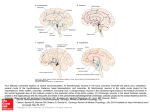
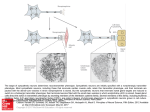
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Holonomic brain theory wikipedia , lookup
Limbic system wikipedia , lookup
Development of the nervous system wikipedia , lookup
Feature detection (nervous system) wikipedia , lookup
Nervous system network models wikipedia , lookup
Cognitive neuroscience wikipedia , lookup
Synaptic gating wikipedia , lookup
Nerve growth factor wikipedia , lookup
Signal transduction wikipedia , lookup
Neuromuscular junction wikipedia , lookup
Activity-dependent plasticity wikipedia , lookup
Haemodynamic response wikipedia , lookup
Synaptogenesis wikipedia , lookup
Environmental enrichment wikipedia , lookup
Neuroanatomy wikipedia , lookup
De novo protein synthesis theory of memory formation wikipedia , lookup
Metastability in the brain wikipedia , lookup
Epigenetics in learning and memory wikipedia , lookup
Endocannabinoid system wikipedia , lookup
Neurogenomics wikipedia , lookup
Aging brain wikipedia , lookup
Molecular neuroscience wikipedia , lookup
Channelrhodopsin wikipedia , lookup
Alzheimer's disease wikipedia , lookup
Optogenetics wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Boston University OpenBU http://open.bu.edu Theses & Dissertations Boston University Theses & Dissertations 2013 Molecular and morphological analyses of basal forebrain cholinergic neurons in mouse models of aging and Alzheimer's disease Norman, Timothy Alfred Jr. Boston University http://hdl.handle.net/2144/12173 Boston University BOSTON UNIVERSITY SCHOOL OF MEDICINE Thesis MOLECULAR AND MORPHOLOGICAL ANALYSES OF BASAL FOREBRAIN CHOLINERGIC NEURONS IN MOUSE MODELS OF AGING AND ALZHEIMER’S DISEASE By TIMOTHY ALFRED NORMAN JR. B.S., Biology, University of North Carolina at Wilmington, 2005 Submitted in partial fulfillment of the requirements for the degree of Master of Arts 2013 Approved by First Reader _____________________________________________ Jan Krzysztof Blusztajn, Ph.D. Professor, Pathology and Laboratory Medicine Second Reader _____________________________________________ Chris Andry, Ph.D. Associate Professor, Pathology and Laboratory Medicine ACKNOWLEDGEMENTS Many sincere thanks to my PI and mentor, Dr. Jan Blusztajn for the opportunity to conduct research in his laboratory. Through his excellent instruction and example, I am confident in my ability to become an independent scientist and make future discoveries and contributions to the scientific community. I would also like to acknowledge Dr. Blusztajn and Dr. Chris Andry for their advice, support, guidance and encouragement throughout my master’s research and didactic studies. A special thanks to Dr. Nader Rahimi for inspiring vision and emphasizing critical thinking and for going the extra mile to challenge his students. These lessons will undoubtedly aid me in my future endeavors. Most certainly without Dr. Tarik Haydar and Dr. William Tyler, Joe Goodliffe and Nadine Aziz, my experiments would not have been possible. They generously provided technical assistance with confocal imaging, as well as with the Volocity data analysis software. For this I am extremely grateful. Last but not least, I would like to sincerely thank my best friend and wife, Laura Neubauer-Norman. She has proven her love, her loyalty and her patience in supporting me over these last nine years. Thanks for believing in me and I enjoy each day that we get to discover life together. I wish you all the best that life has to offer. iii MOLECULAR AND MORPHOLOGICAL ANALYSES OF BASAL FOREBRAIN CHOLINERGIC NEURONS IN MOUSE MODELS OF AGING AND ALZHEIMER’S DISEASE TIMOTHY ALFRED. NORMAN JR. Boston University School of Medicine, 2013 Major Professor: Jan Krzysztof Blusztajn, PhD., Professor of Pathology and Laboratory Medicine ABSTRACT Basal forebrain cholinergic neurons (BFCNs) of the medial septal nuclei, the diagonal bands of Broca and the nucleus basalis magnocellularis synthesize acetylcholine (ACh) and their projections extend to the cerebral cortex, hippocampus and the amygdala. ACh neurotransmission is essential for learning, attention, memory, arousal and sleep. BFCNs are dependent on a regulated neurochemical environment for the induction, development, maturation and maintenance of their phenotype and viability. However, events that compromise this neurochemical environment can contribute to BFCN dysfunction and/or degeneration, decreased ACh levels and disrupted brain function. During normal aging and Alzheimer’s disease (AD) BFCNs become more vulnerable to dysfunction due to trophic factor withdrawal, cell signaling impairments and other cytopathologic changes. AD is characterized by the deposition of Amyloid-beta (Aβ) plaques and neurofibrillary Tau tangles (NFTs) in the cortex and hippocampus. These pathological AD hallmarks overlap with cortical and iv hippocampal cholinergic dysfunction, implicating both as the drivers of cognitive and behavioral decline associated with AD. Since, BFCNs are highly vulnerable to AD pathophysiology, factors that support the BFCN phenotype may have practical use in preserving neuronal networks and cognitive function. There is now strong evidence that bone morphogenetic protein-9 (BMP9 also known as growth/differentiating factor 2, GDF2) acts as an induction and maintenance factor that regulates BFCN differentiation in-vitro and in-vivo. Here we used transgenic mice that express green fluorescent protein (GFP) in BFCNs to make observations concerning the BFCN phenotype in aging and AD. We found qualitative evidence that BFCNs of the 24-month old WT/ChAT-GFP mice were smaller and more rounded with shorter processes when compared to 6month old mouse BFCNs. We analyzed the effects of intracerebroventricular infusion of BMP9 on BFCN projection fibers in the APPswe/PS1dE9 mouse model of AD using laser scanning confocal microscopy. BMP9 not only increased the density of cholinergic projection fibers in the hippocampus of wild type and AD model APPswe/PS1dE9 mice, but it also reduced the plaque burden in the hippocampus of the AD mouse model. These data indicate that BMP9 ameliorated two major pathophysiologic hallmarks of AD, observable in these transgenic APPswe/PS1dE9 mice. BMP9 reduced Aβ plaque burden in this AD model, and enhanced the outgrowth and viability of cholinergic fibers within the hippocampus of both wild-type and APPswe/PS1dE9 mice. v Prior studies in our laboratory identified 300 genes differentially expressed in the BFCNs purified from APPswe/PS1dE9 mice as compared to their wild type littermates. One of these, the cellular prion protein (PrPc), displayed a 2.8-fold increase in mRNA expression in APPswe/PS1dE9 mice over wild-type controls. Our analysis of PrPc expression by quantitative immunohistochemistry revealed a 46% increase in the number of BFCNs expressing PrPc protein medial septum and vertical and horizontal limbs of the diagonal band of Broca (p=0.0007), but not in the striatum of the APPswe/PS1dE9 model of AD. Recently, studies have shown that direct binding of PrPc is required for Aβ neurotoxicity, synaptic impairment and cognitive dysfunction. Based on our results as well as others, it is possible that high PrPc expression in the BFCNs could be responsible for at least part of the cholinergic deficits observed in AD. Studies have also shown that PrPc is a receptor for Aβ and that this Aβ-PrPc interaction leads to pathologic tau phosphorylation and toxicity providing evidence that all three major hypotheses of AD: the Aβ-hypothesis, the tau hypothesis and the cholinergic hypothesis converge toward PrPc. vi PREFACE The brain is composed of billions of highly organized glial cells and neurons that receive, integrate, store and export information to interconnected subpopulations of neurons. It is the command center of the organism that processes a wide variety of sensorimotor stimuli and higher executive functions such as cognition, memory, and emotions that govern behavior. Brain regions that control these functions and the subpopulations of neurons residing therein can be distinguished by their molecular and morphological characteristics. These phenotypic traits are established by specific gene expression profiles which direct the differentiation and maturation of individual neuronal subclasses. For example, basal forebrain cholinergic neurons can be distinguished by a specific structural morphology and molecular phenotype. The functional extent of the cholinergic system can be observed in many diverse locales throughout the vertebrate organism including the central nervous, cardiovascular, digestive, respiratory and motor systems. Thus a comprehensive understanding of the cholinergic network is essential in understanding organismal biology. Here, this study focuses on the basal forebrain cholinergic neurons and their fibers located in an area of central nervous system long known to be affected by AD. Initially, I will synthesize a comprehensive background review on the developmental, anatomical and molecular phenotype of cholinergic neurons in the basal forebrain. Results pertaining to the molecular and morphological analyses of BFCNs as a function of aging and AD-like pathology, were obtained using a vii combination of immunohistochemistry and confocal microscopic techniques. If these initial discoveries are consistent and repeatable, they may contribute to the development and implementation of a safe and useful intervention for AD. viii TABLE OF CONTENTS Title Page i Approval Page ii Acknowledgements iii Abstract iv Preface vii List of Figures xi List of Abbreviations xii Introduction 1 BFCN Development 3 BMP9 Regulates the BFCN Phenotype 7 BFCN Phenotype 12 Nerve Growth Factor and its Receptors 19 Alzheimer’s Disease 26 Aβ Hypothesis of AD 31 Aβ Targeted Therapeutics 32 Aβ Disrupts Multiple Networks in AD 34 Tau Hypothesis of AD 37 Cholinergic Hypothesis of AD 39 Cholinergic Therapeutics 42 Materials and Methods 44 Transgenic mice 44 ix Tissue Processing 45 Immunofluorescence 46 Confocal Microscopy 48 Image Analysis 48 Results 50 BMP9 Improves AD-like Pathology In-vivo 50 BFCN Gene Expression in APP/CHGFP mice 58 PrPc is Upregulated in BFCNs of APP/CHGFP mice 65 Discussion 70 List of Journal Abbreviations 79 References 80 Curriculum Vita 91 x LIST OF FIGURES Figure 1. Factors influencing BFCN development 10 Figure 2. Basal forebrain and cholinergic projection anatomy 11 Figure 3. Cholinergic gene locus 13 Figure 4. BFCN morphological changes in aged WT/CHGFP mice 17 Figure 5. BFCN morphological changes in APP/CHGFP mice 18 Figure 6. p75/NTR and ChATGFP expression in MSN/VDB 21 Figure 7. Quantification of p75/NTR and ChAT colocalization in WT/CHGFP 25 Figure 8. PET scan image of Aβ in AD brain 29 Figure 9. Aβ disrupts multiple networks in the brain 35 Figure 10. Amyloidosis and Cholinergic deficit in APP/CHGFP 41 Figure 11. Abnormal BFCN morphology in APP/CHGFP 42 Figure 12. BMP9 infusion increases HPC cholinergic innervation 52 Figure 13. BMP9 infusion increases CA1 and hDG cholinergic innervation 53 Figure 14. BMP9 may increase cholinergic branching and extention 56 Figure 15. BMP9 infusion improves the cholinergic network in HPC 57 Figure 16. FACS purification of BFCNs 60 Figure 17. Microarray cluster analysis of WT and APP.PS1 BFCNs 63 Figure 18. Differential mRNA expression in WT and APP.PS1 BFCNs 64 Figure 19. Orthoslice analysis of PrPc/CHGFP colocalization method 69 Figure 20. PrPc is upregulated in APP/CHGFP BFCNs of MSN/VDB/HDB 73 Figure 21. PrPc at the center of AD 77 xi LIST OF ABBREVIATIONS Aβ Amyloid-Beta ACh Acetylcholine AD Alzheimer’s disease APP Amyloid Precursor Protein BACE Beta-Amyloid Converting Enzyme BFCN Basal Forebrain Cholinergic Neurons BLA Basolateral Amygdala BMP9 Bone Morphogenetic Protein 9 BSA Bovine Serum Albumin CA Cornu Ammonis CGL Cholinergic Gene Locus ChAT Choline Acetyltransferase ChT Choline Transporter 1 CNS Central Nervous System CTX Cortex FGF Fibroblast Growth Factor GABA Gamma-Aminobutyric Acid hDG (hilus of) Dentate Gyrus hESC (human) Embryonic Stem Cells DMSO Dimethyl Sulfoxide eGFP enhanced Green Fluorescent Protein xii FACS Flourescence Activated Cell Sorting HDB Horizontal Limb of the Diagonal Band of Broca HPC Hippocampus ICV Intracerebroventricular LGE Lateral Ganglionic Eminence MGE Medial Ganglionic Eminence mRNA (messenger) Ribonucleic Acid MSN Medial Septal Nucleus NBM Nucleus Basalis of Meynert NGF Nerve Growth Factor NT-3 Neurotrophin 3 OB Olfactory Bulb PBS Phosphate Buffered Saline PS1 Presenilin 1 pAB Polyclonal Antibody POA Preoptic Area PrPc Cellular Prion Protein PrPsc Scrapie Prion Protein RE Response Element SHH Sonic Hedgehog SR Stratum Radiatum Moleculare (of the Hippocampus) STR Striatum xiii VAChT Vesicular Acetylcholine Transporter VDB Vertical Limb of the Diagonal Band of Broca WT Wild-Type xiv INTRODUCTION The neuroanatomical development of the basal forebrain cholinergic nuclei and their projection pathways result from highly orchestrated morphogenetic, cell-to-cell and autocrine/paracrine signaling mechanisms (Dale et al., 1997; Sussel et al., 1999). As a result, cholinergic neurons in the basal forebrain differentiate and project their axon terminals to the hippocampus and cerebral cortex where they release acetylcholine to modulate neuronal activity. Thus the development, maturation and maintenance of the cholinergic system is critical for the processes of attention, learning and memory. AD leads to dysfunction and degeneration of multiple neuronal subclasses in specific brain regions by disrupting normally functioning molecular, cellular, local circuits and neuronal networks, including the basal forebrain cholinergic network (Palop and Mucke, 2010). Three AD hypotheses have been described and each additively contributes to the pathological hallmarks of AD: 1) Aβ deposition, 2) neurofibrillary tau tangle formation and 3) the cholinergic deficits. Accumulating evidence implicates the cellular prion protein, PrPc as a factor that promotes AD pathophysiology and that all three hypotheses of AD converge at PrPc. We provide evidence that PrPc is upregulated specifically in BFCNs which may increase their neuronal susceptibility during AD pathological progression. 