1025. Gross PM (1992). Circumventricular organ capillaries. Prog Brain Res 91, 219-233. Gross PM, Wainman DS, Shaver SW, Wall KM, & Ferguson AV (1990). Metabolic activation of efferent pathways from the rat area postrema. Am J Physiol 258, R788R797. Gross PM & Weindl A (1987). Peering through the windows of the brain. J Cereb Blood Flow Metab 7, 663-672. Gruber K, McRae-Degueurce A, Wilkin LD, Mitchell LD, & Johnson AK (1987). Forebrain and brainstem afferents to the arcuate nucleus in the rat: potential pathways for the modulation of hypophyseal secretions. Neurosci Lett 75, 1-5. Gutman MB, Ciriello J, & Mogenson GJ (1988a). Effects of plasma angiotensin II and hypernatremia on subfornical organ neurons. Am J Physiol 254, R746-R754. Gutman MB, Jones DL, & Ciriello J (1988b). Effect of paraventricular nucleus lesions on drinking and pressor responses to ANG II. Am J Physiol 255, R882-R887. Halaas JL, Boozer C, Blair-West J, Fidahusein N, Denton DA, & Friedman JM (1997). Physiological response to long-term peripheral and central leptin infusion in lean and obese mice. Proc Natl Acad Sci U S A 94, 8878-8883. Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, & Friedman JM (1995a). Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269, 543-546. 174 Halaas JL, Gajiwala KS, Maffei M, Cohen SL, Chait BT, Rabinowitz D, Lallone RL, Burley SK, & Friedman JM (1995b). Weight-reducing effects of the plasma protein encoded by the obese gene. Science 269, 543-546. Harris RB, Kelso EW, Flatt WP, Bartness TJ, & Grill HJ (2006). Energy expenditure and body composition of chronically maintained decerebrate rats in the fed and fasted condition. Endocrinology 147, 1365-1376. Harris RB & Martin RJ (1984). Specific depletion of body fat in parabiotic partners of tube-fed obese rats. Am J Physiol 247, R380-R386. Hayes MR, Skibicka KP, Leichner TM, Guarnieri DJ, DiLeone RJ, Bence KK, & Grill HJ (2010). Endogenous leptin signaling in the caudal nucleus tractus solitarius and area postrema is required for energy balance regulation. Cell Metab 11, 77-83. Haynes WG, Sivitz WI, Morgan DA, Walsh SA, & Mark AL (1997). Sympathetic and cardiorenal actions of leptin. Hypertension 30, 619-623. Hervey GR (1959). The effects of lesions in the hypothalamus in parabiotic rats. J Physiol 145, 336-352. Hetherington AW & Ranson SW (1940). Hypothalamic lesions and adiposity in the rat. Anat Rec 78, 149-172. Hindmarch C, Fry M, Yao S, Smith PM, Murphy D, & Ferguson AV (2008). Microarray Analysis of the Transcriptome of the Subfornical Organ in the Rat: Regulation by Fluid and Food Deprivation. Am J Physiol Regul Integr Comp Physiol. Hindmarch C, Yao S, Beighton G, Paton J, & Murphy D (2006). A comprehensive description of the transcriptome of the hypothalamoneurohypophyseal system in euhydrated and dehydrated rats. Proc Natl Acad Sci U S A 103, 1609-1614. Hommel JD, Trinko R, Sears RM, Georgescu D, Liu ZW, Gao XB, Thurmon JJ, Marinelli M, & DiLeone RJ (2006). Leptin receptor signaling in midbrain dopamine neurons regulates feeding. Neuron 51, 801-810. 175 Hosoi T, Kawagishi T, Okuma Y, Tanaka J, & Nomura Y (2002). Brain stem is a direct target for leptin's action in the central nervous system. Endocrinology 143, 3498-3504. Hosoi T, Okuma Y, & Nomura Y (2000). Expression of leptin receptors and induction of IL-1beta transcript in glial cells. Biochem Biophys Res Commun 273, 312-315. Hoyda TD, Smith PM, & Ferguson AV (2009). Gastrointestinal hormone actions in the central regulation of energy metabolism: potential sensory roles for the circumventricular organs. Int J Obes (Lond) 33 Suppl 1, S16-S21. Hu L, Fernstrom JD, & Goldsmith PC (1998). Exogenous glutamate enhances glutamate receptor subunit expression during selective neuronal injury in the