Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Feature detection (nervous system) wikipedia , lookup
Multielectrode array wikipedia , lookup
Alzheimer's disease wikipedia , lookup
Premovement neuronal activity wikipedia , lookup
Optogenetics wikipedia , lookup
Neuromuscular junction wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Action potential wikipedia , lookup
Single-unit recording wikipedia , lookup
Holonomic brain theory wikipedia , lookup
Neurotransmitter wikipedia , lookup
Activity-dependent plasticity wikipedia , lookup
Biological neuron model wikipedia , lookup
End-plate potential wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Chemical synapse wikipedia , lookup
Biochemistry of Alzheimer's disease wikipedia , lookup
Development of the nervous system wikipedia , lookup
Neuroanatomy wikipedia , lookup
Stimulus (physiology) wikipedia , lookup
Synaptic gating wikipedia , lookup
Molecular neuroscience wikipedia , lookup
Nervous system network models wikipedia , lookup
Nonsynaptic plasticity wikipedia , lookup
Node of Ranvier wikipedia , lookup
Neuroregeneration wikipedia , lookup
Axon guidance wikipedia , lookup
+ Models PRONEU-845; No of Pages 18 Progress in Neurobiology xxx (2007) xxx–xxx www.elsevier.com/locate/pneurobio Mechanisms of axon degeneration: From development to disease Smita Saxena, Pico Caroni * Friedrich Miescher Institute, Maulbeerstrasse 66, CH-4058 Basel, Switzerland Received 10 January 2007; received in revised form 31 March 2007; accepted 20 July 2007 Abstract Axon degeneration is an active, tightly controlled and versatile process of axon segment self-destruction. Although not involving cell death, it resembles apoptosis in its logics. It involves three distinct steps: induction of competence in specific neurons, triggering of degeneration at defined axon segments of competent neurons, and rapid fragmentation and removal of the segments. The mechanisms that initiate degeneration are specific to individual settings, but the final pathway of pruning is shared; it involves microtubule disassembly, axon swellings, axon fragmentation, and removal of the remnants by locally recruited phagocytes. The tight regulatory properties of axon degeneration distinguish it from passive loss phenomena, and confer significance to processes that involve it. Axon degeneration has prominent roles in development, upon lesions and in disease. In development, it couples the progressive specification of neurons and circuits to the removal of defined axon branches. Competence might involve transcriptional switches, and local triggering can involve axon guidance molecules and synaptic activity patterns. Lesion-induced Wallerian degeneration is inhibited in the presence of WldS fusion protein in neurons; it involves early local, and later, distal degeneration. It has recently become clear that like in other settings, axon degeneration in disease is a rapid and specific process, which should not be confused with a variety of disease-related pathologies. Elucidating the specific mechanisms that initiate axon degeneration should open up new avenues to investigate principles of circuit assembly and plasticity, to uncover mechanisms of disease progression, and to identify ways of protecting synapses and axons in disease. # 2007 Elsevier Ltd. All rights reserved. Keywords: Axon degeneration; Development; Neurodegenerative disease; Wallerian degeneration; Proteasome; Synaptic activity; SOD1 Contents 1. 2. 3. 4. 5. 6. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . Axon degeneration versus axon retraction . . . . . . . . . . . . . . . . . . . . . 2.1. Definitions and regulation . . . . . . . . . . . . . . . . . . . . . . . . . . . 2.2. Differential recruitment, and comparison to synapse elimination The diverse settings involving axon degeneration. . . . . . . . . . . . . . . . 3.1. Developmental axon degeneration . . . . . . . . . . . . . . . . . . . . . 3.2. Lesions: Wallerian degeneration . . . . . . . . . . . . . . . . . . . . . . . 3.3. Axon degeneration in disease. . . . . . . . . . . . . . . . . . . . . . . . . Shared sequence of events in axon degeneration . . . . . . . . . . . . . . . . Mechanisms of axon degeneration . . . . . . . . . . . . . . . . . . . . . . . . . . 5.1. Wallerian degeneration . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 5.2. Developmental axon degeneration . . . . . . . . . . . . . . . . . . . . . 5.3. Mechanisms of axon degeneration in FALS . . . . . . . . . . . . . . . Shared mechanisms and outstanding issues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 000 000 000 000 000 000 000 000 000 000 000 000 000 000 Abbreviations: AAD, acute axonal degeneration; APP, Alzheimer precursor protein; CNTF, ciliary neurotrophic factor; FALS, familial amyotrophic lateral sclerosis; FF motoneuron, fast-fatiguable motoneuron; FR motoneuron, fast fatigue-resistant motoneuron; MB, mushroom body; Nmnat1, nicotinamide mononucleotide adenylytransferase 1; P15, postnatal day 15; S motoneuron, slow motoneuron; SOD1, superoxide dismutase 1; UPS, ubiquitin-proteasome system; VCP, valosin containing protein; WldS, Wallerian degeneration slow. * Corresponding author. Tel.: +41 61 6973727; fax: +41 61 6973976. E-mail address: [email protected] (P. Caroni). 0301-0082/$ – see front matter # 2007 Elsevier Ltd. All rights reserved. doi:10.1016/j.pneurobio.2007.07.007 Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 2 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx 7. 8. Pathology versus axon degeneration in neurodegenerative diseases 7.1. ‘‘Dying-back’’ diseases . . . . . . . . . . . . . . . . . . . . . . . . . . 7.2. Axonal swellings and spheroids . . . . . . . . . . . . . . . . . . . . Outlook . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 1. Introduction Recent mechanistic studies of axon degeneration have highlighted how this is an active process of controlled axon self-destruction similar in many ways to the active selfdestruction of cells during apoptosis (Coleman, 2005; Low and Cheng, 2005; Luo and O’Leary, 2005; Raff et al., 2002). Although the molecular mechanisms involved are different (but see Williams et al., 2006; Kuo et al., 2006), the principles underlying the triggering and execution of these two selfdestruction processes do exhibit similarities. Thus, apoptosis and axon degeneration both seem to involve tight and versatile control pathways culminating into the disinhibition of ubiquitous intrinsic destruction machinery, and the efficient recruitment of local phagocytes to ensure removal of any trace that might activate autoimmune responses. The available evidence further suggests that the local self-destruction of axons and dendrites underlie related mechanisms (Luo and O’Leary, 2005; Williams and Truman, 2005), suggesting that the degeneration process reflects a general intrinsic mechanism to actively remove, and sometime replace, defined parts of a neuron’s processes. In view of the well-defined properties and significant explanatory value of these active degeneration processes, it seems to us important to reserve the term axon (or dendrite) degeneration for self-destruction processes of entire neurite segments, distinguishing them from local neuritic and organelle pathology. According to this definition, diseases of the nervous system can thus involve axonal and/or dendritic degeneration processes, but these likely only represent a rather small fraction of the pathology that can be associated with disease and/or age. As will become clear in the last part of this review (Sections 7.1 and 7.2), such a distinction is particularly important in order to avoid confusions that arise from our limited understanding of the causal relationships between neuronal pathology, neuronal dysfunction and disease progression. In contrast, the tightly regulated and active aspects of the degeneration processes, together with their dramatic outcome make it likely that, where they can be unambiguously identified, axon and/or dendrite degeneration processes will serve as valuable markers of disease onset and progression. As discussed in the final part of this review (Sections 7.1 and 7.2), this sets axon degeneration processes apart from less well-delineated local pathology and dysfunction phenomena. This review elaborates on some of the major recent advances in understanding axon degeneration and its regulation. We will first define the process, comparing and contrasting it to axon retraction. We will then discuss examples involving axon degeneration during development, upon lesions, and in disease. These distinct scenarios share a set of characteristic features . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 000 000 000 000 000 that define the degeneration process and provide first insights into its mechanisms. We will then discuss what is currently known about the specific mechanisms that control axon degeneration during development, upon lesions and in disease. A comparison of these specific mechanisms suggests a tentative model of how axon degeneration induced in these distinct settings might involve dedicated pathways of degeneration derepression. Finally, we will discuss the evidence for the presence of axon degeneration processes in major nervous system disease settings. Based on this analysis, we identify a number of issues that, in our opinion, are currently unresolved, and make suggestions for experimental systems and approaches that will likely advance our understanding of the mechanisms of axon loss in neurodegenerative diseases. 2. Axon degeneration versus axon retraction 2.1. Definitions and regulation There are two opposite mechanisms through which sections of axons are removed: retraction and degeneration (Low and Cheng, 2005; Luo and O’Leary, 2005). In retraction, the axonal process is gradually pulled backward, and the corresponding axonal material is transported back to more proximal sections of the axon. In contrast, degeneration involves rapid blebbing, and fragmentation of an entire axonal stretch into short segments, which are then removed by phagocytic cells such as locally activated glia or macrophages. Both processes are efficient and tightly regulated, and both can be followed by regrowth of the axon from its proximal end. However, although their ultimate outcome is similar, the two processes differ in that all cellular material, including intracellular signaling organelles (e.g. signaling endosomes), is conserved inside the neuron in retraction, whereas degeneration involves shedding of the axonal material, and its uptake by local phagocytes. Axon retraction has only been documented for modest local axon shortening events during axon outgrowth, whereas larger removal events all appear to involve axon degeneration. In keeping with the recycling of distal axonal material, axonal microtubules are not disrupted during retraction (Luo and O’Leary, 2005), where they continue to carry out their essential role as the predominant transport track for intracellular motor machinery (Gunawardena and Goldstein, 2004). Instead, actin filaments are the cytoskeletal system that mediates the retraction process. This mainly involves activation of the small GTPase RhoA, followed by activation of the major microfilament contracting component myosin light chain through phosphorylation by the Rho effector ROCK (Bito et al., 2000; Kozma et al., 1997; Luo and O’Leary, 2005). Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx Interestingly, activation of RhoA to initiate retraction involves a de-repression mechanism: in several cases, inactivation of the predominant inhibitor of Rho p190RhoGAP initiates retraction (Billuart et al., 2001; Lamprecht et al., 2002). As will become apparent below, this mode of activation through de-repression of pre-existing machinery appears to be a common theme in the regulation of regressive processes in neurons, including axon retraction, degeneration and of course apoptosis. One obvious advantage of this mode of regulation is that in de-repression the regressive machinery is already in place and poised to function throughout the axonal (and dendritic) tree, allowing for rapid and local triggering of the retraction (or degeneration) process (Luo and O’Leary, 2005; Portera-Cailliau et al., 2005). A further advantage is that systems operating based on derepression mechanisms allow for particularly tight and versatile regulation of the triggering process. In contrast to axon retraction, and as will be discussed in more detail below (Section 5), axon degeneration involves disassembly of microtubule filaments as an early, and possibly critical, process (Luo and O’Leary, 2005). Although the issue has not been addressed directly yet, it is therefore possible that, like in axon retraction, the regulation of axon degeneration might impinge onto the regulation of a major cytoskeletal element in neurons (Yamada et al., 1970). Accordingly, mechanisms regulating microtubule stability may not only play important roles in axon guidance, but also in axon degeneration. Candidate microtubule regulatory components of established neuronal processes include Microtubule Associated Proteins (MAPs) such as tau and MAP2, tubulin acetylating and de-acetylating proteins such as HDAC6, and filament severing proteins such as Katanin (Baas and Qiang, 2005; King et al., 2006; Qiang et al., 2006; Yu et al., 2005). 