1 Since BFCNs are essential for memory and cognitive function, exogenous application of factors that regulate BFCN development may prove useful in generating BFCNs from pluripotent stem cells or by promoting their viability and physiological function in brain regions affected by AD. In support of these ideas, we provide strong in-vivo evidence that BMP9, a factor that promotes the cholinergic neuronal phenotype, can ameliorate two pathophysiological hallmarks of AD by reducing Aβ plaques and by increasing cholinergic density in the hippocampus. These data describe an important concept in potential AD therapies by focusing on factors that influence multiple aspects of AD pathology. Given the genetic, environmental and pathophysiological complexities of AD it is highly unlikely that there is one magic bullet to slow, stop or prevent the progressive nature of the disease. Therapeutic candidates should include those factors with potential to reduce more than one AD pathophysiological mechanism. Since PrPc interacts with Aβ, is expressed by BFCNs and may lead to tau phosphorylation targeting this protein may ultimately improve AD from three angles. Likewise, by increasing cholinergic innervation and decreasing plaque load in the hippocampus, BMP9 was shown to improve AD-like pathology in-vivo and highlights the utility of this developmental regulator of the BFCNs as a potential AD therapy. 2 BFCN DEVELOPMENT BFCN origination and development begins with regional specification of the early telencephalon, proliferation and differentiation is then initiated through induction, maintenance and maturation signaling mechanisms. These highly conserved patterns of vertebrate telencephalic development are established by morphogenetic gradients regulated by growth factors and cytokines which modify gene transcription to determine body axes, regional boundaries and the differentiation of forebrain cell types (Shimamura et al., 1995; Rubenstein and Beachy, 1998). In general, current developmental models are based on the idea that neuronal populations arise from spatio-temporally regulated gene expression programs fine-tuned by concentration-dependent patterns or signaling ‘pulses’ within the neurochemical niche. Competent embryonic tissues are organized into distinct molecular domains to establish transverse and longitudinal borders along morphogenetic gradients. The establishment of the caudal-rostral, dorsalventral, medial-lateral axes by distinct morphological and molecular boundaries creates an organized “grid-like” prosomeric model of forebrain development (Rubenstein et al., 1994). This grid-like patterning of forebrain anatomical structure is evolutionarily conserved across multiple species of organisms including lower mammals, i.e., mice to more complex non-human primates and humans from embryogenesis into adult forebrain development (Wedeen et al., 2012). 3 The axis of patterning in the rostrobasal telencephalon gives rise to subpallial proliferative zones of the medial ganglionic eminence (MGE), lateral ganglionic eminence (LGE) and the preoptic area (POA) which contribute to nearly all forebrain structures such as the cortex, striatum, hippocampus, amygdala and septum (Marin et al., 2000; Gelman et al., 2009; Nobrega-Pereira et al., 2010). Migratory cells from each of these three embryonic proliferative zones are further classified into septal progenitor domains, SE1-6 (Flames et al., 2007), which develop into the heterogeneously populated neuronal nuclei located in the basal forebrain, in which there are several cholinergic nuclei present. Basal forebrain cholinergic neurogenesis in mice occurs between embryonic days (E)11-18 along a caudo-rostral gradient, i.e. the more caudolaterally located nucleus basalis of meynert (NBM) arises prior to the more rostromedial diagonal bands of Broca (DBB) and the medial septum (MS) (Schwab et al., 1988) (Brady et al., 1989; Schambra et al., 1989). Interestingly, some of the first studies identified that the progressive cholinergic degeneration in AD began in the nucleus basalis of Meynert (NBM) and followed along the same caudorostral gradient (Doucette and Ball, 1987). Before the proliferative MGE, LGE, POA areas arise, the early forebrain developmental pathway is initiated by prechordal mesoderm induction of ventral midline cells (rostral diencephalon) by cooperative signaling through contactmediated Sonic hedgehog (SHH) and diffusible bone morphogenetic protein 7 (BMP7) gradients. These morphogenic gradients establish the rostro-ventral 4 regionalization of the prospective forebrain around the 10-12 somite stage. At this time, Nkx2.1 is expressed in progenitor cells of the MGE in the developing basal telencephalon and anti-BMP7 antibody blocks normal Nkx2.1 expression in the MGE (Dale et al., 1997). Nkx2.1 is a homeobox transcription factor whose expression is spatially restricted to the rostrobasal forebrain during development (Price et al., 1992) and Nkx2.1 repression in the developing dorsal telencephalon specifies ventral forebrain development (Sussel et al., 1999). In Nkx2.1 mutant mice, Sussel et al, observed structural and cytochemical abnormalities in the MGE induced by disrupted progenitor cell production and migration around E12.5. Of major importance, by E18.5 the Nkx2.1 mutants displayed a complete loss of TrkApositive cells, an early cholinergic neuronal marker (Sobreviela et al., 1994) throughout the entire basal forebrain (Sussel et al., 1999). From these studies it is apparent that normal Nkx2.1 expression within a highly regulated spatiotemporal manner is essential for all cells originating and migrating through the MGE, including BFCNs. In contrast to other MGE progenitor cells, BFCN Nkx2.1 expression is maintained and can be observed in postmitotic BFCNs into adulthood (Wei et al., 2012). Other functions ascribed to Nkx2.1 pertain to neuronal connectivity and migration of cells from the basal forebrain to the neocortex and striatum; however the role of Nkx2.1 in adult BFCNs has not been elucidated. 5 Whereas Nkx2.1 expression leads to regional specification of the MGE and initiates neuronal progenitor subtypes, LIM-homeobox (Lhx) genes are transcription factors which are expressed in parallel with Nkx2.1 in the developing basal forebrain (Wanaka et al., 1997). Mice expressing mutant Lhx8 (LIM-homeobox gene Lhx8/L3/Lhx7) also lacked cholinergic neurons in the telencephalic areas of the striatum, septum, and diagonal bands, but not in the nucleus accumbens or the magnocellular preoptic nucleus (Zhao et al., 2003). This demonstrates some heterogeneity in the cholinergic induction pathways and presents evidence for alternate mechanisms for cholinergic specification. Importantly, the loss of functional Lhx8 at E13.5 specifically blocked the formation of BFCNs in these areas without affecting other neuronal types. When quantified, mutant Lhx8 resulted in ~75% reduction in cholinergic cell counts in the telencephalon and a significant loss of hippocampal cholinergic inputs. Subsequent studies by Mori confirmed that Lhx8 plays a pivotal role in the development and maintenance of BFCNs by targeting the expression of choline acetyltransferase (ChAT), vesicular acetylcholine transporter (VAChT) and the 75kD/low affinity NGF receptor (p75/NTR), all of which are biochemical markers of the cholinergic phenotype (Mori et al., 2004). 6 BMP9 REGULATES THE BFCN PHENOTYPE Bone morphogenetic proteins are known to play a major role in body axis patterning during embryogenesis. BMP9 is abundantly expressed in the embryonic basal forebrain and spinal cord at E14 (Lopez-Coviella et al., 2000) and BMP9 induces Lhx8 expression (Bissonnette et al., 2011). The complete mechanism from Nkx2.1 to BMP9 induction of Lhx8 remains to be determined. The temporal expression of BMP9 and its receptor ALK1, have been shown to overlap with the critical period for BFCN differentiation. E14 primary septal neurons transiently treated with BMP9 undergo dynamic morphological and molecular changes in vitro (Lopez-Coviella et al., 2000). Instead of growing uniform monolayers, BMP9 treated septal cells organized into clusters and extended long TuJ1-positive projections. These same neurons were also induced by BMP9 to express ChAT, VAChT and p75/NTR and displayed a 20fold increase in ACh production over PBS treated controls (Lopez-Coviella et al., 2000). Induction of the BFCN transcriptome and cholinergic phenotype by BMP9 supports the idea that it acts as the differentiating factor responsible for the induction and maintenance of BFCNs (Lopez-Coviella et al., 2000; LopezCoviella et al., 2005). Moreover, intracerebroventricular (icv) infusion of BMP9 in mice with a unilateral septohippocampal lesion preserved the ChAT expression and prevented most of the cholinergic defects due to reduced hippocampal ACh levels. BMP9 also caused increased expression of nerve growth factor (NGF) 7 and its receptors, p75/NTR and TrkA in the hippocampus ipsilateral to the lesion. BMP9 activates signaling pathways via serine-threonine kinase BMP receptors which phosphorylate Smad1/5/8 proteins. BMP9 was identified to be developmentally regulated in parallel with various types of BMP receptors whose expression were also tightly regulated between E14 and perinatal time points in the mouse basal forebrain (Lopez-Coviella et al., 2006). BmpR-Ib and Smad1 proteins were highly expressed during E14-18 and BmpR-II protein levels increased around postnatal day 1 (P1) which were maintained into adulthood. Messenger RNA levels for another BMP receptor, Alk1 increased from E17 into adulthood and ALK1 was proposed as the maintenance receptor for BFCNs after differentiation. In E14 cultured basal forebrain cells, Smad1/5 phosphorylation was quickly induced after BMP9 treatment, which also increased their translocation to the nucleus to complex with Smad4 for up to 1 hr. This Smad1/5-Smad4 complex leads to activation of specific target genes (Massague and Wotton, 2000). Once in the nucleus, gene targets of the TGF-β/BMP signal transduction require several coregulators for transcription initiation. Smadinteracting zinc finger protein, Sizn1, is a coregulator of BFCN differentiation at the transcription level. Immunoprecipitation experiments showed that Sizn1 directly interacts with Smad1 and recruits transcriptional machinery such as CBP/p300 to increase expression from the cholinergic gene locus coding for ChAT and VAChT. Knockdown of Sizn1 in E13.5 primary septal cell cultures decreased ChAT and VAChT expression after BMP2 treatment (Cho et al., 8 2008). Not only does this emphasize the ability of Sizn1 to activate the cholinergic gene locus but it also suggests overlapping signaling mechanisms between BMP2 and BMP9 to induce cholinergic phenotypic expression. Indeed, BMP2 treatment of E14 septal cell cultures increased ACh production 15-fold, whereas BMP9 increased ACh production 20-fold as compared to PBS controls (Lopez-Coviella et al., 2002). Given that BMP9 promotes Smad1/5 activation and nuclear translocation with Smad4, along with other undiscovered comodulators it is reasonable to hypothesize that Sizn1 may also mediate BMP9 induced transcription from the cholinergic gene locus. These in vitro and in vivo experiments demonstrate that BMP signaling acts as a differentiation and maintenance pathway for the BFCN phenotype. Elucidation and confirmation of these developmental pathways has enabled the in vitro differentiation, maturation, transplantation and functional integration of BFCNs derived from pluripotent stem cells. After stimulation with fibroblast growth factor 8 (FGF8) and SHH to produce neuronally committed progenitors competent to BMP9 treatment, human embryonic stem cells (hESCs) were differentiated into BFCNs in-vitro, grafted into ex-vivo mouse brain slices and then confirmed to be electrophysiologically active with voltage-clamp recordings (Bissonnette et al., 2011). Exogenously applied BMP9 induced the BFCN phenotype and a dramatic increase in Lhx8 and Gbx1 mRNA transcripts were observed (Bissonnette et al.). Overexpression of these two transcription factors was more efficient (94% vs. 85%) than BMP9 at producing BFCNs from hESCs. 