ventral arcuate nucleus of postnatal mice. Neuroendocrinology 68, 77-88. Huang XF, Koutcherov I, Lin S, Wang HQ, & Storlien L (1996). Localization of leptin receptor mRNA expression in mouse brain. Neuroreport 7, 2635-2638. Hubschle T, Thom E, Watson A, Roth J, Klaus S, & Meyerhof W (2001). Leptin-induced nuclear translocation of STAT3 immunoreactivity in hypothalamic nuclei involved in body weight regulation. J Neurosci 21, 2413-2424. Huo L, Maeng L, Bjorbaek C, & Grill HJ (2007). Leptin and the control of food intake: neurons in the nucleus of the solitary tract are activated by both gastric distension and leptin. Endocrinology 148, 2189-2197. Hyland NP, Pittman QJ, & Sharkey KA (2007). Peptide YY containing enteroendocrine cells and peripheral tissue sensitivity to PYY and PYY(3-36) are maintained in dietinduced obese and diet-resistant rats. Peptides 28, 1185-1190. Ingalls AM, Dickie MM, & Snell GD (1950). Obese, a new mutation in the house mouse. J Hered 41, 317-318. Ingves MV & Ferguson AV (2010). Prokineticin 2 modulates the excitability of area postrema neurons in vitro in the rat. Am J Physiol Regul Integr Comp Physiol 298, R617R626. 176 Inoue W, Poole S, Bristow AF, & Luheshi GN (2006). Leptin induces cyclooxygenase-2 via an interaction with interleukin-1beta in the rat brain. Eur J Neurosci 24, 2233-2245. Iovino M, Papa M, Monteleone P, & Steardo L (1988). Neuroanatomical and biochemical evidence for the involvement of the area postrema in the regulation of vasopressin release in rats. Brain Res 447, 178-182. Ishibashi S & Nicolaidis S (1981). Hypertension induced by electrical stimulation of the subfornical organ (SFO). Brain Res Bull 6, 135-139. Itoh K, Imai K, Masuda T, Abe S, Tanaka M, Koga R, Itoh H, Matsuyama T, & Nakamura M (2002). Relationship between changes in serum leptin levels and blood pressure after weight loss. Hypertens Res 25, 881-886. Janigro D, West GA, Nguyen TS, & Winn HR (1994). Regulation of blood-brain barrier endothelial cells by nitric oxide. Circ Res 75, 528-538. Jequier E (2002). Leptin signaling, adiposity, and energy balance. Ann N Y Acad Sci 967, 379-388. Jobst EE, Enriori PJ, & Cowley MA (2004). The electrophysiology of feeding circuits. Trends Endocrinol Metab 15, 488-499. Kalia M & Sullivan JM (1982). Brainstem projections of sensory and motor components of the vagus nerve in the rat. J Comp Neurol 211, 248-264. Kastin AJ, Pan W, Maness LM, & Banks WA (1999). Peptides crossing the blood-brain barrier: some unusual observations. Brain Res 848, 96-100. Kennedy GC (1953). The role of depot fat in the hypothalamic control of food intake in the rat. Proc R Soc Lond B Biol Sci 140, 578-596. Kerkerian L & Pelletier G (1986). Effects of monosodium L-glutamate administration on neuropeptide Y-containing neurons in the rat hypothalamus. Brain Res 369, 388-390. 177 Kishi T, Aschkenasi CJ, Choi BJ, Lopez ME, Lee CE, Liu H, Hollenberg AN, Friedman JM, & Elmquist JK (2005). Neuropeptide Y Y1 receptor mRNA in rodent brain: distribution and colocalization with melanocortin-4 receptor. J Comp Neurol 482, 217243. Kloek C, Haq AK, Dunn SL, Lavery HJ, Banks AS, & Myers MG, Jr. (2002). Regulation of Jak kinases by intracellular leptin receptor sequences. J Biol Chem 277, 41547-41555. Knigge KM, Joseph SA, Sladek JR, Notter MF, Morris M, Sundberg DK, Holzwarth MA, Hoffman GE, & O'Brien L (1976). Uptake and transport activity of the median eminence of the hypothalamus. Int Rev Cytol 45, 283-408. Krisch B & Leonhardt H (1978). The functional and structural border of the neurohemal region of the median eminence. Cell Tissue Res 192, 327-339. Krisch B, Leonhardt H, & Buchheim W (1978). The functional and structural border between the CSF- and blood-milieu in the circumventricular organs (organum vasculosum laminae terminalis, subfornical organ, area postrema) of the rat. Cell Tissue Res 195, 485-497. Landt M, Gingerich RL, Havel PJ, Mueller WM, Schoner B, Hale JE, & Heiman ML (1998). Radioimmunoassay of rat leptin: sexual dimorphism reversed from humans. Clin Chem 44, 565-570. Landt M, Horowitz JF, Coppack SW, & Klein S (2001). Effect of short-term fasting on free and bound leptin concentrations in lean and obese women. J Clin Endocrinol Metab 86, 3768-3771. Leshan RL, Bjornholm M, Munzberg H, & Myers MG, Jr. (2006). Leptin receptor signaling and action in the central nervous system. Obesity (Silver Spring) 14 Suppl 5, 208S-212S. Levin BE & Dunn-Meynell AA (2002). Reduced central leptin sensitivity in rats with diet-induced obesity. Am J Physiol Regul Integr Comp Physiol 283, R941-R948. 178 Levin BE, Dunn-Meynell AA, & Banks WA (2004). Obesity-prone rats have normal blood-brain barrier transport but defective central leptin signaling before obesity onset. Am J Physiol Regul Integr Comp Physiol 286, R143-R150. Levin BE, Hogan S, & Sullivan AC (1989). Initiation and perpetuation of obesity and obesity resistance in rats. Am J Physiol 256, R766-R771. Levine AS & Morley JE (1984). Neuropeptide Y: a potent inducer of consummatory behavior in rats. Peptides 5, 1025-1029. Li G, Zhang Y, Rodrigues E, Zheng D, Matheny M, Cheng KY, & Scarpace PJ (2007). Melanocortin activation of nucleus of the solitary tract avoids anorectic tachyphylaxis and induces prolonged weight loss. Am J Physiol Endocrinol Metab 293, E252-E258. Li J, Ma W, & Wang S (2011). Slower gastric emptying in high-fat diet induced obese rats is associated with attenuated plasma ghrelin and elevated plasma leptin and cholecystokinin concentrations. Regul Pept 171, 53-57. Li Z & Ferguson AV (1993). Angiotensin II responsiveness of rat paraventricular and subfornical organ neurons in vitro. Neuroscience 55, 197-207. Lin S, Thomas TC, Storlien LH, & Huang XF (2000). Development of high fat dietinduced obesity and leptin resistance in C57Bl/6J mice. Int J Obes Relat Metab Disord 24, 639-646. Lind RW (1986). Bi-directional, chemically specified neural connections between the subfornical organ and the midbrain raphe system. Brain Res 384, 250-261. Lind RW, Ohman LE, Lansing MB, & Johnson AK (1983). Transection of subfornical organ neural connections diminishes the pressor response to intravenously infused angiotensin II. Brain Res 275, 361-364. Lind RW, Van Hoesen GW, & Johnson AK (1982). An HRP study of the connections of the subfornical organ of the rat. J Comp Neurol 210, 265-277. 179 Liu C, Liu XJ, Barry G, Ling N, Maki RA, & De Souza EB (1997). Expression and characterization of a putative high affinity human soluble leptin receptor. Endocrinology 138, 3548-3554. Lu H, Duanmu Z, Houck C, Jen KL, Buison A, & Dunbar JC (1998). Obesity due to high fat diet decreases the sympathetic nervous and cardiovascular responses to intracerebroventricular leptin in rats. Brain Res Bull 47, 331-335. Lutz TA, Mollet A, Rushing PA, Riediger T, & Scharrer E (2001). The anorectic effect of a chronic peripheral infusion of amylin is abolished in area postrema/nucleus of the solitary tract (AP/NTS) lesioned rats. Int J Obes Relat Metab Disord 25, 1005-1011. Lutz TA, Senn M, Althaus J, DelPrete E, Ehrensperger F, & Scharrer E (1998). Lesion of the area postrema nucleus of the solitary tract (AP/NTS) attenuates the anorectic effects of amylin and calcitonin gene-related peptide (CGRP) in rats. Peptides 19, 309-317. Mangiapane ML & Brody MJ (1983). Electrical stimulation of subfornical organ (SFO) increases arterial pressure and regional vascular resistance. Fed Proc 42, 584. Mangiapane ML & Simpson JB (1980a). Subfornical organ lesions reduce the pressor