2.2. Differential recruitment, and comparison to synapse elimination When are axon segments eliminated by retraction and when by degeneration? As a simple rule, retraction seems to involve the elimination of shorter branches, whereas degeneration tends to affect longer axonal segments (Low and Cheng, 2005; Luo and O’Leary, 2005). That might in part reflect the fact that degeneration seems to be the more economical and mechanistically simpler solution to the problem of rapidly eliminating larger sections of a process. However, there are exceptions to this simple size rule, and the issue is likely more interesting and complex. Thus, retraction can involve comparatively long axonal branches such as collaterals of hippocampal mossy fibers that initially form extended infrapyramidal projections in CA3, and are pruned through an Ephrin-dependent retraction process during hippocampal development (Bagri et al., 2003; Gao et al., 1999). Likewise, although the removal of short axonal sidebranches during synapse elimination in the development of motor nerve-muscle innervation does not proceed through a canonical degeneration process, it is clearly more reminiscent of degeneration than retraction (Bernstein and Lichtman, 1999; Bishop et al., 2004). As will be discussed (Section 5), developmental axon degeneration (i.e. axon degeneration in 3 the absence of lesions) seems to require transcriptional processes in neurons that confer intrinsic competence for degeneration. In contrast, although intrinsic factors could also be involved, retraction might predominantly involve local regulation. Accordingly, retraction could be viewed as a more contingent, dynamically regulated and potentially reversible local phenomenon, whereas degeneration processes likely reflect more global (e.g. transcriptional) transition states in neurons. Developmental synapse elimination is a local process involving the activity-dependent and competitive elimination of inputs by most axons, and the partial reoccupation of vacated synaptic sites by side-branches of the remaining axons (Bernstein and Lichtman, 1999; Buffelli et al., 2003; Kasthuri and Lichtman, 2003; Lichtman and Colman, 2000; Lohof et al., 1996; Penn et al., 1998; Personius and Balice-Gordon, 2001). Synapse elimination occurs subsequent to naturally occurring neuronal cell death; it does not involve losses of neurons, but instead shrinkage and focusing of their local arborization territories. The process has been analyzed in a series of groundbreaking in vivo imaging studies of postnatal neuromuscular junctions by the laboratory of Jeff Lichtman (BaliceGordon and Lichtman, 1993; Buffelli et al., 2003; Kasthuri and Lichtman, 2003, 2004; Walsh and Lichtman, 2003). These authors have recently shown that short axonal side-branches to individual neuromuscular junctions are eliminated through the formation of swellings (for which they introduced the term axosomes), the thinning and disappearance of the intervening process connecting the axosomes, and the removal of the axosomes by phagocytosis through Schwann cells (Bishop et al., 2004). Axosomes can then be detected as doublemembrane systems within Schwann cell cytosol, and some of the axosome content eventually gets access to Schwann cell cytosol. Interestingly, and in further analogy to axon degeneration, swellings and axosomes contain very few intact microtubules. Similar double-membrane engulfment systems have been reported during the synapse elimination process of climbing fibers in the cerebellum (Eckenhoff and Pysh, 1979), where entire climbing fiber inputs are eliminated from individual Purkinje cells, to produce the mature innervation pattern of one climbing fiber per Purkinje cell (Lohof et al., 1996; Mason and Gregory, 1984). Although no corresponding in vivo imaging data have been reported yet for this synapse elimination process on a larger terminal arborization scale than that of short side-branches to individual neuromuscular junctions, it seems likely that it will again involve an axosome shedding process. It will be of particular interest to determine the extent to which synapse elimination is mechanistically similar to developmental axon degeneration. The two processes might well be closely related, and their comparison might provide insights into the regulatory mechanisms that initiate and regulate synapse elimination, a process about which comparatively little is known so far; a comparison to developmental axon degeneration and synapse elimination might also provide mechanistic insights into side-branch and synapse elimination processes mediating structural plasticity in the adult (De Paola et al., 2006; Galimberti et al., 2006; Lichtman and Colman, 2000). Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 4 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx 3. The diverse settings involving axon degeneration 3.1. Developmental axon degeneration All studies of neuronal circuit assembly during development have highlighted how axon guidance and target innervation proceed with high specificity and accuracy, producing a very low degree of errors (Katz and Shatz, 1996). Why then should axon degeneration be a significant feature in neural development? The answers to this simple-minded question are interesting, and often provide unexpected insights into the logics underlying the evolution and development of nervous systems. Not surprisingly, and like synapse elimination, axon degeneration does not primarily serve the purpose of improving accuracy during circuit assembly (Low and Cheng, 2005; Luo and O’Leary, 2005). Instead, it seems to reflect processes of progressive neuronal specification, and to provide flexibility for circuit assembly mechanisms among individuals and across species (Luo and O’Leary, 2005). Due to the fact that it represents a natural process uninfluenced by artificial manipulations or pathology and due to its high predictability in time and space, developmental axon degeneration is particularly well suited to investigate the mechanisms underlying axon degeneration. Perhaps the best-known examples for developmental processes involving axon degeneration can be found in the development of cortical layer 5 projection neurons, a system that has been thoroughly investigated by the laboratory of Dennis O’Leary (Luo and O’Leary, 2005; O’Leary, 1992; O’Leary and Koester, 1993). These neurons project to multiple specific targets as a function of their cortical identity. Thus, for example, motor cortex neurons project, among other targets, to the spinal cord, whereas visual cortex neurons project to the superior colliculus. Remarkably, however, these diverse cortical neurons initially send off axon branches to most or even all potential targets of layer 5 neurons, and subsequently prune those branches that are inappropriate for their identity (Bastmeyer and O’Leary, 1996; O’Leary and Stanfield, 1989; O’Leary and Terashima, 1988; Stanfield and O’Leary, 1985b; Stanfield et al., 1982). This pruning process takes place at a well-defined time in development, and exhibits typical features of axon degeneration, including widespread blebbing and rapid removal of axonal remnants (Hoopfer et al., 2006; Luo and O’Leary, 2005). Transplantation experiments early during the development of the neocortex have established that layer 5 projections degenerate according to the identity of the host region. This suggests that specification processes involving interactions with the local environment play an important role in determining the patterns of branch degeneration of cortical projection neurons (Stanfield and O’Leary, 1985a). The above finding highlights how this late process of axonal specification might provide a high degree of flexibility in matching requirements for early outgrowth and target invasion by cortical projection neurons to protracted processes of neuronal identity plasticity in the development of the neocortex (Luo and O’Leary, 2005). This dissociation between early generic processes of axon guidance, and protracted processes of neuronal specification followed by corresponding axonal refinements might also provide the developing neocortex with the flexibility required to modify neocortical neuron projection patterns among closely related subtypes of cortical projection neurons, and among the same type of neuron in closely related species (Innocenti et al., 1999; Luo and O’Leary, 2005; Zufferey et al., 1999). Similar processes of extensive degeneration have been described for the callosal projections of cortical neurons (Innocenti, 1981, 1995; Innocenti et al., 1983, 1999; Zufferey et al., 1999), and for the fornix projections of hippocampal subiculum neurons beyond the mamillary bodies of the hypothalamus (Stanfield et al., 1987; Stanfield and O’Leary, 1985a). In addition to pruning their transient projections to subsets of targets that have become inappropriate, neurons can prune aspects of their arborization within their target region (Low and Cheng, 2005; Luo and O’Leary, 2005; O’Rourke et al., 1994). This pruning again involves specific axon degeneration; where it has been described so far, it serves the purpose of achieving appropriate topographic termination patterns (Luo and O’Leary, 2005). Degeneration in the target region has been studied most extensively in the retinotopic projection of ganglion cells from distinct positions in the retina to the superior colliculus (mammals) or optic tectum (birds) (Luo and O’Leary, 2005). Ganglion cell axons initially grow throughout the longitudinal extension of their target, extend transverse collaterals from multiple points along the longitudinal projection, and then local arborizations at their topographically correct terminations. This process is followed by degeneration of the longitudinal projection distal to the appropriate collateral, and of all inappropriate proximal collaterals (Luo and O’Leary, 2005). The molecular information for appropriate retinotopic connectivity is based on the graded expression of ephrins and their receptors in the retina and the superior colliculus, suggesting that this repulsive signaling mechanism might be involved in directing the degeneration of inappropriate processes in the colliculus (Hindges et al., 2002; Yates et al., 2001). Degeneration, however, further depends on the expression of correlated synaptic activity among groups of retinal ganglion cells during a critical time window of development (Bansal et al., 2000; Debski and Cline, 2002; Feller, 2002; McLaughlin et al., 2003a,b; Meister et al., 1991; O’Rourke et al., 1994; Tian and Copenhagen, 2003; Wong et al., 1993). In addition to projecting to the superior colliculus, a subset of retinal ganglion cells sends collaterals to the dorsal lateral geniculate nucleus, where they ultimately establish projections in an eye-specific local segregation pattern (Chen and Regehr, 2000). Again, the segregated pattern emerges from local pruning of terminal branches in a process that depends on correlated synaptic activity among ganglion cells (Bansal et al., 2000; Chen and Regehr, 2000; Feller, 2002; Penn et al., 1998; Ruthazer et al., 2003; Stellwagen and Shatz, 2002; Tian and Copenhagen, 2003; Wong et al., 1993). Interestingly, this smaller-scale pruning process can be induced during a longer period of late development than the degeneration of retinotopically inappropriate projections in the superior colliculus (Luo and O’Leary, 2005). At least for certain types of neurons, the local expression levels of target-derived neurotrophic factors such as NGF, Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx BDNF, NT3 or GDNF can influence the extent of terminal arborization within the target region (Alsina et al., 2001; Markus et al., 2002; Patel et al., 2003; Petersson et al., 2003). In experiments that have established this principle, transgenic overexpression of neurotrophins in natural or ectopic targets of sensory neurons expressing corresponding receptors led to higher innervation densities or the establishment of ectopic innervation by these neurons in the adult (Causing et al., 1997; LeMaster et al., 1999). In complementary experiments, sensory neurons lacking neurotrophin receptors, but prevented from dying by deletion of the pro-apoptotic gene Bax, exhibited greatly reduced target arborization sizes (Markus et al., 2002; Patel et al., 2003). The relationship between target-derived neurotrophic factors influencing target innervation densities and axonal degeneration processes has not yet been investigated in much detail. However, the principles revealed by neurotrophic factor-controlled axon growth and innervation density have led to a powerful experimental paradigm to investigate axon degeneration in vitro. Thus, elegant studies by Robert Campenot using a two-compartment in vitro assay, which allows separate access and treatment of the cell body and axonal compartments of sensory neurons, have established that, at least in vitro, neurotrophins acting locally on axons have specific roles in preventing axon degeneration (Campenot, 1982). Neurotrophin depletion in the axonal but not the cell body compartment led to a rapid degeneration of the section of the axon depleted of NGF, but spared the axon stump inside the cell body compartment (MacInnis and Campenot, 2005). These findings have provided compelling evidence that local signaling can direct the patterns of axon degeneration. In addition, they have provided an attractive experimental system to investigate the regulation of axon degeneration in the absence of lesions (see also below). Vertebrate systems have provided a fascinating range of experimental paradigms to investigate the roles of axon degeneration in the assembly of neuronal circuits, but possibly the most powerful experimental systems to investigate mechanisms of axon degeneration in development and upon lesions have been established in Drosophila, mainly in the laboratory of Liqun Luo (Luo and O’Leary, 2005). While many fly neurons function specifically either during larval or during adult stage, some neurons function during both stages and reorganize their projections during metamorphosis to accommodate new connectivity requirements in the adult (Levine et al., 1995; Technau and Heisenberg, 1982; Tissot and Stocker, 2000). This is thus again an example of axonal projections that have become inappropriate upon a switch in neuronal and circuit specification, and are, as a consequence, eliminated through axon degeneration. Mushroom body (MB) g neuron axons prune their larval collaterals and termination regions by a typical degeneration process at the onset of metamorphosis, but retain the initial segment of the axon, which then re-grows to form the adult projection (Watts et al., 2003). Interestingly, unlike g neurons, MB a and b neurons do not remove parts of their projection through retraction, but inhibition of p190RhoGAP does cause retraction in these neurons, underscoring the exquisite specificity of degeneration- and retraction-mediated 5 regressive events (Billuart et al., 2001; Luo and O’Leary, 2005). The degeneration process of MB g neuron axons depends on the onset of ecdysone hormone signaling (a system related to retinoic acid signaling in vertebrates) at metamorphosis (Schubiger et al., 1998; Technau and Heisenberg, 1982), and on the cell autonomous expression and function of corresponding nuclear hormone receptor pairs in MB g neurons (Lee et al., 2000; Schubiger et al., 1998). The degeneration process proceeds in a temporally well-defined manner, so that its stages could be characterized in great detail (Luo and O’Leary, 2005; Watts et al., 2003, 2004; Zhai et al., 2003). The analysis revealed that axonal degeneration first involves microtubule depolymerization, followed by axonal blebbing, axon fragmentation, neurofilament degradation, and removal of the axonal swellings (Watts et al., 2003; Zhai et al., 2003). Interestingly, microtubule depolymerization and axonal blebbing are not restricted to the sections of distal axons that would degenerate, but also extend into the proximal section of the axon peduncle (i.e. the section that persists, and will grow again after the degeneration of the distal parts). In contrast, fragmentation is restricted to the axonal sections destined for removal (Watts et al., 2003). It might be significant that the proximal axon peduncle, which does not fragment, does thin out substantially before starting to grow (Watts et al., 2003). These results suggest the existence of separate control mechanisms to initiate degeneration in specific neurons, and to spatially restrict the process to well-defined sections of their axonal arbor. As discussed below (Section 6), the mechanisms that assign defined axon segments for degeneration, ensuring that degeneration is restricted spatially to those defined axonal sections remain a major open issue in understanding axonal degeneration. 