9 Progenitor State BFCN Differentiation BMP9 E10 Lhx8 Gbx1 Nkx2 .1 ? Maturation BFCN E18 Smad1/5 E14 ? NGF ChAT VAChT 75/NTR TrkA Figure 1 BFCN differentiation from Nkx 2.1 expressing progenitors in the MGE and LGE at E10, to early BFCN induction and specification at E14. The morphogenic gradient induces transcription of BFCN cholinergic markers, ChAT, VAChT and the NGF/neurotrophin receptors at E18 which is maintained into adulthood. This is possibly due to BMP9 mediated regulatory feedback loops to modulate its own inductive signaling capacity in conjunction with Lhx8/Gbx1 transcriptional activation. Furthermore, BMP9 treatment with Lhx8 knockdown by siRNA abrogated BFCN differentiation, providing further evidence for BMP9 downstream signaling through Lhx8/Gbx1 to activate transcription of the cholinergic gene locus (Bissonnette et al., 2011). These results confirm and strengthen the role of BMP9 mediated transcriptional activation as a sufficient event for BFCN development and advances the idea that BFCN generation from a patient’s own stem cells may hold therapeutic potential for diseases specifically affecting this neuronal subclass. 10 CTX CA1 HPC CA2 F CA3 OB MSN VDB DG BLA HDB NBM A CTX CA1 HPC HPC CTX CA2 CA3 F MSN BLA BLA VDB HDB B HDB NBM Figure 2 Sagittal (A, top panel) and coronal (B, lower panel) views with basal forebrain nuclei labeled in white and projection targets in yellow and red arrows indicating BFCN projection paths. Modified from Mouse Brain Explorer 2, Allen Brain Institute. 11 BFCN PHENOTYPE In 1933, the neurophysiologist and Nobel Laureate, H.H. Dale proposed the term “cholinergic” for those neural pathways that utilize the neurotransmitter ACh. These pathways are illustrated in the atlas presented in Figure 2. The biochemical basis of the cholinergic system requires macromolecules to mediate ACh synthesis, release, synaptic transmission, degradation and choline reuptake. In the cytoplasm, choline acetyltransferase (ChAT) forms ACh from the precursors, choline and acetyl-CoA (Bielarczyk and Szutowicz, 1989) (Jope and Jenden, 1980) which is essential for survival since transgenic ChAT knockout mice do not survive past postnatal day 1. After ACh synthesis, BFCNs utilize vesicular acetylcholine transporter (VAChT) to sequester ACh into quanta or vesicles to be released from synapses and effect postsynaptic electrochemical signals. Eliminating ACh release in the hippocampus by genetically reducing VAChT results in hyperactivity, impaired long-term potentiation and visuospatial memory (Martyn et al., 2012). ChAT and VAChT represent a unique “gene within a gene” organization on human chromosome10q11 establishing the cholinergic gene locus (CGL) (Eiden, 1998) illustrated in Figure 3. VAChT is located within the first intron of the ChAT gene, allowing coordinate regulation of this gene locus whose organization is conserved through C. elegans, Drosophila and H.sapiens. Coordinate regulation does not necessarily imply coordinate expression. The 12 effects of both CNTF/LIF and retinoic acid receptor-α (RAR-α) stimulation by alltrans-retinoic acid led to proportional increases in ChAT and VAChT mRNA expression, whereas a 2- and 4-fold increase,respectively was observed in a septal cell line after cyclic-AMP treatment (Berse and Blusztajn, 1995). Figure 3 The cholinergic gene locus (CGL) codes for ChAT (its central and peripheral variants) as well as VAChT from chromosome 10. The CGL consists of 3 non-coding (R, N, M) and 15-16 coding exons depending on the species. (*) represent transcriptional start sites for the 5 alternative ChAT variants. The intronless VAChT gene is located within the first intron, between the first two non-coding exons of the full-length ChAT reading frame. 13 Differential expression patterns from a common locus could be accomplished by multiple response elements (RE) in the cholinergic gene locus such as a BMPRE, cAMP-RE, repressor complexes or additional interactions that regulate transcription through epigenetic modification. REST4, a transcriptional activator, can de-repress the gene silencing action of the REST/CoREST complex and activate the cholinergic gene locus in a protein kinase A (PKA) dependent manner (Hersh and Shimojo, 2003). This may explain the coordinate regulation, but differential expression in ChAT and VAChT mRNA when the septal cells were treated with PKA activating cAMP (Berse and Blusztajn, 1995). Epigenetic modifications may promote regulation of expression from the cholinergic gene locus as histone deacetylase 9 (HDAC9), a class IIa enzyme that removes an acetyl group from the histone N-terminal tail and represses the ChAT promoter in neuronal cultures (Aizawa et al., 2012). Multiple layers of complex regulatory mechanisms govern ChAT and VAChT expression from the cholinergic gene locus, to establish the biochemical basis of all properly functioning cholinergic neurons, however the exact mechanisms for the differential expression patterns of ChAT and VAChT from a common locus remains elusive. Postsynaptic molecules such as ionotropic, nicotinic acetylcholine receptors (nAChR), metabotropic muscarinic acetylcholine receptors (mAChR), the hydrolytic enzyme, acetylcholinesterase (AChE) control the electrochemical 14 neurotransmission signals provoked by ACh release. Each of these factors is a salient feature of acetylcholine neurotransmission. VAChT, a twelve membrane domain transporter protein, is localized in the nerve terminals where it transports ACh into synaptic vesicles. Presynaptic excitation of a cholinergic neuron induces calcium-dependent exocytosis of ACh in the synapse (Li et al., 1995a). Exocytosis of ACh is mediated by a series of highly-regulated docking, synaptic membrane-vesicular fusion and lastly, vesicular recycling mechanisms (Sudhof, 1995). Postsynaptic nAChR and mAChRs, as well as mAChR autoreceptors located presynaptically can bind ACh (Blusztajn and Berse, 2000) to initiate temporally-defined patterns of intracellular signaling and gene expression pathways. AChE, a carboxylesterase capable of hydrolyzing 36,000 molecules of ACh into free acetate and choline per second, effectively terminates the neurotransmitter signal. The choline transporter 1 (ChT1) is responsible for the presynaptic, sodium-dependent, high-affinity choline reuptake which recycles choline for another round of ChAT catalyzed ACh synthesis and VAChT sequestration (Eiden, 1998; Okuda et al., 2000). These macromolecules modulate the cholinergic effects to optimize neural circuits throughout the entire brain. ACh is important for mechanisms of synaptic plasticity, which is activity-dependent strengthening or weakening of synapses, thought to be responsible for learning acquisition, memory storage and retrieval, and long term neural circuit maintenance (Picciotto et al., 2012). 15 Both choline and acetyl-CoA are essential for cellular metabolic pathways and the production of acetylcholine in BFCNs. Age induced metabolic stress and this double utilization of acetyl-CoA and choline in cholinergic neurons may increase BFCN susceptibility to dysfunction and degeneration leading to cognitive and memory impairments (Perry et al., 1980; Wurtman, 1992) (Szutowicz et al., 1996). As choline can be metabolized from phosphatidylcholine, a major component of the plasma membrane, diseases that result in cholinergic dysfunction may destabilize the plasma membrane and cytoskeletal dynamics to cause morphological changes in BFCNs. Our initial observations in 6- and 24month old mice indeed revealed morphological changes with age in BFCNs. We observed retracted arborizations as cell bodies appeared more rounded and smaller in size, as illustrated in Figure 4. With AD-like amyloidosis, BFCNs in AD model mice appear smaller and have tortuous, stunted projections as illustrated in Figure 5 and representative of the cholinergic defect the APP/CHGFP mice. These morphological changes in wild type (WT/CHGFP) mice with age and in AD model (APP/CHGFP) mice are in line with previous reports and highlights the susceptibility of BFCNs to morphological changes with aging and AD. 16 24 month 6 month WT/CHGFP Figure 4 Age-induced morphological changes of BFCNs in the MSN/VDB of 6 months (top) and 24 months old (bottom) WT/CHGFP mice (20x objective). Note: Aged BFCNs have shorter processes and have taken on a smaller, more spherical shape in contrast to the ellipsoid somas and long projections evident in the 6-month old BFCNs. 17 APP/CHGFP 8 months WT/CHGFP Figure 5 Reduced BFCN cell size and stunted axonal projections in the MSN of the APP/CHGFP AD mouse model expressing eGFP from the ChAT promoter (40x, objective lens). 18 NERVE GROWTH FACTOR AND ITS RECEPTORS Discovery of nerve growth factor (NGF), the first growth factor, by Rita LeviMontalcini in the 1950s transformed modern neuroscience and earned Dr. LeviMontalcini the 1986 Nobel Prize. Initially, NGF was shown to regulate cell growth, differentiation and survival of noradrenergic neurons. Later it was learned that NGF’s mechanism of action influenced multiple neuronal cell populations in the CNS and PNS (Aloe, 2004). High concentrations of NGF in the terminal cholinergic targets of the cerebral cortex and hippocampus implicated an NGF-cholinergic association (Johnson et al., 1987). Co-expression of the NGF receptors, TrkA and p75/NTR with ChAT is a characteristic only observed in BFCNs and demonstrates a reliance on NGF signaling in the basal forebrain (Sobreviela et al., 1994). NGF and its receptors are now known to be important for the development and maintenance of BFCNs and NGF has been shown by multiple groups to increase ACh levels (Berse et al., 1999; Auld et al., 2001), ChAT (Gnahn et al., 1983) , VAChT (Berse et al., 1999), and TrkA expression (Li et al., 1995b). NGF is also induced by BMP9 in dissociated E14 primary septal cell cultures (Schnitzler et al., 2010). These data suggest that both NGF and BMP9 have overlapping functions to support BFCN development, survival and phenotypic maintenance throughout neurodevelopment into adulthood. In aged rats (26-30 month old), impairments to BFCN NGF retrograde transport resulted in a 60% reduction in 19 cholinergic cell size and a 43% decline in TrkA mRNA leading the authors to propose this as a mechanism for cholinergic hypofunction and degeneration in aging and AD (Cooper et al., 1994). Furthermore, triple colocalization of p75/NTR and TrkA NGF receptors with ChAT is restricted to the cholinergic neurons of the basal forebrain and their respective projection sites in the CNS (Sobreviela et al., 1994) making these molecules markers of the BFCN network. Sobreviela (Sobreviela et al., 1994) performed an extensive quantitative analysis of TrkA and p75/NTR distributions in the CNS and found virtually all (>95%) TrkA+ neurons in the