effect of systemic angiotensin II. Neuroendocrinology 31, 380-384. Mangiapane ML & Simpson JB (1980b). Subfornical organ: forebrain site of pressor and dipsogenic action of angiotensin II. Am J Physiol 239, R382-R389. Mangiapane ML & Simpson JB (1983). Drinking and pressor responses after acetylcholine injection into subfornical organ. Am J Physiol 244, R508-R513. Mark AL, Correia ML, Rahmouni K, & Haynes WG (2002). Selective leptin resistance: a new concept in leptin physiology with cardiovascular implications. J Hypertens 20, 12451250. Mark AL, Shaffer RA, Correia ML, Morgan DA, Sigmund CD, & Haynes WG (1999). Contrasting blood pressure effects of obesity in leptin-deficient ob/ob mice and agouti yellow obese mice. J Hypertens 17, 1949-1953. 180 Marsh AJ, Fontes MA, Killinger S, Pawlak DB, Polson JW, & Dampney RA (2003). Cardiovascular responses evoked by leptin acting on neurons in the ventromedial and dorsomedial hypothalamus. Hypertension 42, 488-493. McKinley MJ, McAllen RM, Davern P, Giles ME, Penschow J, Sunn N, Uschakov A, & Oldfield BJ (2003). The sensory circumventricular organs of the mammalian brain. Adv Anat Embryol Cell Biol 172, III-122, back. Medeiros N, Dai L, & Ferguson AV (2012). Glucose-responsive neurons in the subfornical organ of the rat--a novel site for direct CNS monitoring of circulating glucose. Neuroscience 201, 157-165. Meister B & Hakansson ML (2001). Leptin receptors in hypothalamus and circumventricular organs. Clin Exp Pharmacol Physiol 28, 610-617. Mercer JG, Hoggard N, Williams LM, Lawrence CB, Hannah LT, & Trayhurn P (1996). Localization of leptin receptor mRNA and the long form splice variant (Ob-Rb) in mouse hypothalamus and adjacent brain regions by in situ hybridization. FEBS Lett 387, 113116. Mercer JG, Moar KM, Findlay PA, Hoggard N, & Adam CL (1998). Association of leptin receptor (OB-Rb), NPY and GLP-1 gene expression in the ovine and murine brainstem. Regul Pept 75-76, 271-278. Miller AD & Leslie RA (1994). The area postrema and vomiting. Front Neuroendocrinol 15, 301-320. Millington GW (2007). The role of proopiomelanocortin (POMC) neurones in feeding behaviour. Nutr Metab (Lond) 4, 18. Miselis RR (1981). The efferent projections of the subfornical organ of the rat: a circumventricular organ within a neural network subserving water balance. Brain Res 230, 1-23. Miselis RR (1982). The subfornical organ's neural connections and their role in water balance. Peptides 3, 501-502. 181 Miselis RR, Shapiro RE, & Hand PJ (1979). Subfornical organ efferents to neural systems for control of body water. Science 205, 1022-1025. Montanaro MS, Allen AM, & Oldfield BJ (2005). Structural and functional evidence supporting a role for leptin in central neural pathways influencing blood pressure in rats. Exp Physiol 90, 689-696. Morley JE & Flood JF (1991). Amylin decreases food intake in mice. Peptides 12, 865869. Myers MG, Jr., Munzberg H, Leinninger GM, & Leshan RL (2009). The geometry of leptin action in the brain: more complicated than a simple ARC. Cell Metab 9, 117-123. Nishizawa Y & Bray GA (1980). Evidence for a circulating ergostatic factor: studies on parabiotic rats. Am J Physiol 239, R344-R351. Norgren R (1978). Projections from the nucleus of the solitary tract in the rat. Neuroscience 3, 207-218. Nowak A & Bojanowska E (2008). Effects of peripheral or central GLP-1 receptor blockade on leptin-induced suppression of appetite. J Physiol Pharmacol 59, 501-510. Okuya S, Inenaga K, Kaneko T, & Yamashita H (1987). Angiotensin II sensitive neurons in the supraoptic nucleus, subfornical organ and anteroventral third ventricle of rats in vitro. Brain Res 402, 58-67. Olney JW (1969). Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164, 719-721. Olney JW (1971). Glutamate-induced neuronal necrosis in the infant mouse hypothalamus. An electron microscopic study. J Neuropathol Exp Neurol 30, 75-90. Olney JW & Sharpe LG (1969). Brain lesions in an infant rhesus monkey treated with monsodium glutamate. Science 166, 386-388. 182 Olney JW, Sharpe LG, & Feigin RD (1972). Glutamate-induced brain damage in infant primates. J Neuropathol Exp Neurol 31, 464-488. Pardridge WM, Boado RJ, & Farrell CR (1990). Brain-type glucose transporter (GLUT1) is selectively localized to the blood-brain barrier. Studies with quantitative western blotting and in situ hybridization. J Biol Chem 265, 18035-18040. Paton JF, Waki H, Abdala AP, Dickinson J, & Kasparov S (2007). Vascular-brain signaling in hypertension: role of angiotensin II and nitric oxide. Curr Hypertens Rep 9, 242-247. Paxinos G, Chai SY, Christopoulos G, Huang XF, Toga AW, Wang HQ, & Sexton PM (2004). In vitro autoradiographic localization of calcitonin and amylin binding sites in monkey brain. J Chem Neuroanat 27, 217-236. Paxinos G & Watson C (1982). The rat brain in stereotaxic coordinates Academic Press, New York. Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, Boone T, & Collins F (1995). Effects of the obese gene product on body weight regulation in ob\ob mice. Science 269, 540-543. Perez VJ, Olney JW, Martin JF, & Cannon WO (1979). Minimal tissue concentrations of glutamate required to produce necrosis of hypothalamic neurons in newborn mice. Biol Neonate 35, 17-22. Peruzzo B, Pastor FE, Blazquez JL, Schobitz K, Pelaez B, Amat P, & Rodriguez EM (2000). A second look at the barriers of the medial basal hypothalamus. Exp Brain Res 132, 10-26. Petrov T, Howarth AG, Krukoff TL, & Stevenson BR (1994). Distribution of the tight junction-associated protein ZO-1 in circumventricular organs of the CNS. Molecular Brain Research 21, 235-246. Phillips MI, Hoffman WE, & Bealer SL (1982). Dehydration and fluid balance: central effects of angiotensin. Fed Proc 41, 2520-2527. 183 Piper ML, Unger EK, Myers MG, Jr., & Xu AW (2008). Specific physiological roles for signal transducer and activator of transcription 3 in leptin receptor-expressing neurons. Mol Endocrinol 22, 751-759. Price CJ, Hoyda TD, & Ferguson AV (2008). The area postrema: a brain monitor and integrator of systemic autonomic state. Neuroscientist 14, 182-194. Pulman KJ, Fry WM, Cottrell GT, & Ferguson AV (2006). The subfornical organ: a central target for circulating feeding signals. J Neurosci 26, 2022-2030. Qi Y, Henry BA, Oldfield BJ, & Clarke IJ (2010). The action of leptin on appetiteregulating cells in the ovine hypothalamus: demonstration of direct action in the absence of the arcuate nucleus. Endocrinology 151, 2106-2116. Rahmouni K, Haynes WG, Morgan DA, & Mark AL (2002). Selective resistance to central neural administration of leptin in agouti obese mice. Hypertension 39, 486-490. Rahmouni K & Morgan DA (2007). Hypothalamic arcuate nucleus mediates the sympathetic and arterial pressure responses to leptin. Hypertension 49, 647-652. Rahmouni K, Morgan DA, Morgan GM, Mark AL, & Haynes WG (2005). Role of selective leptin resistance in diet-induced obesity hypertension. Diabetes 54, 2012-2018. Rauch M, Riediger T, Schmid HA, & Simon E (2000). Orexin A activates leptinresponsive neurons in the arcuate nucleus. Pflugers Arch 440, 699-703. Rethelyi M (1984). Diffusional barrier around the hypothalamic arcuate nucleus in the rat. Brain Res 307, 355-358. Riediger T, Rauch M, & Schmid HA (1999a). Actions of amylin on subfornical organ neurons and on drinking behavior in rats. Am J Physiol 276, R514-R521. Riediger T, Schmid HA, Lutz T, & Simon E (2001). Amylin potently activates AP neurons possibly via formation of the excitatory second messenger cGMP. Am J Physiol Regul Integr Comp Physiol 281, R1833-R1843. 