3.2. Lesions: Wallerian degeneration One setting where defining the segment of an axon destined for degeneration does not seem to be a major challenge is the degeneration of the portion of an axon that lies distally from a physical lesion. This lesion-induced degeneration process had already been described by Waller in 1850 for peripheral nerves (Waller, 1850), and is since known as Wallerian degeneration (Coleman, 2005; Raff et al., 2002). The interest in the mechanisms underlying Wallerian degeneration has grown dramatically since the discovery of a mutant mouse strain (WldS mice, for Wallerian degeneration Slow), where Wallerian degeneration is greatly delayed, and possibly suppressed (Coleman, 2005; Glass et al., 1993; Mack et al., 2001). Transplantation experiments established that the alteration was intrinsic to the transected axons (Glass et al., 1993), thus ruling out the possibility that Wallerian degeneration might involve a passive process of starvation caused by the interruption of material delivery from the neuronal cell body to the distal part of the axon. Subsequent cloning of the mutation revealed the presence of a genomic triplication, giving rise to a unique fusion protein (the WldS protein), consisting of the first 70 residues of UFD2a/UBE4B, a polyubiquitination enzyme, full-length nicotinamide mononucleotide adenylytransferase (Nmnat1), an Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 6 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx enzyme involved in NAD synthesis, and a brief linker sequence (Conforti et al., 2000; Mack et al., 2001). The exact mechanism through which WldS suppresses Wallerian degeneration is not entirely clear yet, and, as discussed below (Section 6), is a major challenge for future studies. Nevertheless, by providing the field with a molecular tool to influence axonal degeneration, the discovery, characterization and exploitation of WldS in gain-offunction studies, mainly by Michael Coleman and his collaborators, has been a critical breakthrough in studying the mechanisms of axonal degeneration. Wallerian degeneration can be investigated in the PNS and CNS in vivo, but also in in vitro models of lesion-induced axonal degeneration (Coleman, 2005; Hoopfer et al., 2006; MacInnis and Campenot, 2005). As an experimental system, it has the obvious advantage that the experimenter determines the precise site and initiation time of the process. Its scientific and clinical interest is further enhanced by the fact that, through its removal of axon remnants, debris and inhibitory factors, Wallerian degeneration likely facilitates the regeneration of the proximal stump of the lesioned axon into and along the distal nerve. In adult mice, the distal sections of physically lesioned nerves (crush or transection) do not exhibit obvious structural changes during the first 36 h after the lesion. This period of apparent preservation is followed by a rapid process of axonal degeneration, which is completed within about an hour (Beirowski et al., 2004; Coleman, 2005). In close similarity to developmental axon degeneration, Wallerian degeneration involves microtubule disassembly, blebbing of the distal axon, followed by fragmentation and removal of the remnants by phagocytes (mainly macrophages upon peripheral nerve lesions) (Coleman, 2005; Luo and O’Leary, 2005). Peripheral synapses are removed slightly prior to distal axon segments, but the difference in time is not such that it by itself makes a compelling case for the existence of a separate mechanism of synapse degeneration (Parson et al., 1997; but see also Miledi and Slater, 1970). While very similar processes of Wallerian degeneration have been observed in several organisms including Drosophila, and in animals of different ages including those in which lesions were made during development, the time it takes from the lesion to the first signs of degeneration can vary from 5 h (Drosophila) to up to 2 days (Luo and O’Leary, 2005). Finally, some chemically induced acute lesions such as local lesions of microtubules with colchicine or vincristine induce axon degeneration closely resembling Wallerian degeneration, and should probably be considered as equivalent to physical lesions (Coleman, 2005). A recent in vivo imaging study of lesioned DRG axons in dorsal spinal cord has provided novel and unanticipated insights into processes of Wallerian degeneration (Kerschensteiner et al., 2005). Using Thy1 transgenic mice in which a few DRG neurons and their axons express the Green Fluorescent Protein GFP, and can thus be repeatedly visualized in vivo, Jeff Lichtman and his colleagues have discovered that nerve lesions in vivo are followed by a further axon degeneration process that takes place long before the onset of classical Wallerian degeneration (Kerschensteiner et al., 2005). Thus, about 20 min after the lesion, axonal swellings appear near the lesion site, but at both the proximal and distal stump. These swellings are rapidly followed by fragmentation and removal of about 150 mm of axon from the proximal and from the distal stump; the fragmentation and removal process only takes about 5 min from onset to completion. This striking degeneration process is again suppressed in mice expressing the WldS protein, providing strong evidence that it closely resembles classical Wallerian degeneration (Kerschensteiner et al., 2005). Based on its rapid onset, rapid completion, and restriction to axon segments near the lesion site, Lichtman and colleagues termed the process acute axonal degeneration (AAD). The rapid onset and limited spatial extent of the process might reflect local access by extracellular solutes, including calcium, to axonal cytosol in the proximal and distal stump through the lesion site. The functional consequences of this acute degeneration process are important, and include disconnection from the cell body of any side-branch within 150–200 mm from the lesion site, and the formation of a gap that might significantly hinder regeneration. The in vivo imaging studies also provided novel insights about the Wallerian degeneration process itself (i.e. distally from the lesion site), including the fact that, following a delay time, axonal swellings develop rapidly (minutes to seconds), that fragmentation rapidly follows the formation of the swellings, and that WldS prevents all of these events (Kerschensteiner et al., 2005). Several of these features had not been appreciated in previous studies, due to the fact that the degeneration process does not proceed synchronously in individual lesioned nerves. This elegant study demonstrates vividly how it will be important and rewarding to monitor axon degeneration processes on-line, in their in vivo setting, and at the level of identified axons. 3.3. Axon degeneration in disease It has long been noted that some neurodegenerative diseases involve protracted gradual ‘‘dying-back’’ processes of distal synapses and axons that can precede the loss of neuronal cell bodies by months (Cavanagh, 1964; Griffin et al., 1995). More recently, it has become clear that most, and possibly all, neurodegenerative diseases involve major synapse and axon losses before the appearance of symptoms, and long before the loss of neurons (Bjartmar et al., 2003; Griffin et al., 1995; Bruck, 2005; Fischer et al., 2004; Frey et al., 2000; Raff et al., 2002; Sagot et al., 1996; Schmalbruch et al., 1991). These diseases include Alzheimer’s, Parkinson’s, Huntington’s, motoneuron diseases, and Prion diseases (Coleman and Yao, 2003; Fischer et al., 2004; Frey et al., 2000; Gunawardena and Goldstein, 2005; Li et al., 2001; Luo and O’Leary, 2005; Sagot et al., 1996; Schmalbruch et al., 1991; Stokin et al., 2005). The long prevailing focus of neurodegenerative research on neuronal cell death and apoptosis pathways has tended to distract from these important early observations (Raff et al., 2002). However, recent unambiguous experimental demonstrations that, in at least some neurodegenerative disease models, protecting cell bodies from death has no impact on disease progression and life-span, whereas protecting axons and synapses does, have helped to refocus research on the ‘‘dying-back’’ phenomenon (Gould et al., Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx 2006; Sagot et al., 1995, 1996, 1998; Ferri et al., 2003; see also Libby et al., 2005). Clearly, if a neuron is chronically disconnected from its targets through peripheral axon losses, it will probably make little difference to the organism whether that neuron stays alive or dies. But is it so, that most neurodegenerative diseases involve ‘‘dying-back’’ processes? And, more importantly, do the ‘‘dying-back’’ processes exhibit any relationship to welldefined axon retraction or degeneration processes? These questions are much more difficult to address experimentally than it would seem. The main difficulties arise from the apparently heterogeneous and non-synchronous nature of the pathology processes in neurodegenerative diseases and their animal models. One criterion that has been used in several recent animal model studies has been to determine whether pathology and/or disease progression were slowed down in the presence of WldS protein. These attempts have met with success in some cases, and with no success in others, but the conclusions are not as straightforward as one might have expected (see discussion in Sections 7.1 and 7.2). A fundamentally different approach consists in investigating the progression of ‘‘dying-back’’ pathology in well-defined animal models of neurodegenerative disease as if it was a developmental process, investigating disease progression at the level of identified axons, and attempting to establish a detailed and predictive map of the disease process that would serve as a basis to investigate the properties and mechanisms of the ‘‘dying-back’’ phenomenon. This approach was recently applied successfully to two mouse models of human familial amyotrophic lateral sclerosis (FALS) (Pun et al., 2006), and the main findings are summarized below. ALS is a deadly late-onset degenerative motoneuron disease of high prevalence (life-time risk of ca. 1:1000) (Boillee et al., 2006a). The familial forms of ALS represent about 10% of total cases, and about a fifth of them are due to point mutations in the ubiquitous and abundant cytosolic enzyme superoxide dismutase 1 (SOD1) (Boillee et al., 2006a). The mode of inheritance of mutant SOD1-linked FALS is autosomal dominant. Why inherited mutations in a ubiquitous protein cause late-onset degeneration restricted to specific types of neurons is a conundrum that is not unique to FALS, and is in fact a main mystery in neurodegenerative diseases (Monani, 2005). Disease-linked human SOD1 mutants have been expressed as transgenes in mice, using a human minigene to drive ubiquitous expression resembling that of the protein in humans (Gurney et al., 1994). These studies have established that SOD1 mutants act through toxic gain-of-function mechanisms to cause FALS in humans and mice (Bruijn et al., 1997; Wong et al., 1995). Subsequent detailed studies, mainly from the laboratories of Don Cleveland and Jean-Pierre Julien, have established that axonal transport is a major early target of disease in FALS (Collard et al., 1995; Lobsiger et al., 2005; Xia et al., 2003; Zhang et al., 1997), although the causal relationships between early presymptomatic defects in axonal transport and disease onset and progression are not entirely clear yet (Boillee et al., 2006a). Finally, in a striking and important new development, studies using cell type specific 7 transgenics (Lino et al., 2002), conditional knockouts of mutant transgene (Boillee et al., 2006b), and elegant analysis of chimeric mice (Clement et al., 2003) have established that the disease process in FALS is not cell autonomous to motoneurons (Boillee et al., 2006a). Instead, mutant SOD1 expression in motoneurons is necessary but not sufficient for disease initiation and progression, and expression in microglia drives disease progression; it is not excluded that mutant expression in further cell types also influences the disease process in these FALS models (Boillee et al., 2006a,b). A line of human-minigene transgenic mice, which express high levels of human SOD1(G93A), exhibit first clinical signs of disease at P80–90, first motoneuron losses at P90–100, and die at postnatal day (P) 135 4 (Gurney et al., 1994). The consistency in the timing of disease onset and progression is a striking feature of several animal models of neurodegenerative diseases, and its investigation will likely lead to interesting insights about the relationships between disease and aging. In SOD1(G93A) mice, this tight temporal predictability has made it possible to analyze motoneuron axons and synapses in separate mice of different ages, and to treat the data as if they were