MS/DBB were positive for p75/NTR and ChAT with slightly less colocalization of all three BFCN markers in the NBM (~85%). TrkA, a type-I receptor tyrosine kinase, has high-affinity for the processed, mature form of NGF and initiates cell signaling cascades, receptor mediated endocytosis and retrograde transport of NGF to the BFCN soma for survival and maturation of BFCNs and maintenance of their target innervation (Fagan et al., 1997; Moises et al., 2007; Niewiadomska et al., 2011). Conversely, p75/NTR is a promiscuous neurotrophin receptor that has a lower affinity for NGF and prefers to bind immature proNGF when acting unilaterally (Sofroniew et al., 2001). More specifically, loss of NGF signaling in TrkAnull mice produces significantly smaller and fewer BFCNs (Fagan et al., 1997); whereas deletion of p75/NTR in mice results in increased BFCN counts and cholinergic fiber hyperinnervation of hippocampal targets (Yeo et al., 1997). At baseline, 20 p75/NTR is expressed in a 10:1 ratio with TrkA and the vulnerability of BFCNs to changes in TrkA/NGF signaling may mediate neuronal shrinkage, dysfunction Figure 6 Co-expression of transgenic ChAT-eGFP (green) and endogenous p75/NTR (red) in the MSN/VDB provides a positive control validating eGFP expression in ChAT/p75-positive BFCNs. Note: Compare the anatomical features of MS/VDB in this figure to the atlas in Figure 2B. 21 or death, as in aging and AD when TrkA:p75/NTR expression ratios are altered (Chao and Hempstead, 1995). TrkA downregulation coupled with impaired retrograde axonal transport in the basal forebrain is one of the early findings in AD and correlates significantly with lower cognitive performance (Mufson et al., 1996). p75/NTR has demonstrated pro-survival capabilities by increasing the specificity of TrkA, as well as TrkB and TrkC neurotrophin receptors for their respective growth factors i.e, NGF, BDNF and NT-3 respectively (Kaplan and Miller, 1997). In the absence of ligand, co-immunoprecipitation experiments have demonstrated binding between the p75/NTR and TrkA (Huber and Chao, 1995) which required both the intact extracellular and intracellular domains of both proteins (Bibel et al., 1999). The p75/NTR-TrkA co- receptor increased NGF responsiveness and neuronal survivability (Chao and Hempstead, 1995). Much research regarding BFCN phenotypic changes and receptor expression levels during aging and AD has been carried out, however these studies have not been entirely consistent. In particular, studies on the role of p75/NTR in AD have been controversial with research showing both neuroprotective and neurotoxic effects. p75/NTR has a highly conserved death domain which in certain contexts are known to induce apoptosis. However, in cholinergic neurons p75/NTR downregulates cholinergic markers but does not induce apparent cholinergic cell death (Barrett, 2000). In mice with p75/NTR functional deletion mutation, Greferath (Greferath et al., 2012) found the number 22 and size of ChAT expressing BFCNs increased but the number of p75/NTRpositive neurons in the basal forebrain was unchanged suggesting a nonapoptotic, antineurotrophic role for p75/NTR. Earlier human studies observed a 38% and 43% reductions in p75/NTR expressing neurons in the basal forebrains of humans with mild cognitive impairment and mild AD, respectively (Mufson et al., 2002). The latter found that the reductions of p75/NTR were significantly correlated with cognitive impairment, as determined by the Mini-Mental State Exam and the Global Cognitive Test. These conflicting reports are not surprising, given the complexities surrounding the physiological role of p75/NTR receptor itself. proNGF-p75/NTR can cause caspase activation and lead to apoptosis. Not only can p75/NTR undergo proteolytic cleavage by the Beta-Amyloid Converting Enzyme (BACE) (Zampieri et al., 2005), but it can also promote cleavage of APP to generate insoluble Aβ (Costantini et al., 2005). p75/NTR also binds to Aβ in primary hippocampal cultures (Kuner et al., 1998) and intrahippocampal Aβ injections were dependent on p75/NTR expression in vivo to produce neurotoxic effects (Sotthibundhu et al., 2008). In contrast, there are multiple lines of evidence suggesting that p75/NTR may mediate anti-Aβ aggregation and pro-survival functions. BACE cleavage of the p75/NTR extracellular domain (ECD) can confer protection from neurotoxic amyloid oligomers. Zhang (Zhang et al., 2003) found a dose-dependent 23 upregulation of p75/NTR in primary human neurons exposed to Aβ 1-40 and Aβ1-42. This correlated with increased neuronal survivability in a PI3K-Akt dependent mechanism. Transfecting the same cells with a plasmid encoding anti-sensep75/NTR effectively reduced p75/NTR levels and sensitized these neurons to Aβ1-42 cytotoxicity (Zhang et al., 2003). Another study by Wang et al, showed that p75/NTR increased Aβ production and deposition in the APPswe/PS1dE9 mouse model of AD, but inhibited Aβ aggregation with its extracellular domain (ECD). Injection of the p75/NTR ECD into the hippocampus of p75/NTRnullAPPswe/PS1dE9 mice significantly reduced the area fraction of Aβ plaques in these mice (Wang et al., 2011). Wang et al, also found elevated insoluble Aβ in sera and less Aβ in the brains of p75/NTR+/+ mice, as compared to p75/NTRnullAPPswe/PS1dE9 mice, suggesting that p75/NTR may prevent Aβ aggregation and increase Aβ clearance. p75/NTR expression is consistently increased by BMP9 in-vitro and in-vivo (Lopez-Coviella et al., 2000; Lopez-Coviella et al., 2005; Schnitzler et al., 2008b). Since BMP9 is important for BFCN development, maturation and maintenance, it would be worthwhile to determine whether BMP9 modulates the enzymatic proteolysis of p75/NTR ECD toward the more advantageous, neuroprotective pathway during AD pathology. 24 Figure 7 Colocalization of transgenic ChAT-eGFP and p75/NTR expression in the VDB. Costes Colocalization Coefficient (M1): 0.999; Costes Colocalization Coefficient (M2): 0.957. These colocalization coefficients calculated by Volocity software for the representative image support the findings of (Sobreviela et al., 1994). Note the morphology and the overlapping densities of the p75/NTR and ChAT fiber networks. 25 ALZHEIMER’S DISEASE AD is a complex, neurodegenerative disease that robs humans of their most valuable, personal possession: their mind. AD is diagnosed by the presence of two histopathologic hallmarks: Aβ plaques and neurofibrillary tau tangles. Major brain regions affected by AD are the neocortex, the medial temporal lobe and adjacent limbic structures including the hippocampus and amygdala, as well as the cerebellum. Atrophy and dysfunction in these regions which are responsible for learning, attention, memory, insight, judgment, orientation, emotion and other higher executive abilities lead to global loss of cognitive ability. Included in this global cognitive impairment are memory loss, dementia, psychosis, depression, aggression/agitation and other behavioral detriments. AD is a disorder with a strong genetic component and is divided into familial and sporadic forms.. Approximately 25% of AD is the familial form and the rest fall under sporadic. Late-onset AD (LOAD) is the most common variety of sporadic AD and typically affects those 65 years and older. Sporadic AD is thought to arise due to age-induced exposures and accumulation of environmental risk factors, however very little is known about the etiology of sporadic AD (Bird, 1993). Recently, genome wide association studies have found a strong genetic component leading to increased risk of early-onset, familial AD as well as late-onset (sporadic AD) (Guerreiro et al., 2013). 26 The familial form of AD is much less common and is associated with early onset AD (EOAD) in people less than 65 years old. Familial AD displays an autosomal dominant pattern of inheritance. The genetic variants in the genes coding for APP, PSEN1 and PSEN2 are deterministically associated with earlyonset AD. Late onset-AD is associated with specific genetic variants in the genes coding for TREM2, MTHFD1L, BCHE, CR1, CLU, BIN1, PICALM, MS4A, CD2AP, EPHA1, ABCA7 and CD33. However, aging and inheritance of the apolipoprotein ApoE4 allele have the largest effect of AD risk (Naj et al., 2011; Guerreiro et al., 2013). 27 AD GENETIC VARIANT TYPE OF AD DISEASE IMPLICATION or GENE FUNCTION APP, PSEN1/PSEN2 Deterministic, Early-onset AD Increased production of toxic Aβ intermediates and plaque formation APOE4 Late-onset AD Defective lipid and amyloid clearance TREM2 Late-onset AD Defective microglial phagocytosis EPHA1 Late-onset AD Axon guidance and ADAM10 metalloprotease substrate recognition (OMIM: 179610) CD2AP Late-onset AD Vesicle formation and cytoskeletal polarization (OMIM: 604421) CR1 Late-onset AD Complement receptor and activator of innate immune response (OMIM: 120620) CLU Late-onset AD Clearance of cellular debris and apoptosis (OMIM: 185430) BIN1 Late-onset AD Tumor suppressor that interacts with phospholipases MTHFD1L Late-onset AD Conversion of methionine from homocysteine BCHE Potential association with Early and Late-onset AD Hydrolysis of ACh and correlates to plaque burden. TABLE 1 Selected genetic variants identified as risk factors for early-onset and late onset AD. Gene function listed instead of pathologic contribution since most genetic variants are only associated with AD risk and have not yet been causally linked to AD pathophysiology. 28 Diagnosing Alzheimer’s disease has been challenging because the cognitive dysfunction, memory loss and behavioral changes occur many years after the onset of disease progression (Niedowicz et al., 2012). Until recently, a confirmatory diagnosis of AD was only available after postmortem autopsy analysis for Aβ plaques and neurofibrillary tangles. Advances in PET imaging and biomarker discovery may enable early diagnosis and the initiation of preventative or symptomatic treatments of for humans with preclinical AD. Figure 8 Positron Emission Tomography (PET) scan images of a cognitively normal brain compared to an AD brain. Aβ plaques are radiolabeled with Pittsburgh compound B (PiB) which is now used to help diagnose AD before symptoms are evident. Higher PiB concentration (red and yellow regions) indicates increased presence of Aβ plaques. Image from Alzheimer’s Disease Education and Referral center, National institute of Aging. 29 Research into the genetic components of AD has allowed the generation of transgenic mice to study certain parameters of the disease pathophysiology invivo. Although not a complete recapitulation of human AD pathology, these mice can be engineered as classical knockouts, transgenics or knock-ins to test certain AD genes and gene-gene interactions. Most AD mouse models focus on APP and Aβ plaque formation, however double-, triple-, on up to 5x- transgenic mice have been produced to study narrow or broad aspects of AD pathophysiology. These mice exhibit reactive gliosis and inflammation, dystrophic neurites and cognitive deficits which are also observed in human AD, but the mice lack significant brain atrophy (Mineur et al., 2005; Oakley et al., 2006). Our studies involve the APPswe/PS1dE9 mouse model of AD engineered to express mutated human forms of APP and PSEN1 which are known to be deterministic of AD in humans (see Materials and Methods for more info on APPswe/PS1dE9 mice). Previous studies have found these mice display progressive amyloidosis with a significant cholinergic deficit (Figures 5, 10, 11 and 15) and cognitive impairments (Savonenko et al., 2005) (Gimbel et al., 2010). 30 Aβ HYPOTHESIS OF AD AD is a progressive disease characterized by memory loss and cognitive impairment exacerbated by Aβ-induced synaptic failure (Selkoe, 2002). There is now a large body of evidence implicating Aβ as the molecular pathogenic culprit of AD, which has culminated into the Aβ hypothesis. Aβ monomers, oligomers, fibrils and plaques generated from insoluble fragments of the amyloid precursor protein (APP) cleavage have been extensively investigated and it is widely accepted that Aβ deposition disrupts molecular, cellular and neuronal network dynamics. The Aβ hypothesis provides a mechanistic explanation for progressive AD and according to Walsh (Walsh and Selkoe, 2004) is supported by the following observations: 1) progressively increased Aβ peptide deposition around neuritic and dystrophic neurites in the cortex and hippocampus, 2) synthetic Aβ peptides display in vitro and in vivo toxicity, 3) inherited APP mutations results in higher production of amyloidogenic Aβ , 4) transgenic animals with APP and presenilin PSEN1/PSEN2 mutations generate greater levels of the more toxic Aβ42 peptide, resulting in more severe, rapid onset AD-like pathology, and 5) strong correlations have been identified between Aβ oligomers, cognitive deficits and synaptic retraction. 