184 Riediger T, Schmid HA, Lutz TA, & Simon E (2002). Amylin and glucose co-activate area postrema neurons of the rat. Neurosci Lett 328, 121-124. Riediger T, Schmid HA, Young AA, & Simon E (1999b). Pharmacological characterisation of amylin-related peptides activating subfornical organ neurones. Brain Res 837, 161-168. Rodriguez EM (1976). The cerebrospinal fluid as a pathway in neuroendocrine integration. J Endocrinol 71, 407-443. Rodriguez EM, Blazquez JL, & Guerra M (2010). The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: the former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 31, 757-776. Rodriguez EM, Blazquez JL, Pastor FE, Pelaez B, Pena P, Peruzzo B, & Amat P (2005). Hypothalamic tanycytes: a key component of brain-endocrine interaction. Int Rev Cytol 247, 89-164. Rodriguez EM, Gonzalez CB, & Delannoy L (1979). Cellular organization of the lateral and postinfundibular regions of the median eminence in the rat. Cell Tissue Res 201, 377408. Rogers RC & Hermann GE (1983). Central connections of the hepatic branch of the vagus nerve: a horseradish peroxidase histochemical study. J Auton Nerv Syst 7, 165-174. Rosas-Arellano MP, Solano-Flores LP, & Ciriello J (1996). Arcuate nucleus inputs onto subfornical organ neurons that respond to plasma hypernatremia and angiotensin II. Brain Res 707, 308-313. Rossi M, Kim MS, Morgan DG, Small CJ, Edwards CM, Sunter D, Abusnana S, Goldstone AP, Russell SH, Stanley SA, Smith DM, Yagaloff K, Ghatei MA, & Bloom SR (1998). A C-terminal fragment of Agouti-related protein increases feeding and antagonizes the effect of alpha-melanocyte stimulating hormone in vivo. Endocrinology 139, 4428-4431. 185 Roth JD, Maier H, Chen S, & Roland BL (2009). Implications of amylin receptor agonism: integrated neurohormonal mechanisms and therapeutic applications. Arch Neurol 66, 306-310. Roth JD, Roland BL, Cole RL, Trevaskis JL, Weyer C, Koda JE, Anderson CM, Parkes DG, & Baron AD (2008). Leptin responsiveness restored by amylin agonism in dietinduced obesity: Evidence from nonclinical and clinical studies. PNAS 0706473105. Santini F, Maffei M, Pelosini C, Salvetti G, Scartabelli G, & Pinchera A (2009). Melanocortin-4 receptor mutations in obesity. Adv Clin Chem 48, 95-109. Saper CB, Chou TC, & Elmquist JK (2002). The need to feed: homeostatic and hedonic control of eating. Neuron 36, 199-211. Satoh N, Ogawa Y, Katsuura G, Hayase M, Tsuji T, Imagawa K, Yoshimasa Y, Nishi S, Hosoda K, & Nakao K (1997). The arcuate nucleus as a primary site of satiety effect of leptin in rats. Neurosci Lett 224, 149-152. Schmid HA & Simon E (1992). Effect of angiotensin II and atrial natriuretic factor on neurons in the subfornical organ of ducks and rats in vitro. Brain Res 588, 324-328. Schwartz MW, Seeley RJ, Campfield LA, Burn P, & Baskin DG (1996). Identification of targets of leptin action in rat hypothalamus. J Clin Invest 98, 1101-1106. Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D Jr. (1996). Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med. 2, 589593. Sexton PM, Paxinos G, Kenney MA, Wookey PJ, & Beaumont K (1994). In vitro autoradiographic localization of amylin binding sites in rat brain. Neuroscience 62, 553567. Shapiro RE & Miselis RR (1985a). The central neural connections of the area postrema of the rat. J Comp Neurol 234, 344-364. 