derived from a longitudinal study (i.e. a study of disease progression within the same individual) (Pun et al., 2006). A detailed comparative analysis of neuromuscular innervation patterns in the hindlimb of wildtype and SOD1(G93A) mice revealed that reproducible numbers of axons innervating topographically defined regions of several hindlimb muscles were lost abruptly between P48 and P50 in the mutant mice (Fischer et al., 2004; Frey et al., 2000; Pun et al., 2006). A second group of axons were lost in many muscles at P80–90 and the remaining motoneuron axons still innervated their muscles when the mice died. Further analysis revealed that the axons that were lost at P48–50 invariably belonged to fast-fatiguable (FF) motoneurons, the intermediate axons (lost at P80–90) belonged to fast fatigue-resistant (FR) motoneurons, and the most resistant axons belonged to slow (S) motoneurons (Pun et al., 2006). These motoneuron subtypes exhibit distinct physiological properties, and are recruited under distinct and specific circumstances to produce distinct muscle contraction forces (Burke, 1994). Since individual motor pools (the set of all alpha-motoneurons in the spinal cord innervating a given skeletal muscle) are made of distinct and characteristic combinations of physiological subtypes of motoneurons (Burke, 1994), this selective vulnerability of FF, and then FR motoneuron axons means that in SOD1(G93A) mice certain muscles lose most of their innervation at P48–50, whereas other muscles still maintain a large part of their innervation when the mice die. Importantly, because the distribution of the innervation in individual muscles is highly organized and reproducible among the individuals of a species (Pun et al., 2006), anatomically defined subcompartments of large muscles exclusively innervated by FF motoneurons become completely and reproducibly denervated at P48–50; furthermore, subcompartments innervated by FF and FR motoneurons lose their FF innervation at P48–50, and all remaining innervation at P80–90 (Pun et al., 2006). These results have made it possible to investigate the process of disease-related axon loss with Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 8 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx unprecedented precision and predictability. By focusing on the loss of axons innervating purely FF subcompartments, it was possible to not only monitor the process of axon loss, but also to identify earlier alterations in neuromuscular junctions and axons that were predictive of subsequent axon loss. The analysis established that in SOD1 models of FALS, the peripheral intramuscular part of FF motoneuron axons, with all their peripheral synapses, is lost abruptly (i.e. pruned, see below) between P48–50, and that the pruning process exhibits the characteristic features of axon degeneration, including rapid appearance of swellings, axon fragmentation and rapid removal of the debris. FR axons are pruned at P80–90, through a comparable process (Pun et al., 2006; Schaefer et al., 2005). Starting about 8–10 days prior to axon degeneration, vulnerable FF axons exhibit local accumulations of synaptic vesicles and a loss of synaptic vesicles from neuromuscular junctions, suggesting the presence of a defect in anterograde transport in these vulnerable axons (Pun et al., 2006). Local daily applications to muscle of CNTF, a trophic factor known to protect motoneuron axons in motoneuron diseases (Linker et al., 2005), prevents synaptic vesicle depletion from neuromuscular junctions and FF axon degeneration. Interestingly, applications of CNTF after P45 fails to protect FF motoneuron axons from degeneration, suggesting that CNTF might counteract pathological processes leading up to axon degeneration, but might not inhibit axon degeneration itself (Pun et al., 2006). Very similar, but time-shifted observations were made in SOD1(G85R) mice, a different SOD1 model of FALS in which mice exhibit clinical signs of disease around 7 months, and die at 8–9 months (Bruijn et al., 1997; Pun et al., 2006). These results suggested that selective vulnerability and axonal degeneration of first FF and then FR motoneuron axons is a central feature of disease progression in mouse models of FALS (Pun et al., 2006). More generally, these FALS models in mice provided evidence that regulated, selective and efficient axonal degeneration does take place during the course of at least one neurodegenerative disease (Pun et al., 2006; Schaefer et al., 2005). Together with the detailed anatomical and temporal information about the degeneration process, these models provide an experimental system to investigate the mechanisms that can trigger axon degeneration in the adult, in the absence of acute physical or chemical lesions to axons. 4. Shared sequence of events in axon degeneration As we have seen, the settings in which axon degeneration has been documented are very different. Axon degeneration during development is a tightly regulated and confined selfdestruction process triggered in the absence of lesions or pathology; it depends on mechanisms that define the onset, and the exact location of the destruction. These mechanisms can be very different, ranging from differentiation processes to patterns of synaptic activity during critical periods. The scale of ‘‘programmed’’ axon destruction can vary from most of a neuron’s processes in Drosophila MB g neurons, to the elimination of innervation to entire targets (e.g. layer 5 cortical neurons), and on to the local elimination of collaterals in the target region (e.g. retinal ganglion cell innervation in superior colliculus). Elimination on an even smaller scale might be involved in the segregation of eye inputs in dorsal lateral geniculate nucleus, and in synapse elimination, although these local events based on activity-dependent competition might underlie qualitatively different mechanisms. When comparing the different developmental settings (Fig. 1), it seems clear that regulation type or pruning scale do not make a significant difference with respect to the characteristic temporal and Fig. 1. Schematic representation of the shared (black) and setting-specific (colours) processes involved in axon degeneration upon lesions (Wallerian degeneration) and in development. Although it likely reflects a general feature of axon degeneration, a requirement for phagocyte recruitment (glia) has been demonstrated for developmental degeneration in Drosophila MB g neurons, but not yet in the other settings. Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx morphological features of developmental axon degeneration. In each case, the entire axonal section to be removed is affected in a nearly synchronous manner, and degeneration involves microtubule disassembly, followed by axonal swellings, neurite and neurofilament fragmentation, and removal of the remnants by recruited phagocytes. Furthermore, for any given axon, the process typically only takes a few hours from onset to completion. Significantly, although Wallerian degeneration is brought about by a lesion, which the axons involved sustain passively, the characteristic sequence of events, in vivo or in vitro, does not differ significantly from that in developmental axon degeneration (Coleman, 2005; Luo and O’Leary, 2005) (Fig. 1). Finally, although the information is currently less complete, axonal degeneration associated with disease, i.e. a process that is neither programmed nor induced by an acute lesion, does not seem to be significantly different from degeneration in development or upon lesions. This convergence onto an apparently common pattern of axon selfdestruction suggests that it might be useful to subdivide studies of axon degeneration into three main aspects: the specific mechanisms that activate the pathway of self-destruction, the particular mechanisms that define the exact section of an axon to be destroyed, and the common mechanism of axon destruction. 5. Mechanisms of axon degeneration 5.1. Wallerian degeneration Wallerian degeneration (i.e. lesion-induced axon degeneration) is efficiently blocked in the presence of mouse WldS protein, irrespective of whether it is induced in vertebrates or Drosophila (Mack et al., 2001; MacDonald et al., 2006). In the presence of WldS, lesioned vertebrate axons fail to degenerate and continue to conduct action potentials for 2–3 weeks; the slow degradation that eventually ensues differs from the degeneration process, suggesting that the presence of WldS completely negates Wallerian degeneration (Coleman, 2005). As we will see below (Sections 5.2 and 5.3), this sets Wallerian degeneration apart from developmental axon degeneration (WldS insensitive, Hoopfer et al., 2006), and also, at least in some cases, from disease-related degeneration. WldS protein thus likely affects a reaction specific to lesion-induced degeneration. That reaction must lie at the very onset of the degeneration process or at some point during the delay period that precedes axon degeneration, since WldS protein blocks all steps of the degeneration process, including microtubule disassembly. Since WldS is not detected in the axon, and is detected essentially only in the nucleus, the reaction might involve gene expression processes (Coleman, 2005). That suggests that WldS might affect the expression of proteins important to initiate Wallerian degeneration; indeed, WldS does alter the expression of a specific set of genes in neurons (Gillingwater et al., 2006). Based on these observations, a tentative model for the initiation of Wallerian degeneration would include the constitutive presence (or absence) throughout axons of WldS-sensitive proteins essential to initiate lesion- 9 induced degeneration (Coleman, 2005). In the presence of signals initiated by the lesion process, these targets of WldS would be essential to disinhibit, and thus activate the degeneration machinery (Coleman, 2005). The fact that activation of local degeneration in AAD is dramatically more rapid (about 20 min) than the 36 h needed for the onset of the distal degeneration process in the very same axons, suggests that degeneration might be triggered by a leak process associated with physical disruption, e.g. a leak of ions such as calcium and/or sodium, possibly followed by reverse operation of the sodium-calcium exchanger and the accumulation of calcium, which have been shown to contribute to Wallerian degeneration (Coleman, 2005). The exact mechanism affected by WldS is not clear yet, but it specifically affects axon degeneration and not apoptosis (Finn et al., 2000), does not seem to affect ubiquitination, and also does not seem to represent simple overexpression of Nmnat1 protein (Coleman, 2005; Conforti et al., 2007). One intriguing suggestion is that WldS might interact with the chromatin-deacetylation protein SIRT1, an effector of NAD that has been implicated in the regulation of cellular aging (Araki et al., 2004). A further lead is that its N-terminal end interacts in the nucleus with the key ubiquitin-proteasome system (UPS) component valosin-contaning protein (VCP) (Coleman, 2005; DiAntonio et al., 2001). In addition to WldS protein, Wallerian degeneration depends on the activity of the UPS (Zhai et al., 2003). Blockade of UPS with specific inhibitors, or upon overexpression of interfering mutants, prevents all steps of the degeneration process, including microtubule disassembly. Significantly, and unlike WldS, UPS inhibitors prevent axonal degeneration in all settings where it has been tested (see Sections 5.2 and 5.3) (Hoopfer et al., 2006). UPS activity is thus a general requirement to initiate axon degeneration. That requirement must specifically apply to an upstream process in the initiation of the degeneration pathway because application of UPS inhibitors just prior or subsequent to lesions did not interfere with Wallerian degeneration (Zhai et al., 2003; Coleman, 2005; Hoopfer et al., 2006). Although uncertainty about the exact time when the inhibitors reach sufficient concentrations inside axons precludes a definitive conclusion, these findings suggest that, although distal degeneration might only start 36 h after a lesion, critical reactions are triggered around the time of the actual lesion. A possible and testable model could thus envision UPS-dependent activation of degeneration competence at the time of the lesion or shortly thereafter, and variable initiation of the degeneration process, either 20 min (local AAD; Kerschensteiner et al., 2005) or 5–36 h after the lesion (distal degeneration). In this model, WldS protein might lead to the absence of an axonal component essential to trigger lesioninduced competence (or the presence of an axonal protein preventing this triggering) (Coleman, 2005). Importantly, the model would dissociate the induction of degeneration competence from the initiation of the degeneration process itself, both temporally and spatially. Although speculative at this point, this dissociation would tie in well with the regulatory properties of developmental, and possibly also disease-related axon degeneration (see below). Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 10 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx Research on Wallerian degeneration further uncovered a requirement for the recruitment of phagocytes (e.g. activated glia) in the degeneration process (MacDonald et al., 2006; Awasaki et al., 2006). This recruitment was prevented in the presence of WldS protein in neurons, or of UPS inhibitors, suggesting that the recruitment of phagocytes by lesioned axons depends on the axonal degeneration pathway. Significantly, this pathway depends on the expression and accumulation of Draper in the prospective phagocytes, a receptor also required to recruit phagocytes to rapidly clear cellular remnants in apoptosis (Zhou et al., 2001; Hoopfer et al., 2006; MacDonald et al., 2006). These findings in Wallerian and developmental degeneration (see below) thus establish a striking mechanistic relationship between the late phases of active cell and axonal self-destruction processes. 