31 Aβ TARGETED THERAPEUTICS The Aβ-hypothesis has opened the possibility of immunotherapies for inhibiting amyloidosis in the brains of AD patients. This was first attempted in humans in a clinical trial of intravenously infused bapineuzumab which reduced Aβ burden and tau aggregation, but it failed to demonstrate any improvements in cognition or activities of daily living (Lobello et al., 2012). It is difficult to interpret these results due to the late initiation of treatment in the disease process as plaque accumulation and synaptic degeneration is known to occur decades before symptoms of cognitive dysfunction appear. To address this issue, the very first AD prevention trial was approved by the NIH and will systematically test Genentech’s anti-Aβ antibody crenezumab in a presymptomatic Columbian cohort (n=300) harboring the PSEN1 (E280A) mutation. A smaller group of pre-symptomatic American participants with other presenilin mutations from the DIAN cohort will also receive crenezumab (Selkoe, 2012). The outcome of this preventative study will likely represent the largest and most authoritative challenge to the Aβ-hypothesis. One reason Aβ has garnered so much attention is because the molecular underpinnings of APP catabolism and generation of neurotoxic Aβ is the most understood process in AD pathophysiology. In addition, understanding the pathogenic pathways and molecular regulation of Aβ generation from APP 32 provides multiple targets for prevention and/or treatment. APP is sequentially cleaved by BACE and gamma-secretase to generate Aβ40 and Aβ42 fragments. Pharmacological approaches developed by Eli Lilly, using the gamma-secretase inhibitor semagacestat (LY450139) to prevent Aβ generation showed significant dose-dependent reductions of Aβ40 in human CNS over 12 hours (Bateman et al., 2009), however Phase III clinical trials were abruptly ended as results indicated worse cognitive performance compared to placebo. Further complications included gastrointestinal irritability and increased risks of skin cancer due to interference of signaling by Notch – a known substrate for gamma-secretase (Imbimbo and Giardina, 2011). Elan Pharmaceuticals has since developed and tested ELN475516, a gamma secretase inhibitor that specifically inhibits cleavage of APP and reduces Aβ40 without interfering with Notch signaling in mice (Basi et al., 2010). The safety or effectiveness of ELN475516 remains to be tested in humans. Inhibiting Aβ generation is more appealing than blocking fibrillary plaque formation because the smaller Aβ oligomers are thought to be more toxic to synapses (Lue et al., 1999) and Aβ oligomers are a better determinant of neurodegeneration and cognitive decline than Aβ plaques (McLean et al., 1999). By decreasing the number of plaques without increasing the clearance rate of Aβ oligomers, a potential therapy may increase the number of free oligomers (or dimers and trimers). This might actually induce more harm than good since Aβ 33 oligomers are more hydrophobic and are known to interfere with CNS physiology at multiple levels. Aβ DISRUPTS MULTIPLE NETWORKS IN AD Olzscha et al. (Olzscha et al., 2011) have proposed that amyloid-like aggregates induce a “fatal network collapse” by affecting key cellular processes such as chromatin remodeling, transcription, vesicular transport, signaling and cytoskeletal dynamics. In addition to synaptic retraction and degeneration, multiple groups have shown that Aβ interferes with AMPA-R, NMDA-R, nACh, mACh and mGlu5 receptors at synapses. This would disrupt intracellular functions as well as local circuit connections and ultimately larger neuronal networks (Palop and Mucke, 2010). As mentioned earlier, Aβ oligomers have been shown to bind p75/NTR, nAChRs, mAChRs and PrPc all of which are expressed pre- or post-synaptically as a part of the cholinergic network. This suggests that the Aβ-hypothesis and cholinergic hypothesis of AD may not be mutually exclusive. 34 Figure 9 Aβ disrupts multiple functional networks at the molecular, synaptic, local circuits and neuronal network levels. Additionally, Aβ has been found to bind, sequester and negatively affect intracellular transport by inducing tau hyperphosphorylation via α7-nAChRs, mitochondrial release of cytochrome C to activate apoptotic cascades, and initiating the stress-induced unfolded protein response in the ER. Aβ possesses nanomolar affinity for the PrPc protein and this interaction was essential to mediate Aβ-induced toxicity and cognitive impairment (Lauren et al., 2009). However, less specific interactions with other proteins may be propagated through the disordered, unstructured domains allowing Aβ to evolve an aberrant 35 interactome interfering with essential function and inducing toxicity (Bolognesi et al., 2010). Aβ contains a hydrophilic, extracellular domain and a hydrophobic, intramembrane domain which allows Aβ monomers to act as nucleating centers to create expanded oligomers. These hydrophilic-hydrophobic regions establish structural disorder within Aβ to induce aggregation of individual monomers into βsheet rich oligomers, fibrils and plaques. These pathogenic Aβ units then interact and sequester numerous proteins with essential functions disrupting a host of physiological functions (Olzscha et al., 2011). This amyloid induced fatal network collapse in molecular and cellular networks can be extrapolated to larger neuronal networks that require interconnected brain regions for appropriate social, emotional and behavioral integration. As these interconnections suffer synaptic failure and neurodegeneration, network efficiencies in the brain become compromised then cognitive and behavioral impairments ensue. 36 TAU HYPOTHESIS OF AD Tau (also known as, MAPT) is a member of microtubule associated protein (MAP) family. Tau provides structural integrity and stability to microtubules. In the CNS, microtubules influence neuronal morphology, dendritic scaffolding and intracellular transport of nutrients, neurotransmitters and organelles such as mitochondria and rough endoplasmic reticulum. These processes are essential for cytoskeletal maintenance, synaptic plasticity and cell viability (Savelieff et al., 2013). Various mechanisms resulting in tau hyperphosphorylation induces its dissociation from microtubules yielding “seeds” of intracellular tau monomers to form oligomers, granules and neurofibrillary tangles (NFTs) (Takashima, 2010). Phosphorylated tau is also resistant to both proteolytic cleavage by CAPN2 a calcium-activated neutral protease, as well as polyubiquitin mediated proteosomal degradation (Iqbal et al., 2009). These conditions lead to increased intracellular concentrations of the pathologic tau intermediates which form the NFTs. NFTs can be observed in the frontal and temporal lobes of the neocortex, the entorhinal cortex as well as limbic structures of the AD brain and are correlative of synaptic and neuronal loss (Takashima, 2010) (Braak and Braak, 1991). Evidence for the tau hypothesis of AD include: 1) NFTs, in the absence of amyloid plaques, are found in brains afflicted with other dementia-related pathologies (Ward et al., 2012), 2) Neurodegeneration correlates with phospho- 37 tau/NFT levels but not amyloid plaques (Iqbal et al., 2009) (de Souza et al., 2012), 3) Normal, aged individuals may contain Aβ plaque burdens as high as AD patients, but lack tau-positive dystrophic neurites and neuritic plaques (Dickson et al., 1992), 4) overexpression of the P301S human tau mutation in transgenic mice induces cortical, hippocampal and amydalar NFT formation, inflammatory gliosis and neurodegeneration (Yoshiyama et al., 2007). The extent of NFTs in the brain strongly correlate with neurodegeneration and cognitive impairment in AD (Braak and Braak, 1991). Using NFT immunostaining, Braak and Braak, identified six different stages of AD pathology as defined by the amount of NFT involvement in postmortem human brain samples. Braak and Braak (BB) stages I/II include NFTs confined to the transentorhinal area, BB stages III/IV include NFTs in the hippocampus and other limbic regions and BB stages V/VI refer to extensive NFTs throughout neocortical regions. Tau can be phosphorylated on 38 serine/threonine residues by multiple kinases including glycogen synthase kinase-3 (GSK-3), cyclin-dependent kinase5 (cdk5), protein kinase A (PKA), the extracellular regulated kinases (ERK1/2), stress-activated protein kinases (SAPKs) casein kinase 1 (CK-1) and FYNkinase. Interestingly, a single nucleotide polymorphism (rs7768046) in the FYNkinase and (rs913275) in the PPP2R4 phosphatase are associated with increased total-tau and phosphorylated tau in cerebrospinal fluid from AD patients (Bekris et al., 2012). 38 Whereas multiple kinases facilitate tau hyperphosphorylation, a relatively few number of phosphoprotein phosphatases are able to balance this pathologic mechanism. Only PP-1, PP-2A and PP-2B have been shown to dephosphorylate and inhibit tau’s self-assembly to preserve microtubule stability (Wang et al., 1996). Significantly decreased phosphatase activity has been measured in AD brains, suggestive of a dephosphorylation defect that contributes to the phosphor-tau mediated pathophysiology of AD (Gong et al., 1993; Guadagna et al., 2012). These studies may lead to tau-specific phosphatase activators or kinase inhibitors to reduce tau hyperphosphorylation and preserve the neuronal cytoskeleton, intracellular transport or possibly even cognitive function. CHOLINERGIC HYPOTHESIS OF AD The cholinergic hypothesis of AD proposed by Bartus (Bartus et al., 1982) is supported by a vast body of research pertaining to: 1) alterations in choline uptake, the rate-limiting step for ACh synthesis (Slotkin et al., 1990), 2) reduced ChAT activity and ACh release, 3) reduced (α7)-nAChR expression (Hernandez et al., 2010), 4) dysfunctional or reduced M1/M3-mAChR expression (Terry and Buccafusco, 2003), and 5) impaired cholinergic axonal transport of NGF (Mufson et al., 1996). Dystrophic cholinergic neurites, neuritic plaques and reduced hippocampal and cortical cholinergic innervation as well as reduced BFCN counts and synaptic degeneration have all been observed in human and 39 transgenic mouse models of AD (Mufson et al., 2003; Perez et al., 2007). One of the earliest observed features discovered in postmortem AD brains is the pronounced cholinergic degeneration which is correlated to severity of memory loss and cognitive decline. After Davies (Davies and Maloney, 1976) reported a selective loss of cholinergic neurons in AD patients, it was postulated that AD may be a selective neurotransmitter disease affecting acetylcholine signaling. Subsequent losses of cholinoceptive pyramidal neurons and even greater synaptic degeneration in the cortex as measured by synaptophysin staining are the strongest correlates of dementia and memory loss (Terry et al., 1991; Francis et al., 1999). Further support for the cholinergic hypothesis comes from studies demonstrating various cognitive dysfunction and learning impairments in mice, rats, monkeys and humans with basal forebrain lesions and pharmacological cholinergic blockade (Terry and Buccafusco, 2003). Blocking cholinergic signaling with the mAChR antagonists, scopolamine, atropine or glycopyrrolate produce amnestic effects with age-related sensitivity whereas memory improvements have been observed with specific nAChR and mAChR agonists (Ray et al., 1992; Terry and Buccafusco, 2003). 40 Figure 10 Laser scanning confocal micrograph of Aβ42 immunostaining (red) in the hippocampus (left) and cerebral cortex (right) in APP/CHGFP mice. Note: dystrophic, tortuous neurites adjacent to the plaques and the weak, punctate network of cholinergic fibers (green), (40x objective lens). 