186 Shapiro RE & Miselis RR (1985b). The central neural connections of the area postrema of the rat. J Comp Neurol 234, 344-364. Shaver SW, Pang JJ, Wainman DS, Wall KM, & Gross PM (1992). Morphology and function of capillary networks in subregions of the rat tuber cinereum. Cell Tissue Res 267, 437-448. Shek EW, Brands MW, & Hall JE (1998). Chronic leptin infusion increases arterial pressure. Hypertension 31, 409-414. Shioda S, Funahashi H, Nakajo S, Yada T, Maruta O, & Nakai Y (1998). Immunohistochemical localization of leptin receptor in the rat brain. Neurosci Lett 243, 41-44. Shiraishi T, Sasaki K, Niijima A, & Oomura Y (1999). Leptin effects on feeding-related hypothalamic and peripheral neuronal activities in normal and obese rats. Nutrition 15, 576-579. Simpson JB, Epstein AN, & Camardo JS, Jr. (1978). Localization of receptors for the dipsogenic action of angiotensin II in the subfornical organ of rat. J Comp Physiol Psychol 92, 581-601. Simpson JB & Routenberg A (1973). Subfornical organ: site of drinking elicitation by angiotensin II. Science 181, 1172-1174. Simpson JB & Routtenberg JB (1975). Subfornical organ lesions reduce intravenous angiotensin-induced drinking. Brain Res 88, 154-161. Simpson KA, Martin NM, & Bloom SR (2009). Hypothalamic regulation of food intake and clinical therapeutic applications. Arq Bras Endocrinol Metabol 53, 120-128. Skibicka KP & Grill HJ (2009). Hindbrain leptin stimulation induces anorexia and hyperthermia mediated by hindbrain melanocortin receptors. Endocrinology 150, 17051711. 187 Smith PM, Bains JS, & Ferguson AV (1997). Long duration pressor responses following activation of subfornical organ neurons are the result of increased circulating vasopressin. Neurosci Lett 233, 81-84. Smith PM, Beninger RJ, & Ferguson AV (1995). Subfornical organ stimulation elicits drinking. Brain Res Bull 38, 209-213. Smith PM, Chambers AP, Price CJ, Ho W, Hopf C, Sharkey KA, & Ferguson AV (2009). The subfornical organ: a central nervous system site for actions of circulating leptin. Am J Physiol Regul Integr Comp Physiol 296, R512-R520. Smith PM, Rozanski G, & Ferguson AV (2010). Acute electrical stimulation of the subfornical organ induces feeding in satiated rats. Physiol Behav 99, 534-537. Stellar E (1954). The physiology of motivation. Psychol Rev 61, 5-22. Sun K & Ferguson AV (1997). Cholecystokinin activates area postrema neurons in rat brain slices. Am J Physiol 272, R1625-R1630. Takasaki Y (1978). Studies on brain lesion by administration of monosodium Lglutamate to mice. I. Brain lesions in infant mice caused by administration of monosodium L-glutamate. Toxicology 9, 293-305. Tanaka J, Kaba H, Saito H, & Seto K (1986a). Efferent pathways from the region of the subfornical organ to hypothalamic paraventricular nucleus: an electrophysiological study in the rat. Exp Brain Res 62, 509-514. Tanaka J, Kaba H, Saito H, & Seto K (1986b). Lateral hypothalamic area stimulation excites neurons in the region of the subfornical organ with efferent projections to the hypothalamic paraventricular nucleus in the rat. Brain Res 379, 200-203. Tanaka J, Saito H, & Kaba H (1987). Subfornical organ and hypothalamic paraventricular nucleus connections with median preoptic nucleus neurons: an electrophysiological study in the rat. Exp Brain Res 68, 579-585. 188 Tartaglia LA (1997). The leptin receptor. The Journal of Biological Chemistry 272, 60936096. Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, & et al (1995). Identification and expression cloning of a leptin receptor, OB-R. Cell 83, 1263-1271. Thrasher TN, Simpson JB, & Ramsay DJ (1982). Lesions of the subfornical organ block angiotensin-induced drinking in the dog. Neuroendocrinology 35, 68-72. Trevaskis JL, Coffey T, Cole R, Lei C, Wittmer C, Walsh B, Weyer C, Koda J, Baron AD, Parkes DG, & Roth JD (2008). Amylin-mediated restoration of leptin responsiveness in diet-induced obesity: magnitude and mechanisms. Endocrinology 149, 5679-5687. Tsujii S & Bray GA (1989). Acetylation alters the feeding response to MSH and betaendorphin. Brain Res Bull 23, 165-169. Tumer N, Erdos B, Matheny M, Cudykier I, & Scarpace PJ (2007). Leptin antagonist reverses hypertension caused by leptin overexpression, but fails to normalize obesityrelated hypertension. J Hypertens 25, 2471-2478. Tung YC, Hewson AK, & Dickson SL (2001). Actions of leptin on growth hormone secretagogue-responsive neurones in the rat hypothalamic arcuate nucleus recorded in vitro. J Neuroendocrinol 13, 209-215. Ueda T, Ugawa S, Saishin Y, & Shimada S (2001). Expression of receptor-activity modifying protein (RAMP) mRNAs in the mouse brain. Brain Res Mol Brain Res 93, 3645. Unger RH, Clark GO, Scherer PE, & Orci L (2010). Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochim Biophys Acta 1801, 209-214. van der KD & Koda LY (1983). Organization of the projections of a circumventricular organ: the area postrema in the rat. J Comp Neurol %20;219, 328-338. 189 van Dijk G, Thiele TE, Donahey JCK, Campfield LA, Smith FJ, Burn P, Bernstein IL, Woods SC, & Seeley RJ (1996). Central infusions of leptin and GLP-1-(7-36) amide differentially stimulate c-FLI in the rat brain. American Journal Of Physiology Regulatory Integrative And Comparative Physiology 40, R1096-R1100. Wall KM, Nasr M, & Ferguson AV (1992). Actions of endothelin at the subfornical organ. Brain Res 570, 180-187. Wang J, Obici S, Morgan K, Barzilai N, Feng Z, & Rossetti L (2001). Overfeeding rapidly induces leptin and insulin resistance. Diabetes 50, 2786-2791. Wang QP, Guan JL, Pan W, Kastin AJ, & Shioda S (2008). A diffusion barrier between the area postrema and nucleus tractus solitarius. Neurochem Res 33, 2035-2043. Wardlaw SL (2011). Hypothalamic proopiomelanocortin processing and the regulation of energy balance. Eur J Pharmacol 660, 213-219. Washburn DLS, Smith PM, & Ferguson AV (1999). Control of neuronal excitability by an ion sensing receptor. Eur J Neurosci 11, 1947-1954. Weindl A & Joynt RJ (1973). Barrier properties of the subcommissural organ. Arch Neurol 29, 16-22. World Health Organization (2001). Obesity and Overweight: Fact Sheet number 311. Available from: http://www.who.int/mediacentre/factsheets/fs311/en/index.html Wilkinson M, Brown R, Imran SA, & Ur E (2007). Adipokine gene expression in brain and pituitary gland. Neuroendocrinology 86, 191-209. Williams G, Bing C, Cai XJ, Harrold JA, King PJ, & Liu XH (2001). The hypothalamus and the control of energy homeostasis: different circuits, different purposes. Physiol Behav 74, 683-701. 190 Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF, & Elmquist JK (2010). Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci 30, 2472-2479. Wislocki GB & Leduc EH (1952). Vital staining of the hematoencephalic barrier by silver nitrate and trypan blue, and cytological comparisons of the neurohypophysis, pineal body, area postrema, intercolumnar tubercle and supraoptic crest. J Comp Neurol 96, 371-417. Wolburg H, Liebner S, & Lippoldt A (2003). Freeze-fracture studies of cerebral endothelial tight junctions. Methods Mol Med 89, 51-66. Yang B & Ferguson AV (2003). Adrenomedullin influences dissociated rat area postrema neurons. Regul Pept 112, 9-17. Yang G, Ge H, Boucher A, Yu X, & Li C (2004). Modulation of direct leptin signaling by soluble leptin receptor. Mol Endocrinol 18, 1354-1362. Zardetto-Smith AM & Gray TS (1987). A direct neural projection from the nucleus of the solitary tract to the subfornical organ in the rat. Neurosci Lett 80, 163-166. Zhang W, Telemaque S, Augustyniak RA, Anderson P, Thomas GD, An J, Wang Z, Newgard CB, & Victor RG (2010). Adenovirus-mediated leptin expression normalises hypertension associated with diet-induced obesity. J Neuroendocrinol 22, 175-180. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, & Friedman JM (1994). Positional cloning of the mouse obese gene and its human homologue. Nature 372, 425432. 191