5.2. Developmental axon degeneration The most extensive mechanistic insights about developmental axonal degeneration derive from genetic analyses of MB g neuron axon pruning during Drosophila metamorphosis (Luo and O’Leary, 2005). These studies have established that competence for axonal (and dendritic) degeneration involves transcriptional processes initiated by ecdysone/ecdysone receptor-mediated signaling. While the ecdysone is secreted into the environment of neurons, ecdysone receptor (a nuclear receptor that functions together with retinoic receptor protein) must be expressed in the neurons that will prune their axons (Lee et al., 2000). Ecdysone/ecdysone receptor signaling appears to mediate competence for axonal degeneration in all neurons known to prune their axons during metamorphosis in Drosophila (Luo and O’Leary, 2005; Schubiger et al., 1998). The relevant neuronal products of ecdysone signaling in axon degeneration have not been identified yet, and it is not yet clear how they are eventually required for the initiation of microtubule disassembly in pruning axons. Strikingly, overexpression of dominant-negative ecdysone receptor, which prevents developmental pruning during metamorphosis, did not influence lesion-induced axon degeneration of the very same axons in developing Drosophila (Hoopfer et al., 2006). Even more strikingly, overexpression of WldS protein, which suppresses lesion-induced axon degeneration in Drosophila, had no detectable effect on developmental axon pruning during Drosophila metamorphosis (Hoopfer et al., 2006; Luo and O’Leary, 2005). Furthermore, developmental axon degeneration in vertebrates was also not affected by overexpression of WldS protein (Hoopfer et al., 2006; Luo and O’Leary, 2005). Taken together, these results provide compelling experimental evidence that axonal degeneration induced during development and upon lesions is brought about by distinct mechanisms, and that WldS specifically inhibits Wallerian, but not developmental axon degeneration (Hoopfer et al., 2006; Luo and O’Leary, 2005). Interestingly, competence for developmental degeneration of cortical axon collaterals in vertebrates depends on the expression and nuclear translocation of the transcription factor Otx1 in these neurons (Weimann et al., 1999; Zhang et al., 2002) suggesting that induction of competence through transcriptional processes might be a general feature in developmental axon degeneration. One possibility could be that transcriptional switches during defined periods of development provide a prerequisite to induce competence for axon degeneration, and that additional signals, e.g. through axon guidance molecules, trophic factor deprivation, or signaling initiated in the absence of synchronized synaptic activity might confer competence for degeneration. But this still leaves a need to identify the mechanisms that initiate the degeneration of specific axon branches. In an alternative, and perhaps more parsimonious scenario, neurons might go through prolonged periods of axon degeneration competence induced by transcriptional switches during defined periods of development, and the additional more contingent signals would initiate the pruning at specific locations along the axonal tree. The analysis in Drosophila has further established that, like in Wallerian degeneration, UPS activity is required for developmental axon degeneration (Watts et al., 2003). Genetic inactivation of ubiquitin activating enzyme (Uba1), or of the proteasome itself prevented microtubule fragmentation, axon blebbing and pruning, leading to a persistence of larval MB g axons into adulthood (Watts et al., 2003, 2004). A requirement for proteasome activity was also demonstrated for axon degeneration induced upon NGF deprivation (MacInnis and Campenot, 2005). These experiments thus suggest that UPSdependent degradation is a general prerequisite to confer competence for (or initiate) axon degeneration in development and upon lesions. Proteasome-mediated degradation might for example remove a critical inhibitor of the axon degeneration machinery. The fact that only a restricted set of ubiquitin ligases was critically required to induce axon pruning in Drosophila gives reasons to hope that the specific ubiquitination substrate(s) involved in axon degeneration will be identified soon, thus providing first insights about the axonal components that control the degeneration process (Luo and O’Leary, 2005; Watts et al., 2003). In addition to implicating UPS activity for axon degeneration, the analysis of developmental dendrite degeneration in Drosophila has revealed a specific requirement for local caspase activity in this process (Kuo et al., 2006; Williams et al., 2006). This discovery suggests the exciting possibility that aspects of the molecular machinery involved in cellular apoptosis might also be involved in local neurite degeneration. Finally, the analysis in Drosophila has established how glial recruitment through Draper and ced-6 has a critical role in the execution of developmental axon pruning (Hoopfer et al., 2006; Watts et al., 2004; Awasaki and Ito, 2004; Awasaki et al., 2006). The analysis demonstrated that in addition to its role in neurons, ecdysone also signals through glial ecdysone receptors, and that this signaling is essential for glial recruitment to blebbing axons (Awasaki et al., 2006). In addition, however, glial recruitment depended on a signal from the axons, which was suppressed in the absence of UPS activity (Awasaki et al., 2006; Luo and O’Leary, 2005). This signal seemed to only act over very short ranges, and might be mediated through direct contact between degenerating axons and glial processes; that might involve Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx mechanisms analogous to ‘‘find me’’ and ‘‘eat me’’ signals in apoptosis (Zhou et al., 2001; Awasaki et al., 2006; Luo and O’Leary, 2005). The dual signaling to glia, from ecdysone and from blebbing axons, is reminiscent of the separate signals involved in inducing competence and initiating the actual execution process in axon self-destruction. The signals initiate morphological responses in glia reminiscent of a phagocytic phenotype; in addition, they induce plasmalemmal accumulation of Draper in glial processes. In the absence of glial recruitment, axons still exhibited disrupted microtubules and blebbing, but the fragments failed to clear efficiently. As a consequence, flies with disrupted Draper function in glia exhibited a delay in axon clearance, with some axons being maintained into adulthood (Awasaki et al., 2006; Hoopfer et al., 2006). Glial recruitment is thus required to disrupt and remove degenerating axons, but not to initiate the axon degeneration process. The local signals through which degenerating axons recruit glia have not been identified yet, and their elucidation should be of general interest to understand axon degeneration. 5.3. Mechanisms of axon degeneration in FALS Much less is known about the mechanisms through which axon degeneration is induced in SOD1 mouse models of FALS, or in any other disease setting. In principle, one could imagine that axons get highly damaged in disease, e.g. through near to complete blockades of axonal transport, and that the induction of degeneration ultimately resembles that induced by a chemical lesion of microtubules (a WldS-sensitive process). Disease would then resemble Wallerian degeneration, and axonal pruning would presumably be sensitive to WldS protein. However, although SOD1-linked models of FALS clearly involve axonal degeneration as revealed by the abrupt and characteristic fractionation and removal of particular axons at defined times in disease (Fischer et al., 2004; Pun et al., 2006), disease onset and disease progression in the FALS models are essentially insensitive to WldS protein (Fischer et al., 2005; Vande Velde et al., 2004). It seems unlikely that this failure by WldS to protect in disease would be due to an age-related decline in WldS effectiveness, because the protein does confer strong axonal protection in lesioned axons at ages beyond 3 months, when SOD1(G93A) mice exhibit clinical signs of disease. We will get back to this issue because WldS protein does seem to confer some protection in other disease models, but the conclusion is that degeneration-inducing pathways different from those operating in Wallerian degeneration must be operating in FALS. One possible scenario could be that disease induces a switch in affected neurons comparable to the switches leading to competence for axonal degeneration in development, and that subsequent local signals lead to peripheral synapse and axonal pruning. An alternative scenario could be that there are additional, yet unidentified, ‘‘safety’’ pathways to locally disinhibit the axon degeneration machinery in a pathological setting, and that these ‘‘safety’’ pathways (or at least some of them) are not sensitive to the effects of WldS protein. 11 But to what extent does axon degeneration in FALS resemble its developmental and Wallerian counterparts? Could it for example be that degeneration in FALS simply bypasses the initial competence checkpoints, which are sensitive to WldS? One way of addressing these questions is to determine whether the degeneration process in FALS is sensitive to UPS inhibitors. As discussed in Section 3.3 of this review, the appearance of local axonal swellings and the early depletion of synaptic vesicles from neuromuscular junctions in FALS (from P38 to P45 in FF motoneurons of SOD1(G93A) mice) could be prevented by local applications of CNTF into muscle (Pun et al., 2006). In addition, the axonal pathology in FF motoneurons was accompanied by upregulation of Bcl2A1 mRNA and protein, a member of the Bcl2 family of antiapoptotic proteins, in corresponding motoneurons from P40–42 on, and this upregulation was again suppressed by local applications of CNTF to muscle (Pun et al., 2006). The CNTF treatment was, however, only effective if started not later than P42–43, and applications of CNTF from P45 on, i.e. 3–5 days before pruning of FF motoneurons, did not reverse the synaptic vesicle phenotype, or affect the time course of axon pruning (Pun et al., 2006). In marked contrast, daily local applications of proteasome inhibitor into muscle starting at P45 or P46 prevented FF axon pruning up to at least P60 (Pun et al., 2006). These findings provided evidence that axon degeneration in FALS depends on UPS activity, and demonstrated that UPS blockade can prevent pruning at a time when CNTF is not effective anymore. Significantly, and unlike local applications of CNTF, local blockade of UPS from P38 on failed to influence the appearance of axon pathology, the depletion of synaptic vesicles from neuromuscular junctions, or the upregulation of Bcl2A1 in FF motoneurons (Pun et al., 2006). This was not due to some toxic effect of proteasome inhibitor, because the same treatment did prevent FF axon pruning up to at least P54 (Pun et al., 2006). These results have a number of interesting implications (Fig. 2): The fact that blocking axonal pathology and Bcl2A1 upregulation with CNTF also prevented later pruning of FF motoneuron axons, suggests that an early pathological process specifically in FF motoneurons and their axons sets up the stage for UPS-sensitive triggering of axon degeneration in FF motoneurons. Because UPS activity in axon degeneration appears to be required at the time of competence induction, these findings suggest that the process of axon degeneration in FF motoneurons of SOD1(G93A) mice is initiated between P46 and P48, and not before P46. Perhaps most importantly, the results further suggest that CNTFsensitive axonal pathology and Bcl2A1 upregulation are not part of the axon degeneration process (because they are not affected by UPS inhibitors). Taken together, these results make two important points about axon degeneration in FALS mice: first, and like in developmental and Wallerian degeneration, the axon degeneration process in this chronic neurodegenerative disease model is triggered and completed within 2–3 days at most, and second, the prominent disease-related axonal pathology and the related cell body reactions likely lead up to the triggering of axon degeneration, but they are not part of the axon degeneration process itself. Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 12 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx Fig. 2. Schematic representation of the processes leading up to the early peripheral degeneration of FF motoneuron axons in SOD1(G93A) FALS mice. Briefly, local applications of CNTF to muscle from P38 on prevent axon pathology (synaptic vesicle stalling in axons, and synaptic vesicle depletion from neuromuscular junctions), upregulation of Bcl2A1 in motoneurons and axon degeneration in FF motoneurons. In contrast, when the CNTF treatment is initiated at P45, it fails to protect axons from pathology and degeneration, although it does prevent Bcl2A1 upregulation. Proteasome inhibitor (Lactacystin) applied from P38 or P45 on prevents axon degeneration without alleviation of axon pathology or Bcl2A1 upregulation. These findings suggest that CNTF-sensitive axon pathology leads up to axon degeneration competence at P46, but that the axon pathology is not part of the (Lactacystin-sensitive) degeneration process. Green: synaptic vesicles; red: Bcl2A1. 6. Shared mechanisms and outstanding issues The comparative analysis of axon degeneration mechanisms in development, upon lesions and in a neurodegenerative disease model has not only revealed several shared features, but also a number of aspects unique to the different settings (Fig. 1). The unique aspects involve mechanisms that confer competence for self-destruction, and those that initiate the selfdestruction of defined sections of axons. Shared aspects include the central role of an early competence conferring process, the role of UPS, the unique sequence of structural and anatomical events, and the role of glia (although the latter has not yet been addressed in neurodegeneration) (Figs. 1 and 2). We summarize these comparisons by proposing a model of degeneration regulation that takes into account the findings described in the previous sections of this review (Fig. 3). Although it is speculative at this point, the model might be helpful in devising future experiments. The model has two main features: First, distinct regulatory pathways together with the shared requirement for UPS activity lead to axonal competence to initiate Fig. 3. Proposed model of how the induction of axon degeneration might involve setting-specific regulatory processes, and a shared set of execution reactions. The induction of a neuronal competence state for axon degeneration, and the local triggering of degeneration at defined axon segments would involve context-specific regulation. In contrast, neuronal competence itself might reflect a more general de-repression state that could subsequently lead to local axon degeneration through distinct local mechanisms. Finally, upon local triggering, the degeneration process itself involves a common pathway of microtubule disassembly, axonal swellings, axon fragmentation and removal of the remnants. UPS activity is a general requirement to induce competence for axon degeneration. In contrast, WldS protein suppresses competence induction in Wallerian, but not developmental degeneration. Degeneration induced in disease can be sensitive (e.g. pmn/pmn mice) or insensitive (e.g. FALS mice) to inhibition by WldS. Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx self-destruction. Second, separate processes subsequently initiate self-destruction of defined axonal segments. The selfdestruction process itself then involves the shared sequence of microtubule disassembly, axon blebbing, axon fragmentation, and removal of the fragments by recruited phagocytes. In this model, WldS protein specifically interferes with competence induction upon lesions. The various developmental switches could, for example, lead to changes in the expression of critical components that control the inhibition of degeneration in the axon. The pathways leading up to competence might converge onto a UPS-sensitive step, but that step might also operate in parallel to those pathways. Subsequent to the acquisition of competence for degeneration, the model suggests that the various local triggering pathways that operate in the absence of lesions (e.g. activity patterns, neurotrophin deprivation, axon guidance molecules) might have a role comparable to ion leakage in Wallerian degeneration. In this way, the various local signaling pathways might define the axon segments that initiate self-destruction; these pathways could, for example, all converge onto elevated levels of intra-axonal calcium. Finally, the model suggests that the different competence and triggering pathways might be interchangeable, meaning that competence induced through any pathway might be compatible with any type of triggering mechanism. This seems to us a parsimonious assumption, but it might well be that axons have evolved distinct competence-triggering pathways to ensure more tight and specific regulation of the axon degeneration process. Although research into axon degeneration has progressed dramatically during the last 3–4 years, many important aspects of the axon degeneration process remain unclear. Some of the more fascinating questions are intrinsic to developmental neuroscience or to basic studies of neurodegenerative diseases. However, addressing those questions will also depend on a better mechanistic understanding of the axon degeneration process itself. One major unresolved issue involves the molecular mechanisms that control the degeneration process in the axon. Since this likely involves complex signal transduction pathways, including multiple pathways to confer competence, and checkpoints to initiate the process, these questions might be best addressed in a genetic model system, e.g. Drosophila (Lee and Luo, 1999; Luo and O’Leary, 2005). The substrates of the critical ubiquitin ligases that must be degraded by the proteasome machinery in order to confer competence for axon degeneration could be one specific focus for investigations. Investigating the mechanisms through which WldS protein prevents lesioninduced degeneration will likely lead to important complementary information. Since those studies would eventually have to identify critical downstream targets of WldS regulation in the axon, they might again profit from the powerful forward ands reverse genetics approaches available in Drosophila (Lee and Luo, 1999). The identification of any positive upstream regulator of the degeneration pathway in the axon will hopefully provide much needed tools to dissociate competence and initiation of the degeneration process experimentally. Such dissociations will be important in order to investigate the mechanisms that trigger the degeneration process in development and in disease models. In both settings, one major open question is how defined sections of 13 the axon are targeted for degeneration. Answers to this question should open up new avenues to investigate principles of developmental pruning, and to identify ways of protecting synapses and axons in disease. 7. Pathology versus axon degeneration in neurodegenerative diseases 7.1. ‘‘Dying-back’’ diseases If one defines ‘‘dying-back’’ diseases as those, where progressive and substantial losses of synapses and axons precede neuronal losses, then there is evidence that many neurodegenerative diseases exhibit ‘‘dying-back’’ patterns of losses. Welldocumented cases include motoneuron diseases, peripheral neuropathies, spinocerebellar ataxias, nutritional disorders and AIDS (Berger and Schaumburg, 1995; Bjartmar et al., 2003; Griffin et al., 1995; Martin et al., 2002; Pun et al., 2006). In addition, early losses of synapses have been reported for Alzheimer’s, Parkinson’s, Huntington’s and Prion’s diseases (Bommel et al., 2002; Coleman and Yao, 2003; Gunawardena and Goldstein, 2005; Gunawardena et al., 2003; Li et al., 2001; Tsai et al., 2004). The extent to which ‘‘dying-back’’ processes are the predominant cause of functional losses in those diseases is in most cases less clear. Even less clear is the extent to which ‘‘dying-back’’ reflects axon degeneration processes. Although it does not seem unlikely that axon degeneration is involved in several cases, the possibility exists that just as cells can die through apoptosis or necrosis, axons might be lost through degeneration or through more passive, and potentially more toxic, forms of increasing pathology and dystrophy. For example, dystrophic neurites associated with extracellular amyloid plaques in Alzheimer’s disease definitely seem to have escaped degeneration. On the other hand, there are several neurodegenerative diseases in which there is a strong case for the existence of axon degeneration processes (Bommel et al., 2002; Martin et al., 2002; Pun et al., 2006). Current criteria are of two kinds: rare direct evidence of axon degeneration processes such as in FALS (Pun et al., 2006) and in forms of early-onset motoneuron disease (e.g. pmn/pmn mice, Sagot et al., 1995), and evidence that synapse and/or axon losses are reduced in the presence of WldS protein (Hasbani and O’Malley, 2006; Sagot et al., 1996; Sajadi et al., 2004). A comparison between pmn/pmn mice and the FALS models illustrates how alleviation of pathology by WldS protein does not provide a straightforward discrimination between diseases involving axon degeneration and those that do not. Thus, although in pmn/pmn mice the patterns of axon loss have not been investigated in detail comparable to that in FALS mice, these mice do exhibit abrupt synapse and axon losses suggestive of axon degeneration (Sagot et al., 1995). However, while FALS mice are not sensitive to WldS (Fischer et al., 2005; Vande Velde et al., 2004), pmn/pmn mice are (Ferri et al., 2003). The reasons for this discrepancy are not clear at the moment, but one possibility is that the molecular pathways leading to axon degeneration in the two models are different. A detailed investigation of ‘‘dying-back’’ diseases with respect to patterns of axon degeneration and sensitivity to WldS protein should Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 14 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx provide important insights into the mechanisms leading to axon losses in these diseases. The value of these studies will likely be enhanced by a detailed anatomical analysis of the synapse and axon loss processes, and by progress in understanding the mechanisms through which WldS protein prevents Wallerian and other forms of axonal degeneration. 7.2. Axonal swellings and spheroids A discussion of the axonal pathologies associated with neurodegenerative diseases is clearly beyond the scope of this review. However, in view of the possible relationships between defects in axonal transport, axonal swellings, and reported effects of WldS protein in several neurodegenerative disease models, a brief discussion of how those specific issues might relate to axonal degeneration processes seems important. A compelling body of experimental evidence supports the notion that defects in axonal transport are an early manifestation of pathology in most neurodegenerative diseases (Collard et al., 1995; Gunawardena and Goldstein, 2001, 2004, 2005; Gunawardena et al., 2003; Sagot et al., 1998; Stokin and Goldstein, 2006; Stokin et al., 2005; Williamson and Cleveland, 1999; Zhang et al., 1997). Although the causal relationships between these early transport defects and disease onset and progression are not clear yet, it seems likely that these defects will turn out to have causative roles in disease. To a large extent, this expectation is based on the fact that in several cases loss-of-function mutations in intracellular transport or membrane trafficking components have now been related to familial forms of neurodegenerative diseases (Bommel et al., 2002; Eaton et al., 2002; Garcia-Fresco et al., 2006; Gotz et al., 2006; Gunawardena and Goldstein, 2004; LaMonte et al., 2002; Martin et al., 2002; Stokin and Goldstein, 2006; Xia et al., 2003). There are, furthermore, reasons to believe that the appearance of swellings and spheroids in axons could be linked to defects in axonal transport, possibly as a macroscopic manifestation of ‘‘roadblocks’’ (the swellings typically accumulate the fast axonal transport component amyloid precursor protein (APP)), or perhaps even as a mechanism to alleviate ‘‘roadblocks’’ (Gunawardena and Goldstein, 2001, 2004, 2005; Gunawardena et al., 2003). However, the main issue here is whether and when axonal pathologies such as swellings and larger spheroids are an aspect of the axonal degeneration process in disease, whether they represent local pathologies that can trigger axon degeneration, or whether most of these phenomena are not causally related to axon degeneration in disease (Delio et al., 1992; Tsai et al., 2004). These issues can be confusing because axon degeneration does involve axonal swellings, and because WldS protein has been shown to reduce swellings and spheroids in some disease models (Mi et al., 2005). However, and as discussed in a previous section of this review, although axonal swellings in FALS mice might be part of the processes that lead up to axon degeneration, they are clearly not a manifestation of the degeneration process itself (Pun et al., 2006). Most likely, therefore, there are several distinct forms of axonal swellings, and only a certain type is diagnostic of axonal degeneration. It seems to us reasonable to assume that, wherever it is associated with disease, axonal degeneration will invariably exhibit some of its characteristic hallmarks, including a rapid onset (within a few days at most), and a rapid elimination of axonal remnants. That clearly distinguishes axon degeneration from the gradual accumulation of various forms of axonal pathologies, swellings and spheroids, which can last for months and even years (Gotz et al., 2006; Mi et al., 2005; Pirozzi et al., 2006). This distinction is further underlined by the fact that in some disease models distinct axonal pathologies can progress for many months prior to the first evidence of synapse or axon losses. Taking these considerations into account it is therefore particularly puzzling how WldS protein nevertheless can attenuate the development of these pathologies (Mi et al., 2005; Samsam et al., 2003). One possibility, which seems unlikely to us, is that axon degeneration associated with some neurodegenerative diseases is an unusually protracted process. An alternative possibility is that the fusion protein WldS has additional protective effects not specifically related to axon degeneration. 8. Outlook Main recent developments in studying axon degeneration include the growing appreciation that this is a widespread and tightly regulated axon (and dendrite) self-destruction process reminiscent in its logics and significance to apoptosis, and the innovations in genetic tools and molecular live-imaging techniques that have allowed first glimpses of how these processes happen in vivo. A number of basic issues about mechanisms regulating axon degeneration remain open and provide exciting challenges for future research. These include the specific mechanisms leading up to competence for axon degeneration in its different settings, and the molecular mechanisms of this regulation in neuronal cell bodies and at axons. A second set of challenges involves elucidating the mechanisms that determine which specific segments of axons (and dendrites) are targeted for degeneration in the different settings. Progress on these mechanistic issues will likely fuel significant advances in developmental neuroscience, in studies of plasticity in the adult, and in studies of neurodegenerative diseases. Equally exciting advances can be expected from detailed longitudinal live-imaging studies of axon degeneration processes during development and in disease. The few initial in vivo imaging studies of Wallerian degeneration, motoneuron disease and Alzheimer’s disease have given us a taste of things to come (Kerschensteiner et al., 2005; Schaefer et al., 2005; Tsai et al., 2004). One can safely assume that combinations of high-resolution longitudinal analyses in vivo and mechanistic insights of degeneration processes at the molecular level will have a major impact on studies of development, plasticity and disease in the nervous system. References Alsina, B., Vu, T., Cohen-Cory, S., 2001. Visualizing synapse formation in arborizing optic axons in vivo: dynamics and modulation by BDNF. Nat. Neurosci. 4, 1093–1101. Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx Araki, T., Sasaki, Y., Milbrandt, J., 2004. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 305, 1010– 1013. Awasaki, T., Ito, K., 2004. Engulfing action of glial cells is required for programmed axon pruning during Drosophila metamorphosis. Curr. Biol. 14, 668–677. Awasaki, T., Tatsumi, R., Takahashi, K., Arai, K., Nakanishi, Y., Ueda, R., Ito, K., 2006. Essential role of the apoptotic cell engulfment genes draper and ced-6 in programmed axon pruning during Drosophila metamorphosis. Neuron 50, 855–867. Baas, P.W., Qiang, L., 2005. Neuronal microtubules: when the MAP is the roadblock. Trends Cell. Biol. 15, 183–187. Bagri, A., Cheng, H.J., Yaron, A., Pleasure, S.J., Tessier-Lavigne, M., 2003. Stereotyped pruning of long hippocampal axon branches triggered by retraction inducers of the semaphorin family. Cell 113, 285–299. Balice-Gordon, R.J., Lichtman, J.W., 1993. In vivo observations of preand postsynaptic changes during the transition from multiple to single innervation at developing neuromuscular junctions. J. Neurosci. 13, 834– 855. Bansal, A., Singer, J.H., Hwang, B.J., Xu, W., Beaudet, A., Feller, M.B., 2000. Mice lacking specific nicotinic acetylcholine receptor subunits exhibit dramatically altered spontaneous activity patterns and reveal a limited role for retinal waves in forming ON and OFF circuits in the inner retina. J. Neurosci. 20, 7672–7681. Bastmeyer, M., O’Leary, D.D., 1996. Dynamics of target recognition by interstitial axon branching along developing cortical axons. J. Neurosci. 16, 1450–1459. Beirowski, B., Berek, L., Adalbert, R., Wagner, D., Grumme, D.S., Addicks, K., Ribchester, R.R., Coleman, M.P., 2004. Quantitative and qualitative analysis of Wallerian degeneration using restricted axonal labelling in YFP-H mice. J. Neurosci. Methods 134, 23–35. Berger, A.R., Schaumburg, H.H., 1995. Human peripheral nerve disease (peripheral neuropathies). In: Waxman, S.G., Jeffery, D.K., Stys, P.K. (Eds.), The Axon: Structure, Function and Pathophysiolgy. Oxford University Press, New York, pp. 648–660. Bernstein, M., Lichtman, J.W., 1999. Axonal atrophy: the retraction reaction. Curr. Opin. Neurobiol. 9, 364–370. Billuart, P., Winter, C.G., Maresh, A., Zhao, X., Luo, L., 2001. Regulating axon branch stability: the role of p190 RhoGAP in repressing a retraction signaling pathway. Cell 107, 195–207. Bishop, D.L., Misgeld, T., Walsh, M.K., Gan, W.B., Lichtman, J.W., 2004. Axon branch removal at developing synapses by axosome shedding. Neuron 44, 651–661. Bito, H., Furuyashiki, T., Ishihara, H., Shibasaki, Y., Ohashi, K., Mizuno, K., Maekawa, M., Ishizaki, T., Narumiya, S., 2000. A critical role for a Rhoassociated kinase, p160ROCK, in determining axon outgrowth in mammalian CNS neurons. Neuron 26, 431–441. Bjartmar, C., Wujek, J.R., Trapp, B.D., 2003. Axonal loss in the pathology of MS: consequences for understanding the progressive phase of the disease. J. Neurol. Sci. 206, 165–171. Boillee, S., Vande Velde, C., Cleveland, D.W., 2006a. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron 52, 39–59. Boillee, S., Yamanaka, K., Lobsiger, C.S., Copeland, N.G., Jenkins, N.A., Kassiotis, G., Kollias, G., Cleveland, D.W., 2006b. Onset and progression in inherited ALS determined by motor neurons and microglia. Science 312, 1389–1392. Bommel, H., Xie, G., Rossoll, W., Wiese, S., Jablonka, S., Boehm, T., Sendtner, M., 2002. Missense mutation in the tubulin-specific chaperone E (Tbce) gene in the mouse mutant progressive motor neuronopathy, a model of human motoneuron disease. J. Cell Biol. 159, 563–569. Bruck, W., 2005. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J. Neurol. 252 (Suppl 5), v3–v9. Bruijn, L.I., Becher, M.W., Lee, M.K., Anderson, K.L., Jenkins, N.A., Copeland, N.G., Sisodia, S.S., Rothstein, J.D., Borchelt, D.R., Price, D.L., Cleveland, D.W., 1997. ALS-linked SOD1 mutant G85R mediates damage to astrocytes and promotes rapidly progressive disease with SOD1-containing inclusions. Neuron 18, 327–338. 15 Buffelli, M., Burgess, R.W., Feng, G., Lobe, C.G., Lichtman, J.W., Sanes, J.R., 2003. Genetic evidence that relative synaptic efficacy biases the outcome of synaptic competition. Nature 424, 430–434. Burke, R.E., 1994. Physiology of motor units. In: Engel, A.G., FranziniArmstrong, C. (Eds.), Myology. McGraw-Hill, New York, pp. 464–484. Campenot, R.B., 1982. Development of sympathetic neurons in compartmentalized cultures. II. Local control of neurite survival by nerve growth factor. Dev. Biol. 93, 13–21. Causing, C.G., Gloster, A., Aloyz, R., Bamji, S.X., Chang, E., Fawcett, J., Kuchel, G., Miller, F.D., 1997. Synaptic innervation density is regulated by neuron-derived BDNF. Neuron 18, 257–267. Cavanagh, J.B., 1964. The significance of the ‘‘dying back’’ process in experimental and human neurological disease. Int. Rev. Exp. Pathol. 3, 219–267. Chen, C., Regehr, W.G., 2000. Developmental remodeling of the retinogeniculate synapse. Neuron 28, 955–966. Clement, A.M., Nguyen, M.D., Roberts, E.A., Garcia, M.L., Boillee, S., Rule, M., McMahon, A.P., Doucette, W., Siwek, D., Ferrante, R.J., Brown Jr., R.H., Julien, J.P., Goldstein, L.S., Cleveland, D.W., 2003. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 302, 113–117. Coleman, M., 2005. Axon degeneration mechanisms: commonality amid diversity. Nat. Rev. Neurosci. 6, 889–898. Coleman, P.D., Yao, P.J., 2003. Synaptic slaughter in Alzheimer’s disease. Neurobiol. Aging 24, 1023–1027. Collard, J.F., Cote, F., Julien, J.P., 1995. Defective axonal transport in a transgenic mouse model of amyotrophic lateral sclerosis. Nature 375, 61–64. Conforti, L., Tarlton, A., Mack, T.G., Mi, W., Buckmaster, E.A., Wagner, D., Perry, V.H., Coleman, M.P., 2000. A Ufd2/D4Cole1e chimeric protein and overexpression of Rbp7 in the slow Wallerian degeneration (WldS) mouse. Proc. Natl. Acad. Sci. U.S.A. 97, 11377–11382. Conforti, L., Fang, G., Beirowski, B., Wang, M.S., Sorci, L., Asress, S., Adalbert, R., Silva, A., Bridge, K., Huang, X.P., Magni, G., Glass, J.D., Coleman, M.P., 2007. NAD(+) and axon degeneration revisited: Nmnat1 cannot substitute for Wld(S) to delay Wallerian degeneration. Cell Death Differ. 14, 116–127. De Paola, V., Holtmaat, A., Knott, G., Song, S., Wilbrecht, L., Caroni, P., Svoboda, K., 2006. Cell type-specific structural plasticity of axonal branches and boutons in the adult neocortex. Neuron 49, 861–875. Debski, E.A., Cline, H.T., 2002. Activity-dependent mapping in the retinotectal projection. Curr. Opin. Neurobiol. 12, 93–99. Delio, D.A., Fiori, M.G., Lowndes, H.E., 1992. Motor unit function during evolution of proximal axonal swellings. J. Neurol. Sci. 109, 30–40. DiAntonio, A., Haghighi, A.P., Portman, S.L., Lee, J.D., Amaranto, A.M., Goodman, C.S., 2001. Ubiquitination-dependent mechanisms regulate synaptic growth and function. Nature 412, 449–452. Eaton, B.A., Fetter, R.D., Davis, G.W., 2002. Dynactin is necessary for synapse stabilization. Neuron 34, 729–741. Eckenhoff, M.F., Pysh, J.J., 1979. Double-walled coated vesicle formation: evidence for massive and transient conjugate internalization of plasma membranes during cerebellar development. J. Neurocytol. 8, 623–638. Feller, M.B., 2002. The role of nAChR-mediated spontaneous retinal activity in visual system development. J. Neurobiol. 53, 556–567. Ferri, A., Sanes, J.R., Coleman, M.P., Cunningham, J.M., Kato, A.C., 2003. Inhibiting axon degeneration and synapse loss attenuates apoptosis and disease progression in a mouse model of motoneuron disease. Curr. Biol. 13, 669–673. Finn, J.T., Weil, M., Archer, F., Siman, R., Srinivasan, A., Raff, M.C., 2000. Evidence that Wallerian degeneration and localized axon degeneration induced by local neurotrophin deprivation do not involve caspases. J. Neurosci. 20, 1333–1341. Fischer, L.R., Culver, D.G., Davis, A.A., Tennant, P., Wang, M., Coleman, M., Asress, S., Adalbert, R., Alexander, G.M., Glass, J.D., 2005. The WldS gene modestly prolongs survival in the SOD1G93A fALS mouse. Neurobiol. Dis. 19, 293–300. Fischer, L.R., Culver, D.G., Tennant, P., Davis, A.A., Wang, M., CastellanoSanchez, A., Khan, J., Polak, M.A., Glass, J.D., 2004. Amyotrophic lateral Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 16 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx sclerosis is a distal axonopathy: evidence in mice and man. Exp. Neurol. 185, 232–240. Frey, D., Schneider, C., Xu, L., Borg, J., Spooren, W., Caroni, P., 2000. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J. Neurosci. 20, 2534–2542. Galimberti, I., Gogolla, N., Alberi, S., Santos, A.F., Muller, D., Caroni, P., 2006. Long-term rearrangements of hippocampal mossy fiber terminal connectivity in the adult regulated by experience. Neuron 50, 749–763. Gao, P.P., Yue, Y., Cerretti, D.P., Dreyfus, C., Zhou, R., 1999. Ephrin-dependent growth and pruning of hippocampal axons. Proc. Natl. Acad. Sci. U.S.A. 96, 4073–4077. Garcia-Fresco, G.P., Sousa, A.D., Pillai, A.M., Moy, S.S., Crawley, J.N., Tessarollo, L., Dupree, J.L., Bhat, M.A., 2006. Disruption of axo-glial junctions causes cytoskeletal disorganization and degeneration of Purkinje neuron axons. Proc. Natl. Acad. Sci. U.S.A. 103, 5137–5142. Gillingwater, T.H., Wishart, T.M., Chen, P.E., Haley, J.E., Robertson, K., MacDonald, S.H., Middleton, S., Wawrowski, K., Shipston, M.J., Melmed, S., Wyllie, D.J., Skehel, P.A., Coleman, M.P., Ribchester, R.R., 2006. The neuroprotective WldS gene regulates expression of PTTG1 and erythroid differentiation regulator 1-like gene in mice and human cells. Hum. Mol. Genet. 15, 625–635. Glass, J.D., Brushart, T.M., George, E.B., Griffin, J.W., 1993. Prolonged survival of transected nerve fibres in C57BL/Ola mice is an intrinsic characteristic of the axon. J. Neurocytol. 22, 311–321. Gotz, J., Ittner, L.M., Kins, S., 2006. Do axonal defects in tau and amyloid precursor protein transgenic animals model axonopathy in Alzheimer’s disease? J. Neurochem. 98, 993–1006. Gould, T.W., Buss, R.R., Vinsant, S., Prevette, D., Sun, W., Knudson, C.M., Milligan, C.E., Oppenheim, R.W., 2006. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J. Neurosci. 26, 8774–8786. Griffin, J.W., Georges, E.B., Hsieh, S.T., Glass, J.D., 1995. Axonal degeneration and disorders of the axonal cytoskeleton. In: Waxman, S.G., Jeffery, D.K., Stys, P.K. (Eds.), The Axon: Structure, Function and Pathophysiology. Oxford University Press, New York, pp. 375–390. Gunawardena, S., Goldstein, L.S., 2001. Disruption of axonal transport and neuronal viability by amyloid precursor protein mutations in Drosophila. Neuron 32, 389–401. Gunawardena, S., Goldstein, L.S., 2004. Cargo-carrying motor vehicles on the neuronal highway: transport pathways and neurodegenerative disease. J. Neurobiol. 58, 258–271. Gunawardena, S., Goldstein, L.S., 2005. Polyglutamine diseases and transport problems: deadly traffic jams on neuronal highways. Arch. Neurol. 62, 46– 51. Gunawardena, S., Her, L.S., Brusch, R.G., Laymon, R.A., Niesman, I.R., Gordesky-Gold, B., Sintasath, L., Bonini, N.M., Goldstein, L.S., 2003. Disruption of axonal transport by loss of huntingtin or expression of pathogenic polyQ proteins in Drosophila. Neuron 40, 25–40. Gurney, M.E., Pu, H., Chiu, A.Y., Dal Canto, M.C., Polchow, C.Y., Alexander, D.D., Caliendo, J., Hentati, A., Kwon, Y.W., Deng, H.X., et al., 1994. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 264, 1772–1775. Hasbani, D.M., O’Malley, K.L., 2006. Wld(S) mice are protected against the Parkinsonian mimetic MPTP. Exp. Neurol. 202, 93–99. Hindges, R., McLaughlin, T., Genoud, N., Henkemeyer, M., O’Leary, D.D., 2002. EphB forward signaling controls directional branch extension and arborization required for dorsal-ventral retinotopic mapping. Neuron 35, 475–487. Hoopfer, E.D., McLaughlin, T., Watts, R.J., Schuldiner, O., O’Leary, D.D., Luo, L., 2006. Wlds protection distinguishes axon degeneration following injury from naturally occurring developmental pruning. Neuron 50, 883–895. Innocenti, G.M., 1981. Growth and reshaping of axons in the establishment of visual callosal connections. Science 212, 824–827. Innocenti, G.M., Clarke, S., Koppel, H., 1983. Transitory macrophages in the white matter of the developing visual cortex. II. Development and relations with axonal pathways. Brain Res. 313, 55–66. Innocenti, G.M., 1995. Exuberant development of connections, and its possible permissive role in cortical evolution. Trends Neurosci. 18, 397–402. Innocenti, G.M., Kiper, D.C., Knyazeva, M.G., Deonna, T.W., 1999. On nature and limits of cortical developmental plasticity after an early lesion, in a child. Restor. Neurol. Neurosci. 15, 219–227. Kasthuri, N., Lichtman, J.W., 2003. The role of neuronal identity in synaptic competition. Nature 424, 426–430. Kasthuri, N., Lichtman, J.W., 2004. Structural dynamics of synapses in living animals. Curr. Opin. Neurobiol. 14, 105–111. Katz, L.C., Shatz, C.J., 1996. Synaptic activity and the construction of cortical circuits. Science 274, 1133–1138. Kerschensteiner, M., Schwab, M.E., Lichtman, J.W., Misgeld, T., 2005. In vivo imaging of axonal degeneration and regeneration in the injured spinal cord. Nat. Med. 11, 572–577. King, M.E., Kan, H.M., Baas, P.W., Erisir, A., Glabe, C.G., Bloom, G.S., 2006. Tau-dependent microtubule disassembly initiated by prefibrillar {beta}amyloid. J. Cell Biol. 175, 541–546. Kozma, R., Sarner, S., Ahmed, S., Lim, L., 1997. Rho family GTPases and neuronal growth cone remodelling: relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell. Biol. 17, 1201–1211. Kuo, C.T., Zhu, S., Younger, S., Jan, L.Y., Jan, Y.N., 2006. Identification of E2/ E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron 51, 283–290. LaMonte, B.H., Wallace, K.E., Holloway, B.A., Shelly, S.S., Ascano, J., Tokito, M., Van Winkle, T., Howland, D.S., Holzbaur, E.L., 2002. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing lateonset progressive degeneration. Neuron 34, 715–727. Lamprecht, R., Farb, C.R., LeDoux, J.E., 2002. Fear memory formation involves p190 RhoGAP and ROCK proteins through a GRB2-mediated complex. Neuron 36, 727–738. Lee, T., Luo, L., 1999. Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461. Lee, T., Marticke, S., Sung, C., Robinow, S., Luo, L., 2000. Cell-autonomous requirement of the USP/EcR-B ecdysone receptor for mushroom body neuronal remodeling in Drosophila. Neuron 28, 807–818. LeMaster, A.M., Krimm, R.F., Davis, B.M., Noel, T., Forbes, M.E., Johnson, J.E., Albers, K.M., 1999. Overexpression of brain-derived neurotrophic factor enhances sensory innervation and selectively increases neuron number. J. Neurosci. 