41 A Figure 11 B C Abnormal cholinergic morphologies in the vertical limb of the diagonal band (A), striatum (B) and cerebral cortex (C) of 8 month old APP/CHGFP mice, (20x objective lens). CHOLINERGIC THERAPEUTICS The only approved therapeutic drugs for AD block AChE to inhibit ACh hydrolysis thereby potentiating ACh neurotransmission at the synapse. The first generation AChE inhibitor, tacrine showed significant improvement in cognition and quality of life as measured by Alzheimer’s Disease Assessment Scalecognitive subscale (ADAS-cog) scores and Clinician Interview Based Impression of Change plus caregiver input (CIBIC+) tests. However, given that ACh has widespread modulatory effects in the gastrointestinal and cardiovascular systems, multiple side effects involving gastrointestinal discomfort, nausea, vomiting, cardiovascular complications and hepatotoxicity reduced long-term 42 patient compliance and the effectiveness of tacrine (Terry and Buccafusco, 2003). Second generation AChE inhibitors such as donepezil have reduced side effects while demonstrating similar improvements in cognitive function as well as delaying reductions in activities of daily living (ADLs) by up to a year (Rogers et al., 1998). Early to mild AD patients receiving donepezil demonstrated improved quality of life, reduced amount of direct supervision and delayed institutionalization. However not every AD patient responded equally to donepezil treatment and none of the AChE inhibitors improve cognitive performance to normal levels (Mesulam, 2004). The cholinergic hypothesis merely explains the insidious loss of attention and working memory during aging and AD (Schliebs and Arendt, 2011). Potentiating cholinergic neurotransmission to improve cognition simply treats the symptoms of AD and not the underlying cause, making it difficult, if not impossible to pharmacologically counterbalance the cognitive assault of the causative agent with AChE inhibitors or AChR agonists. The causative agent for AD has remained elusive, however various cellular and molecular biological approaches have yielded tremendous gains in the understanding of AD pathophysiology. Multiple studies indicate that BFCN dysfunction and/or degeneration contributes to the memory deficits in advanced aging and AD (Mufson et al., 2008; Grothe et al., 2012; Haense et al., 2012). 43 Therefore, interventions that inhibit Aβ-deposition or tau-phosphorylation while protecting BFCN viability may preserve ACh neurotransmission thus preserving memory function and improving cognition. MATERIALS AND METHODS Transgenic Mice The following strains were used: 1) AD model mice B6C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax and 2) transgenic mice expressing the enhanced green fluorescent protein in cholinergic cells: B6.Cg-Tg(RP23-268L19-EGFP)2Mik/J. Briefly, the AD model mice express two separate transgenes that inserted into a single locus, both under control of the prion promoter. The first transgene is the murine APP protein harboring the human Aβ sequence with Swedish mutations, K595N/M596L (APPswe), associated with the familial form of AD. The second transgenic insert harbors the human presenilin 1 (PS1) protein with a deleted ninth axon (PS1dE9), which was discovered in individuals with early-onset familial AD. In order to study the cholinergic phenotypic changes during AD-like progression in the APPswe/PS1dE9 model, these mice were crossed with transgenic C57BL/6J mice expressing GFP under the control of the Chat promoter. The triple transgenic offspring of APPswe/PS1dE9 (APP.PS1) x CHAT-GFP (CHGFP) will be referred to throughout the paper as APP/CHGFP. All mice were kept on a 12 hour light-dark cycle and given food and water ad 44 libitum. This study used 6- and 8-month-old AD/CHGFP and WT/CHGFP mice for the microarray study and PrPc immunofluorescence, respectively. Genotypes were confirmed with PCR primers specific for human APP. APP/PS1null homozygous ChAT-GFP+/+ transgenic mice expressing GFP from the ChAT promoter, were used as littermate controls for the AD study (WT/ChGFP). Tthese WT/CHGFP mice were used separately for analysis of BFCNs in the 6month old to 24-month old aging profile. Tissue Processing Mice were euthanized with CO2 and decapitated. Brains were placed in an icecold metal brain mold containing 1 mm slots. The medial septum along with the vertical and horizontal diagonal bands of Broca was isolated by cutting a 2 mm section rostral to the optic chiasm. The remaining frontal cortex and olfactory bulbs, rostral to the septal slice, were bisected down the midline and then dissected to isolate each region. The remaining caudal tissue, was bisected down the midline. The left olfactory bulb, left frontal cortex, left hippocampus was placed into small eppendorf tubes on dry ice and then stored at -70°C for biochemical analysis. The right olfactory bulb, right frontal cortex and right hemibrain were drop fixed in 4% paraformaldehyde, 75 mM lysine, 10 mM sodium periodate; pH 7.4 solution for 24 hours at 4 °C and subsequently cryoprotected in a 10% glycerol/2%DMSO solution in 0.1M phosphate buffered saline (PBS, pH 7.4) for 24 hours, afterward the tissues were placed into 20% glycerol/2%DMSO 45 in 0.1M PBS solution and stored at 4°C. The 2 mm forebrain slice containing the basal forebrain and the right hemi-brain containing the hippocampus, were serially sectioned in the coronal plane on a freeze sliding microtome at 40 μm. Free floating sections were then stored in 0.02% sodium azide solution in 0.1M PBS at 4°C. Immunofluorescence For Aβ42 immunofluorescence, free floating sections were removed from 0.02% sodium azide/PBS storage solution and rinsed with PBS. First, the sections were incubated for 3 hr in a blocking buffer solution consisting of 10% normal donkey serum and 0.3% Triton X-100 in PBS. The sections were then incubated overnight with an antibody buffer solution of 1% BSA, 0.3% Triton X-100 in PBS supplemented with recombinant affinity-purified, primary rabbit anti-Aβ42 monoclonal antibody (Life Technologies, 1:2500; cat.# 700254). The next day, sections were rinsed with PBS, blocked in the aforementioned blocking buffer for 3 hrs then incubated in the dark for 6 hrs in the antibody buffer solution supplemented with secondary Alexa Fluor-594 donkey anti-rabbit IgG or donkey anti-goat IgG antibody (Life Technologies,1:1000). After the final PBS rinse, the sections were allowed to dry on SuperfrostPlus slides (Fisher) then coverslipped with VectaShield and allowed to dry at room temperature in the dark, afterwards the slides were stored at -20 °C. Each PBS rinse step consisted of 3-10 min washes and all rinses and incubations were performed at room temperature on a 46 rotating shaker. No Aβ42 fluorescence was observed in wild-type mice or in APP/PS1dE9 mice incubated with secondary antibody alone. For PrPc and p75/NTR immunostaining, tissue sections were washed in PBS (pH 7.4), incubated in blocking buffer solution consisting of 1%BSA and 0.3% Triton X-100 in PBS for two hours to reduce nonspecific binding. Goat anti-mouse PrPc primary antibody (1:750, Abcam) or rabbit anti-mouse p75/NTR primary antibody (Millipore, 1:500) were added to an antibody buffer solution of 10% appropriate serum, 0.3% Triton X-100, 0.02% sodium azide in 0.1 M PBS and the sections were incubated overnight at room temperature on a rotating shaker. Sections were rinsed in PBS and blocked in the aforementioned blocking buffer for 2-3 hours. Sections were then incubated for 6 hr in the dark on a shaker at room temperature, in the aforementioned antibody buffer solution containing secondary donkey anti-rabbit IgG (H+L) or donkey anti-goat IgG (H+L) conjugated to AlexaFluor-594 (1:1000, Molecular Probes). After 3x10 min PBS rinses, the sections were incubated with DAPI (1:1000, Invitrogen) in 0.1M PBS for 10-15 minutes on a shaker in the dark then washed for 15 minutes in PBS. The sections were placed on subbed SuperFrostPlus slides (Fisher) and allowed to dry. Sections were then mounted with ProLong Gold (Invitrogen) or VectaShield (Vector Labs) mounting media and coverslipped. Slides were allowed to harden overnight at 4°C and then sealed with nail polish. 47 Confocal Microscopy Imaging was performed on an LSM710 NLO laser scanning confocal microscope (Zeiss) using Argon/Krypton and Helium/Neon lasers set to 488nm and 594nm, respectively. Z-stack images of the entire MS, VDB and HDB were scanned. Imaging parameters such as pinhole size, detectors, and laser intensity were optimized and held constant in all experiments. Minor heterogeneities from section to section were adjusted using gain and digital offset settings. To minimize crosstalk and bleed-through during imaging, each fluorescent signal was imaged in sequential scanning mode with an EC-Plan NeoFluar 40x/1.30 Oil DIC M27 objective (Zeiss). Image Analysis PrPc Quantification Three coronal sections from 8 female mice (WT; n=4, APP/CHGFP; n=4) spanning the basal forebrain were selected for semiquantitative analyses of PrPc expression in ChAT+ cell bodies. Images were coded and counted in a blinded fashion. In Volocity 3D image analysis software (Perkin Elmer), the orthoslice tool was used to determine the total number of BFCNs expression PrPc in the XZ and Y-Z dimension as shown in Figure 19. Statistical analysis performed with SYSTAT by one-way ANOVA. Data are presented as mean ± SEM. 48 Hippocampal Cholinergic Fiber Quantification after BMP9 Treatment All fluorescence imaging of cholinergic fibers and amyloid plaques was performed on an LSM710 laser scanning confocal microscope (Zeiss) with an EC-Plan NeoFluar 40x/1.30 Oil DIC M27 oil immersion objective at 1.2x optical zoom for final magnification of 48x. Laser settings were optimized and held constant as follows: laser intensity, 2% for Alexa-Fluor 488 and 594; aperture/pinhole, 1.23AU; averaging speed, 4; scan speed, 6; image resolution, 512 x 512. For every mouse (n=29) belonging to one of four groups (APP/BMP, n=7; APP/PBS, n=7; WT/BMP, n=8; WT/PBS, n=7) two coronal hippocampal sections (1 anterior, 1 posterior) were imaged. A total of 263, 10μm z-stacks taken at 1μm intervals were scanned in the hilus of the DG or the stratum radiatum layer of CA1, CA2, and CA3. Images were coded by an individual blinded to the experimental parameters and then transferred to Volocity® 3D analysis software (PerkinElmer). An empirically optimized batch protocol function was established to: 1) find objects of interest using threshold intensities between 20-255, and 2) exclude objects less than 0.3μm3 and greater than 100μm3. The batch protocol was then automatically applied to all images and the total object volume (in μm3) per image/per individual was then exported to an Excel spreadsheet. After a human operator “data-validation” step to confirm Volocity® object recognition [see (Piltti et al., 2011)], a total of 8 images (of 263) required manual correction. Object volumes were accepted as an index of cholinergic fibers present in the hippocampus. Statistical analysis of differences 49 in cholinergic fiber volumes between genotype- and BMP9 treatment groups was performed with SigmaPlot software by two-way ANOVA. Data are expressed as mean ± SEM. RESULTS The aim of these experiments was to test the utility of confocal microscopy to image cholinergic neurons and cholinergic nerve fibers in transgenic mice engineered to express enhanced green fluorescent protein in cholinergic cells. These techniques were applied to studies of aged mice (Figure 4) as well as mice engineered to develop the pathological changes associated with AD for the purpose of understanding the molecular and morphological changes incurred by the basal forebrain cholinergic system during these two processes. In order to study the effects of BMP9 infusion on AD-like pathology, we studied animals at 10-months of age because they display significant amyloid deposition, reduced ChAT activity and BFCN impairments exemplified by neuritic plaques and dystrophic neurites in the cholinergic projection targets of the hippocampus and cerebral cortex (Perez et al., 2007). BMP9 INFUSION IMPROVES AD-LIKE PATHOLOGY IN VIVO We imaged GFP-positive cholinergic fibers in the hippocampus using laser scanning confocal microscopy and quantified z-stacks in the hippocampal areas of the stratum radiatum moleculare of CA1, CA2, CA3 fields and in the hilus of 50 the dentate gyrus. This analysis revealed that BMP9 infusion significantly increased cholinergic fiber densities in WT/CHGFP and APP/CHGFP mice for the combined regions of interest through the hippocampus (Figure 12) and separately within the CA1-stratum radiatum moleculare and hilus of the dentate gyrus (Figures 13A & B). BMP9 infusion dramatically increased ChAT protein in APP/PS1 mice above normal WT levels in the HPC as detected by ELISA and also induced modest but significant increases of hippocampal NGF and NT-3 protein expression levels (Burke et. al, manuscript in preparation). These data provide further evidence that BMP9 supports a neurochemical niche sufficient for BFCN trophic support and may represent a therapy for AD patients by increasing cholinergic neurotransmission to potentially maintain memory or cognitive performance. The observed increase in hippocampal cholinergic density could be attributed to increased axonal branching of existing cholinergic neurons. One interesting observation is that BMP9 increased NT-3 levels which has been shown to increase synaptic connections during development by promoting cholinergic branching and axonal extention to the hippocampus and cerebral cortex (Robertson et al., 2006). A qualitative analysis of BMP9 vs. PBS infused WT/CHGFP mice support the notion of increased cholinergic extentions and possibly branching mediated by neurotrophin signaling possibly via NT-3 (Figure 14). 51 CA1 CA2 Cholinergic fiber volume, µm3 CA3 12000 PBS BMP9 10000 8000 6000 4000 2000 0 Figure 12 hDG WT/CHGFP APP.PS1/CHGFP Quantification of total hippocampal cholinergic fiber densities within the stratum radiatum moleculare (SR) region of CA1, CA2, CA3 and in the hilus of the dentate (hDG) gyrus among the four groups, expressed as mean ± SEM. 52 Cholinergic fibers avoided Aβ42 plaques and in PBS-infused APP/CHGFP mice neuritic plaques and dystrophic neurites were prominent as previously described by (Perez et al., 2007). APP/CHGFP mice that received BMP9 infusion displayed fewer dystrophic neurites adjacent to Aβ42 plaques, as depicted in Figure 15 and there was an enhancement in the cholinergic fiber network in these animals. Visualizaton of Aβ42 immunofluorescence at higher magnifications revealed numerous, diffuse plaques as well as large plaques in the hippocampus of PBS-infused mice while BMP9 infused mice contained mostly large, dense plaques. As Aβ42 is more hydrophobic than Aβ40, it is postulated that its oligomers are the most toxic Aβ species (Selkoe, 1997). Figure 13 (Next two pages) Confocal images from each of the four experimental groups and a scatter plot (n=27) of the cholinergic densities in the hilus of the dentate gyrus (A) and CA1/stratum radiatum (B) regions of the hippocampus. This analysis revealed that BMP9 effectively increased cholinergic innervation to the hippocampus of WT/CHGFP and APP/CHGFP mice. Red bars are the mean for each group; scale bar, 50 μm. 53 Mean DG Fiber Counts 13A. Mean Fiber Density in DG (μm3) 25000 20000 Y Data 15000 10000 5000 0 Wt/Pbs Wt/Bmp App/Pbs App/Bmp X Data Group vs FIber Counts/Objects BMP9 APP/PS1 WT PBS 54 Mean CA1 Counts Mean Fiber Density in CA1 (um3) 13B. 18000 16000 14000 Y Data 12000 10000 8000 6000 4000 2000 0 Wt/Pbs Wt/Bmp App/Pbs App/Bmp X Data Group vs FIber Counts/Objects BMP9 APP/PS1 WT PBS 55 BMP9 WT/CHGFP PBS Figure 14 Cholinergic neurites in the granular layer of CA1 appeared longer in WT/CHGFP mice infused with BMP9 versus PBS (40x). 56 PBS Ab42 GFP Ab42 GFP BMP9 Figure 15 Confocal images of immunofluorescent staining of Aβ42 (red) in the hippocampus also showing disrupted cholinergic network (green). PBS- infused (controls) contained weak, inconsistent fibers and more dystrophic neurites (white arrows) whereas BMP9 infusion ameliorated these cholinergic deficits in APP/CHGFP animals. 57 The apparent BMP9 mediated reductions in the number of smaller, diffuse plaques may effectively reduce the total plaque surface area and minimize disruptive Aβ42 interactions with proteins of essential function. The mechanism by which BMP9 reduces Aβ42 plaque burden is a prospect for further research to determine whether BMP9 reduces plaque deposition or increases plaque clearance. The ability of BMP9 to induce exuberant cholinergic growth in the hippocampus of an AD-like model compels future studies to determine if the improved cholinergic network improves cognition in the APPswe/PS1dE9 mice. This could be achieved using well characterized cognitive-behavioral assays, e.g. the Barnes maze to test spatial memory capabilities of the mouse to locate a dark escape box for safety, or the Morris water maze which also measures visuospatial memory as a function of latent time to locate a platform and escape from the stress of swimming. These data demonstrate that administration of BMP9 ameliorates two overt pathophysiological components of AD by reducing amyloid plaque burden and by increasing the ChAT-positive cholinergic network in the hippocampus. BFCN GENE EXPRESSION IN APP/CHGFP MICE Of approximately 30,000 genes in the human genome, different cell types express only an individual subset of these genes which permit their specification and function. Cells that are selectively affected by disease, are known to 58 undergo homeostatic or pathologic changes in gene expression, a paradigm that may provide diagnostic value or biomarker identification and/or medical targets of therapy. The molecular analysis of specific cell types in the brain is hampered by the brain’s heterogeneity of densely packed neuronal subclasses and glial cells. Furthermore, a given neuronal population within a specific brain region likely represents a minority subset of cells within that region. Despite this, whole cell populations dissociated from a resected area of the brain have enabled a variety of in vitro studies of cell-function and cell-cell interactions but a thorough transcriptomic analysis of a specific cell type remains challenging with this methodology. Importantly, fluorescence-activated cell sorting (FACS) has been used to identify subsets of cells in mixed populations. This technique has provided major advances in cancer and immunology research but is underutilized in studying the CNS. Originally, a method for the generation of healthy, purified BFCN cultures by FACS using a fluorescent antibody against p75/NTR produced cultures enriched in the cholinergic markers p75/NTR, ChAT and ChT (Schnitzler et al., 2008a). This method has been simplified with the use of transgenic mice expressing GFP from the ChAT promoter and can be a powerful tool for purifying BFCNs to study their gene expression profiles in normal, aging and diseased states. 59 Figure 16 FACS purified BFCNs from WT/ChAT-eGFP. Note: 3.7% of cells from E18 mice septae express ChAT-eGFP and these cells have enriched mRNA expression levels for BFCN markers; Chat, p75, Cht. Loading control: Actb; beta-actin. Adapted from: A.C. Schnitzler et. al, J. Neurosci. (2010). 60 To study the gene expression profile of BFCNs in the APPswe/PS1dE9, a mouse model with AD-like pathology, the septal region was harvested from 6 month old WT/CHGFP and APP/CHGFP mice expressing GFP from the ChAT promoter. BFCNs constitute approximately 3-5% of the entire cell population in the septum as indicated in Figure 16 and previous experiments. FACS purified BFCN mRNA was extracted using the guanidiniumthiocyanate-phenol/chloroform method, after a cRNA amplification step and a spiked standard curve was used to normalize RNA frequencies. Next, mRNA reaction mixtures from WT/CHGFP and APP/CHGFP BFCNs obtained by FACS were enriched in mRNA for the cholinergic markers p75/NTR, ChAT and ChT and then subjected to Affymetrix Mouse Genome 430 2.0. These arrays were stained with Streptavidin R-phyocoerythrin using the GeneChip Fluidics Station 400 and scanned with a Hewlett-Packard GeneArray Scanner. The data collected was then analyzed by Microarray Suite 4.0 software. This analysis identified some 300 differentially expressed genes in WT/CHGFP and APP/CHGFP mice BFCNs which clearly formed separate clusters as shown in Figure 17. Some of these genes as shown in Figure 18 have been previously implicated in the pathophysiology of AD and amyloidosis. In particular one of these genes is LIMK1. 61 LIMK1 mRNA levels were reduced 50% below WT expression levels. LIMK1 is cytoplasmic protein kinase regulating cytoskeletal structure and function and is important during neurodevelopment by promoting axonal extention. It is very interesting that LIMK1 has been shown to directly interact with the BMP type II receptors to deregulate actin dynamics (Foletta et al., 2003). LIMK1 also interacts with neuregulins which are known to be important for the development of hippocampal-dependent visuospatial cognition (Wang et al., 1998). Furthermore, inhibition of Aβ-induced LIMK1 phosphorylation of Cofilin with a specific peptide competitor abrogated hippocampal neuronal dystrophy and degeneration (Heredia et al., 2006). In light of these data, our observation that LIMK1 mRNA is reduced in BFCNs of AD-like mice may validate the use of this model to advance the study of human AD, since as altered levels of phospho-LIMK1 have been reported in human brain regions affected by AD (Heredia et al., 2006). 62 Figure 17 Gene expression analysis with Affymetrix microarrays in FACS-purified BFCN from 6 month old wild type (WT) and APP.PS1 male mice. Graphical representation of cluster analysis of 300 differentially-expressed genes is shown. Note that the WT and APP.PS1 BFCN genes organize clusters. 63 into clearly separate mRNA levels, % of wild type 175 150 WT APP.PS1 125 100 75 50 25 0 Limk1 Map2 Capn2 Il33 Notch3 FIGURE 18 Candidate genes with differential mRNA expression in BFCN purified from 6-month old APP.PS1 male mice as compared with the WT littermates. APP/PS1 mRNA expression levels in the graph above were normalized to WT expression levels derived from Figure 17. 64 PrPc IS UPREGULATED IN BFCNs OF APP/CHGFP MICE The mRNA coding for the cellular prion protein (PrPc) was observed to be upregulated in BFCNs of APPswe/PS1dE9 mice. PrPc is a glycosyl phosphatidylinositol (GPI) anchored-cell surface protein which localizes to lipid rafts in the extracellular leaflet of the plasma membrane. It is expressed in the peripheral tissues as well as in neuronal cell bodies and at post-synaptic densities. In its misfolded form, PrPc is converted to amyloidogenic PrPSc and is responsible for the fatal neurodegenerative disorders classified as transmissible spongiform encephalopathies, including Creutzfeldt-Jakob disease, kuru and Gersmann-Straussler-Scheinker syndrome (Prusiner and DeArmond, 1990)). Also, a D178N mutation and homozygosity for methionine at PrPc codon 129 are known to cause fatal familial insomnia (Montagna et al., 2003). Interestingly, a single nucleotide polymorphism that causes a M129V mutation in PrP c has also been associated with late-onset Alzheimer’s disease (Gunther and Strittmatter, 2010). Much more is known about the pathogenic actions of PrPc however new research is beginning to shed light onto the physiological roles of this protein. PrPc functions as a copper-dependent NMDA-R ion channel regulator (You et al., 2011), as a cell adhesion molecule by interacting with integrins, NCAM and laminin, as reviewed in (Biasini et al., 2012) and as a. It has been argued that Aβ oligomers are dependent on PrPc expression to mediate LTP inhibition and 65 synaptic dysfunction in pyramidal neurons of the hippocampus (Gimbel et al., 2010), whereas the physiological and pathological function of PrPc expression within BFCNs is largely unknown. Research has demonstrated two possible ways that Aβ/PrPc interactions can cause excitotoxicity in neurons. First, Aβ treatment of dissociated GABAergic and cholinergic forebrain neurons caused reductions in calcium-activated potassium conductance in a PrPc dependent fashion (Alier et al., 2011). This scenario could effectively reduce the refractory period of membrane repolarization, extend action potentials and excitability and facilitate BFCN excitotoxicity. Secondly, coimmunoprecipation of PrP c with NR2D subunits of NMDA receptors suggests a direct interaction with this glutamate activated non-selective cation channel. Hippocampal neurons treated with copper chelators, Aβ42 or PrPc inactivation resulted in nondesensitized NMDA-induced currents and neurotoxicity (You et al., 2011). This suggests that PrPc may normally modulate ion channels concentrated at synapses whereas Aβ binding to PrPc interferes with this essential regulatory function