19, 5919–5931. Levine, R.B., Morton, D.B., Restifo, L.L., 1995. Remodeling of the insect nervous system. Curr. Opin. Neurobiol. 5, 28–35. Li, H., Li, S.H., Yu, Z.X., Shelbourne, P., Li, X.J., 2001. Huntingtin aggregateassociated axonal degeneration is an early pathological event in Huntington’s disease mice. J. Neurosci. 21, 8473–8481. Libby, R.T., Li, Y., Savinova, O.V., Barter, J., Smith, R.S., Nickells, R.W., John, S.W., 2005. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 1, 17–26. Lichtman, J.W., Colman, H., 2000. Synapse elimination and indelible memory. Neuron 25, 269–278. Linker, R.A., Sendtner, M., Gold, R., 2005. Mechanisms of axonal degeneration in EAE–lessons from CNTF and MHC I knockout mice. J. Neurol. Sci. 233, 167–172. Lino, M.M., Schneider, C., Caroni, P., 2002. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 22, 4825–4832. Lobsiger, C.S., Garcia, M.L., Ward, C.M., Cleveland, D.W., 2005. Altered axonal architecture by removal of the heavily phosphorylated neurofilament tail domains strongly slows superoxide dismutase 1 mutant-mediated ALS. Proc. Natl. Acad. Sci. U.S.A. 102, 10351–10356. Lohof, A.M., Delhaye-Bouchaud, N., Mariani, J., 1996. Synapse elimination in the central nervous system: functional significance and cellular mechanisms. Rev. Neurosci. 7, 85–101. Low, L.K., Cheng, H.J., 2005. A little nip and tuck: axon refinement during development and axonal injury. Curr. Opin. Neurobiol. 15, 549–556. Luo, L., O’Leary, D.D., 2005. Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci. 28, 127–156. MacDonald, J.M., Beach, M.G., Porpiglia, E., Sheehan, A.E., Watts, R.J., Freeman, M.R., 2006. The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50, 869–881. Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx MacInnis, B.L., Campenot, R.B., 2005. Regulation of Wallerian degeneration and nerve growth factor withdrawal-induced pruning of axons of sympathetic neurons by the proteasome and the MEK/Erk pathway. Mol. Cell. Neurosci. 28, 430–439. Mack, T.G., Reiner, M., Beirowski, B., Mi, W., Emanuelli, M., Wagner, D., Thomson, D., Gillingwater, T., Court, F., Conforti, L., Fernando, F.S., Tarlton, A., Andressen, C., Addicks, K., Magni, G., Ribchester, R.R., Perry, V.H., Coleman, M.P., 2001. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 4, 1199–1206. Markus, A., Patel, T.D., Snider, W.D., 2002. Neurotrophic factors and axonal growth. Curr. Opin. Neurobiol. 12, 523–531. Martin, N., Jaubert, J., Gounon, P., Salido, E., Haase, G., Szatanik, M., Guenet, J.L., 2002. A missense mutation in Tbce causes progressive motor neuronopathy in mice. Nat. Genet. 32, 443–447. Mason, C.A., Gregory, E., 1984. Postnatal maturation of cerebellar mossy and climbing fibers: transient expression of dual features on single axons. J. Neurosci. 4, 1715–1735. McLaughlin, T., Hindges, R., O’Leary, D.D., 2003a. Regulation of axial patterning of the retina and its topographic mapping in the brain. Curr. Opin. Neurobiol. 13, 57–69. McLaughlin, T., Torborg, C.L., Feller, M.B., O’Leary, D.D., 2003b. Retinotopic map refinement requires spontaneous retinal waves during a brief critical period of development. Neuron 40, 1147–1160. Meister, M., Wong, R.O., Baylor, D.A., Shatz, C.J., 1991. Synchronous bursts of action potentials in ganglion cells of the developing mammalian retina. Science 252, 939–943. Mi, W., Beirowski, B., Gillingwater, T.H., Adalbert, R., Wagner, D., Grumme, D., Osaka, H., Conforti, L., Arnhold, S., Addicks, K., Wada, K., Ribchester, R.R., Coleman, M.P., 2005. The slow Wallerian degeneration gene, WldS, inhibits axonal spheroid pathology in gracile axonal dystrophy mice. Brain 128, 405–416. Miledi, R., Slater, C.R., 1970. On the degeneration of rat neuromuscular junctions after nerve section. J. Physiol. 207, 507–528. Monani, U.R., 2005. Spinal muscular atrophy: a deficiency in a ubiquitous protein; a motor neuron-specific disease. Neuron 48, 885–896. O’Leary, D.D., 1992. Development of connectional diversity and specificity in the mammalian brain by the pruning of collateral projections. Curr. Opin. Neurobiol. 2, 70–77. O’Leary, D.D., Koester, S.E., 1993. Development of projection neuron types, axon pathways, and patterned connections of the mammalian cortex. Neuron 10, 991–1006. O’Leary, D.D., Stanfield, B.B., 1989. Selective elimination of axons extended by developing cortical neurons is dependent on regional locale: experiments utilizing fetal cortical transplants. J. Neurosci. 9, 2230–2246. O’Leary, D.D., Terashima, T., 1988. Cortical axons branch to multiple subcortical targets by interstitial axon budding: implications for target recognition and ‘‘waiting periods’’. Neuron 1, 901–910. O’Rourke, N.A., Cline, H.T., Fraser, S.E., 1994. Rapid remodeling of retinal arbors in the tectum with and without blockade of synaptic transmission. Neuron 12, 921–934. Parson, S.H., Mackintosh, C.L., Ribchester, R.R., 1997. Elimination of motor nerve terminals in neonatal mice expressing a gene for slow Wallerian degeneration (C57Bl/Wlds). Eur. J. Neurosci. 9, 1586–1592. Patel, T.D., Kramer, I., Kucera, J., Niederkofler, V., Jessell, T.M., Arber, S., Snider, W.D., 2003. Peripheral NT3 signaling is required for ETS protein expression and central patterning of proprioceptive sensory afferents. Neuron 38, 403–416. Penn, A.A., Riquelme, P.A., Feller, M.B., Shatz, C.J., 1998. Competition in retinogeniculate patterning driven by spontaneous activity. Science 279, 2108–2112. Personius, K.E., Balice-Gordon, R.J., 2001. Loss of correlated motor neuron activity during synaptic competition at developing neuromuscular synapses. Neuron 31, 395–408. Petersson, P., Waldenstrom, A., Fahraeus, C., Schouenborg, J., 2003. Spontaneous muscle twitches during sleep guide spinal self-organization. Nature 424, 72–75. Pirozzi, M., Quattrini, A., Andolfi, G., Dina, G., Malaguti, M.C., Auricchio, A., Rugarli, E.I., 2006. Intramuscular viral delivery of paraplegin rescues 17 peripheral axonopathy in a model of hereditary spastic paraplegia. J. Clin. Invest. 116, 202–208. Portera-Cailliau, C., Weimer, R.M., De Paola, V., Caroni, P., Svoboda, K., 2005. Diverse modes of axon elaboration in the developing neocortex. PLoS Biol. 3, e272. Pun, S., Santos, A.F., Saxena, S., Xu, L., Caroni, P., 2006. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat. Neurosci. 9, 408–419. Qiang, L., Yu, W., Andreadis, A., Luo, M., Baas, P.W., 2006. Tau protects microtubules in the axon from severing by katanin. J. Neurosci. 26, 3120– 3129. Raff, M.C., Whitmore, A.V., Finn, J.T., 2002. Axonal self-destruction and neurodegeneration. Science 296, 868–871. Ruthazer, E.S., Akerman, C.J., Cline, H.T., 2003. Control of axon branch dynamics by correlated activity in vivo. Science 301, 66–70. Sagot, Y., Dubois-Dauphin, M., Tan, S.A., de Bilbao, F., Aebischer, P., Martinou, J.C., Kato, A.C., 1995. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J. Neurosci. 15, 7727–7733. Sagot, Y., Rosse, T., Vejsada, R., Perrelet, D., Kato, A.C., 1998. Differential effects of neurotrophic factors on motoneuron retrograde labeling in a murine model of motoneuron disease. J. Neurosci. 18, 1132–1141. Sagot, Y., Tan, S.A., Hammang, J.P., Aebischer, P., Kato, A.C., 1996. GDNF slows loss of motoneurons but not axonal degeneration or premature death of pmn/pmn mice. J. Neurosci. 16, 2335–2341. Sajadi, A., Schneider, B.L., Aebischer, P., 2004. Wlds-mediated protection of dopaminergic fibers in an animal model of Parkinson disease. Curr. Biol. 14, 326–330. Samsam, M., Mi, W., Wessig, C., Zielasek, J., Toyka, K.V., Coleman, M.P., Martini, R., 2003. The Wlds mutation delays robust loss of motor and sensory axons in a genetic model for myelin-related axonopathy. J. Neurosci. 23, 2833–2839. Schaefer, A.M., Sanes, J.R., Lichtman, J.W., 2005. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 490, 209–219. Schmalbruch, H., Jensen, H.J., Bjaerg, M., Kamieniecka, Z., Kurland, L., 1991. A new mouse mutant with progressive motor neuronopathy. J. Neuropathol. Exp. Neurol. 50, 192–204. Schubiger, M., Wade, A.A., Carney, G.E., Truman, J.W., Bender, M., 1998. Drosophila EcR-B ecdysone receptor isoforms are required for larval molting and for neuron remodeling during metamorphosis. Development 125, 2053–2062. Stanfield, B.B., Nahin, B.R., O’Leary, D.D., 1987. A transient postmamillary component of the rat fornix during development: implications for interspecific differences in mature axonal projections. J. Neurosci. 7, 3350– 3361. Stanfield, B.B., O’Leary, D.D., 1985a. Fetal occipital cortical neurones transplanted to the rostral cortex can extend and maintain a pyramidal tract axon. Nature 313, 135–137. Stanfield, B.B., O’Leary, D.D., 1985b. The transient corticospinal projection from the occipital cortex during the postnatal development of the rat. J. Comp. Neurol. 238, 236–248. Stanfield, B.B., O’Leary, D.D., Fricks, C., 1982. Selective collateral elimination in early postnatal development restricts cortical distribution of rat pyramidal tract neurones. Nature 298, 371–373. Stellwagen, D., Shatz, C.J., 2002. An instructive role for retinal waves in the development of retinogeniculate connectivity. Neuron 33, 357–367. Stokin, G.B., Goldstein, L.S., 2006. Axonal transport and Alzheimer’s disease. Annu. Rev. Biochem. 75, 607–627. Stokin, G.B., Lillo, C., Falzone, T.L., Brusch, R.G., Rockenstein, E., Mount, S.L., Raman, R., Davies, P., Masliah, E., Williams, D.S., Goldstein, L.S., 2005. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 307, 1282–1288. Technau, G., Heisenberg, M., 1982. Neural reorganization during metamorphosis of the corpora pedunculata in Drosophila melanogaster. Nature 295, 405–407. Tian, N., Copenhagen, D.R., 2003. Visual stimulation is required for refinement of ON and OFF pathways in postnatal retina. Neuron 39, 85–96. Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007 + Models PRONEU-845; No of Pages 18 18 S. Saxena, P. Caroni / Progress in Neurobiology xxx (2007) xxx–xxx Tissot, M., Stocker, R.F., 2000. Metamorphosis in Drosophila and other insects: the fate of neurons throughout the stages. Prog. Neurobiol. 62, 89–111. Tsai, J., Grutzendler, J., Duff, K., Gan, W.B., 2004. Fibrillar amyloid deposition leads to local synaptic abnormalities and breakage of neuronal branches. Nat. Neurosci. 7, 1181–1183. Vande Velde, C., Garcia, M.L., Yin, X., Trapp, B.D., Cleveland, D.W., 2004. The neuroprotective factor Wlds does not attenuate mutant SOD1-mediated motor neuron disease. Neuromol. Med. 5, 193–203. Waller, A., 1850. Experiments on the sections of glossopharyngeal and hypoglossal nerves of the frog and observations of the alterations produced thereby in the structure of their primitive fibers. Phil. Trans. R. Soc. Lond. 140, 423–429. Walsh, M.K., Lichtman, J.W., 2003. In vivo time-lapse imaging of synaptic takeover associated with naturally occurring synapse elimination. Neuron 37, 67–73. Watts, R.J., Hoopfer, E.D., Luo, L., 2003. Axon pruning during Drosophila metamorphosis: evidence for local degeneration and requirement of the ubiquitin-proteasome system. Neuron 38, 871–885. Watts, R.J., Schuldiner, O., Perrino, J., Larsen, C., Luo, L., 2004. Glia engulf degenerating axons during developmental axon pruning. Curr. Biol. 14, 678–684. Weimann, J.M., Zhang, Y.A., Levin, M.E., Devine, W.P., Brulet, P., McConnell, S.K., 1999. Cortical neurons require Otx1 for the refinement of exuberant axonal projections to subcortical targets. Neuron 24, 819–831. Williams, D.W., Truman, J.W., 2005. Cellular mechanisms of dendrite pruning in Drosophila: insights from in vivo time-lapse of remodeling dendritic arborizing sensory neurons. Development 132, 3631–3642. Williams, D.W., Kondo, S., Krzyzanowska, A., Hiromi, Y., Truman, J.W., 2006. Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci. 9, 1234–1236. Williamson, T.L., Cleveland, D.W., 1999. Slowing of axonal transport is a very early event in the toxicity of ALS-linked SOD1 mutants to motor neurons. Nat. Neurosci. 2, 50–56. Wong, P.C., Pardo, C.A., Borchelt, D.R., Lee, M.K., Copeland, N.G., Jenkins, N.A., Sisodia, S.S., Cleveland, D.W., Price, D.L., 1995. An adverse property of a familial ALS-linked SOD1 mutation causes motor neuron disease characterized by vacuolar degeneration of mitochondria. Neuron 14, 1105– 1116. Wong, R.O., Meister, M., Shatz, C.J., 1993. Transient period of correlated bursting activity during development of the mammalian retina. Neuron 11, 923–938. Xia, C.H., Roberts, E.A., Her, L.S., Liu, X., Williams, D.S., Cleveland, D.W., Goldstein, L.S., 2003. Abnormal neurofilament transport caused by targeted disruption of neuronal kinesin heavy chain KIF5A. J. Cell Biol. 161, 55–66. Yamada, K.M., Spooner, B.S., Wessells, N.K., 1970. Axon growth: roles of microfilaments and microtubules. Proc. Natl. Acad. Sci. U.S.A. 66, 1206– 1212. Yates, P.A., Roskies, A.L., McLaughlin, T., O’Leary, D.D., 2001. Topographicspecific axon branching controlled by ephrin-As is the critical event in retinotectal map development. J. Neurosci. 21, 8548–8563. Yu, W., Solowska, J.M., Qiang, L., Karabay, A., Baird, D., Baas, P.W., 2005. Regulation of microtubule severing by katanin subunits during neuronal development. J. Neurosci. 25, 5573–5583. Zhai, Q., Wang, J., Kim, A., Liu, Q., Watts, R., Hoopfer, E., Mitchison, T., Luo, L., He, Z., 2003. Involvement of the ubiquitin-proteasome system in the early stages of Wallerian degeneration. Neuron 39, 217–225. Zhang, B., Tu, P., Abtahian, F., Trojanowski, J.Q., Lee, V.M., 1997. Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J. Cell Biol. 139, 1307–1315. Zhang, Y.A., Okada, A., Lew, C.H., McConnell, S.K., 2002. Regulated nuclear trafficking of the homeodomain protein otx1 in cortical neurons. Mol. Cell. Neurosci. 19, 430–446. Zhou, Z., Hartwieg, E., Horvitz, H.R., 2001. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell 104, 43–56. Zufferey, P.D., Jin, F., Nakamura, H., Tettoni, L., Innocenti, G.M., 1999. The role of pattern vision in the development of cortico-cortical connections. Eur. J. Neurosci. 11, 2669–2688. Please cite this article in press as: Saxena, S., Caroni, P., Mechanisms of axon degeneration: From development to disease, Prog. Neurobiol. (2007), doi:10.1016/j.pneurobio.2007.07.007
























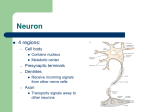
![Neuron [or Nerve Cell]](http://s1.studyres.com/store/data/000229750_1-5b124d2a0cf6014a7e82bd7195acd798-150x150.png)