preventing excitotoxicity. Interestingly, PrPc has been shown to undergo α- and γ-secretase cleavage, similar to APP (Checler, 2012) and provides further evidence for common mechanisms in neurodegenerative diseases. Previous members in our lab enabled the microarray analysis of BFCN mRNA (Schnitzler et al., 2008a). This analysis implicated PrPc as a gene displaying a dysregulated expression profile in the APPswe/PS1dE9 mouse 66 model of AD. BFCNs from 6-month old APP/CHGFP mice exhibited a 2.8-fold increase in PrPc mRNA levels as compared to WT/CHGFP BFCNs (p = 0.02). In light of this, we sought to determine the percentage of CHATGFPpositive BFCNs expressing PrPc protein using semi-quantitative immunofluorescence and laser scanning confocal microscopy. After immunofluorescence and imaging, the Volocity software orthoslice tool was used to quantify the total number of of cholinergic neurons in the basal forebrain and in the striatum that coexpressed PrPc as illustrated in Figure 19. The striatum (STR) is a forebrain region associated with planning, anticipating and modulating pathways that control movement and procedural memory. The striatum is comprised of medium spiny neurons (96%), Dieter’ neurons (2%), parvalbumin-, calretinin-, somatostatin-positive GABAergic interneurons as well as cholinergic interneurons (1%). Cholinergic interneurons in the striatum did not demonstrate any major difference in PrPc protein expression among APP/CHGFP as compared to WT/CHGFP mice. At older ages, the APPswe/PS1dE9 mice develop overt cholinergic deficits and Aβ-plaque deposition the striatum (Perez et al., 2007). Consistent with these reports, we did not see any amyloidosis in the striatum of these 8 month old mice, however signs of cellular blebbing and dystrophic cholinergic neurons in the striatum were observed at this time point. This result suggests that cholinergic degeneration in the striatum may precede Aβ deposition which points to trophic withdrawal, 67 excitotoxicity or other uncharacterized mechanisms as the cause of cholinergic dysfunction. Consistent with other studies in mice and in humans, immunofluorescence for Aβ plaques did not reveal any staining in the MSN, VDB and HDB. However, the percentage of BFCNs expressing PrPc in these regions revealed a 46% increase in PrPc protein expression as shown in Figure 20. The functional significance of increased PrPc expression in APPswe/PS1dE9 BFCNs remains to be determined although it provides fertile ground to study the role of PrP c as a possible pathogenic factor mediating cholinergic dysfunction in AD. 68 Figure 19 An orthoslice screenshot from the quantitative analysis procedure used to determine PrPc and ChATGFP overlap. Sections through the basal forebrain cholinergic nuclei and striatum were immunostained for PrPc (red). 3D Z-stack images were imported to a Volocity library and analyzed in the X, Y, and Z planes using the orthoslice tool. This analysis enabled counting of total number of neurons positive for both ChATGFP and PrPc in both the MS/VDB/HDB and the STR areas (40x objective). 69 DISCUSSION These results support our underlying hypotheses that as a result of aging and AD,that BFCNs acquire altered morphological features, and BFCNs in AD display a differential gene-protein expression profile. Secondly, we hypothesized that BMP9, an induction factor sufficient for BFCN differentiation in-vitro and invivo, would support BFCN maintenance would promote BFCN growth and survivability, in a model of AD characterized by heavy Aβ-amyloidosis and a pronounced cholinergic deficit. Our data reveal that a 7-day BMP9 infusion induced an exuberant improvement in the cholinergic network in the hippocampus of WT/CHGFP and APP/CHGFP mice. We detected a quantitative increase in cholinergic fiber density within the DG (hilus) and the stratum radiatum layers of CA1, CA2 and CA3 with BMP9 infusion. Consistent with previous reports, BMP9 also increased expression of the NGF receptors, p75/NTR and TrkA (data not shown; Burke et. al, manuscript in preparation). BMP9 induced the BFCN phenotype and an optimized trophic environment for BFCNs by increasing levels of NGF and NT-3 (Burke et. al, manuscript in preparation) which possibly contributed to the robust upregulation of cholinergic innervation in the hippocampus of APP/CHGFP mice. Since ACh neurotransmission acts as a neuromodulator, the regrowth and replacement of the exact cholinergic synapses may not be necessary to enhance memory and cognition (Bissonnette et al., 2011). 70 BMP9 infusion quantitatively reduced Aβ plaque levels in the anterior and posterior hippocampal sections of APP/CHGFP mice (Burke et. al, manuscript in preparation). BMP9 also reduced the number of dystrophic cholinergic neurites surrounding Aβ plaques in the hippocampus and shifted plaque immunofluorescent staining patterns from scattered-diffuse to dense-compact plaque morphologies. These data demonstrate that brains affected by AD-like amyloidosis may be primed for BMP9 signaling since a short 7-day infusion resulted in a dramatic increase in cholinergic fiber density and a reduction of Aβ in APP/CHGFP mice. The data from this AD-like mouse model suggest that BMP9 may possess some therapeutic value to counteract the two major pathophysiological hallmarks of AD, Aβ -amyloidosis and the cholinergic deficit. With aging and AD, BFCNs undergo morphological changes. Qualitatively, we observed that BFCNs in 24-month old WT/CHGFP mice appeared to have retracted arborizations and their cell somas were more rounded in shape and smaller in size. These alterations may be due to ageinduced disruptions of specific cytoskeleton networks leading to reduced stability, cell shrinkage and axonal retraction. Not surprisingly, APP/CHGFP BFCNs were also observed to display smaller soma sizes and shorter projections as compared to healthy WT/CHGFP mice. Based on the microarray data for APP/CHGFP BFCNs, proteins associated with cytoskeletal architecture appear to be involved in the AD-like pathophysiology affecting the form and function of 71 BFCNs. MAP2 mRNA was reduced by 50% and CAPN2 levels were increased by more than 75% in BFCNs from APP/CHGFP mice. Calpain 2 (CAPN2) is a calcium-activated neutral protease located to lipid rafts where it regulates cytoskeleton turnover by cleaving actin, microtubuleassociated proteins, spectrins and neurofilaments (Adamec et al., 2002). CAPN2 has been associated in a variety of neurodegenerative disorders including AD. Using confocal immunofluorescence, the active form of CAPN2 was shown to colocalize with 50-75% of hyperphosphorylated, neurofibrillary tau tangles (NFTs) in AD brain tissue (Adamec et al., 2002). These studies as well as our microarray gene expression analysis of APPswe/PS1dE9 BFCNs implicate the proteolytic mechanism of CAPN2 as a potential driver of cytoskeletal disruptions of cholinergic morphology during AD-like pathophysiology. MAP2 is another cytoskeletal protein which crosslinks and stabilizes microtubules to microtubules and microtubules to intermediate filaments. Dendritic localized MAP2 and axonal localized Tau (MAPT) are in the same microtubule-associated protein (MAP) family and hyperphosphorylation of these proteins leads to microtubule destabilization, aggregation and NFT formation. The APPswe/PS1dE9 AD-like model do not produce NFTs whereas NFTs are a hallmark feature observed in post-mortem human AD brain tissue. 72 PrPc APP/CHGFP WT/CHGFP ChAT-GFP 73 Merge Figure 20 Representative images and quantification of PrPc expression (red) in BFCNs (green). APP/CHGFP (APP.PS1) basal forebrain cholinergic neurons reveal a significant increase in the percentage of BFCNs expressing PrPc as compared to WT/CHGFP BFCNs (p=0.007). White arrows in upper panel indicate PrPc+ and CHATGFP+ cells. Note that APP/CHGFP BFCNs (left, bottom panel) consistently appear smaller than WT BFCNs (left,top panel). 74 Based on PrPc mRNA levels from the microarray analysis, we compiled z-stacks of BFCNs expressing GFP and used a specific immunofluorescent staining to detect PrPc protein expression in BFCNs. When compared to WT/CHGFP control littermates, the percentage of BFCNs in APP/CHGFP BFCNs expressing PrPc increased by 46% (Figure 20). It should be noted that the engineered APPswe and PS1dE9 transgenes are under the transcriptional control of an upstream (Prnp) prion promoter in order to drive high transgene expression in the brain and highest expression is observed in the cortex and hippocampus (Perez et al., 2007) however our studies focused on the basal ganglia cholinergic nuclei in the medial septum and the diagonal bands of Broca. It is highly unlikely that the integration of the exogenous Prnp promoter would increase the transcriptional activity of the endogenous promoter leading to increased PrPc mRNA or protein expression in basal forebrain cholinergic neurons. Additionally, transcription preinitiation complexes formed by TFIIA, -IIB, -IID, -IIE, -IIF, -IIH and RNA polymerase II at transcription start sites specify basal level gene expression and are found to be in general abundance within the nucleus (Thomas and Chiang, 2006). However specific coactivators and cofactors which fine-tune gene-specific promoter activity may be in shorter supply. In this scenario, introduction of the exogenous, transgenic Prnp promoter could reduce the endogenous Prnp promoter activity and PrPc levels by roughly half. Furthermore, there was no difference between PrPc expression in striatal cholinergic interneurons of WT and APPswe/PS1dE9 75 mice, indicating that PrPc up-regulation demonstrated some cell-type specificity. These data lead us to reason that the measured increase of PrPc expression in BFCNs is not due to the effects of the exogenous Prnp promoter but is a byproduct of the underlying pathophysiology that this AD model phenotypically expresses. The upregulation of PrPc is interesting due to the fact that Aβ-PrPc was shown to inhibit synaptic plasticity in the hippocampus (Lauren et al., 2009). Aβ binding to PrPc may interfere with its physiological functions at the synapse, such as NMDA-R regulation and cell adhesion or it may activate aberrant signaling cascades. In support of this idea, two independent groups have proposed a pathologic mechanism for Aβ oligomer signaling via PrPc leading to activated FYN kinase (Larson et al., 2012; Um et al., 2012). Um et al, (Um et al., 2012) determined that oligomeric Aβ-PrPc-FYN kinase signaling reduced synaptic levels of NMDA-R/NR2B subunits, induced dendritic spine retraction and neuronal impairment. Larson et al, (Larson et al., 2012) used size exclusion chromatography to show that Aβ dimers from human AD brain extracts bind PrPc, leading to phosphorylation and activation of FYN kinase which resulted in tau phosphorylation and toxicity in vivo. 76 Cholinergic Hypothesis PrP c Aβ-Hypothesis Tau Hypothesis Figure 21 PrPc at the center of the AD Triad. This proposed model shows how PrPc contributes to all three AD hypotheses. This model is based on results from multiple labs demonstrating the role of PrPc in various in-vitro and in-vivo experiments, however the contribution of PrPc to human AD is still uncertain. 77 We present the first examination of PrPc expression levels in BFCNs of this AD-like mouse model. This increase in PrPc mRNA expression in 6-month old and PrPc protein levels in 8-month old APPswe/PS1dE9 mice suggests that PrPc upregulation may potentially be an early event during AD-like pathogenesis in this model. The relationship of PrPc to all three major AD hypotheses is especially intriguing. The convergence of three pathophysiological mechanisms observed in AD onto PrPc implicates it as a target for AD treatment through the use of a small peptidomimetic competitor of Aβ-PrPc interactions which would be able to cross the blood-brain barrier. Finally, PrPc is known to undergo proteolytic cleavage by ADAM17/TACE to generate an N1 terminal fragment that mediates neuroprotection (Checler, 2012). Fluharty et al, have also demonstrated that this PrPc-N1 fragment binds Aβ to prevent neurotoxicity (Fluharty et al., 2013). Since PrPc was elevated in the APPswe/PS1dE9 model of AD the next studies should investigate PrPc levels in basal forebrain cholinergic neurons of human AD brain samples to determine if this protein is relevant to human disease. Since the upregulation of PrPc may also lead to increased N1 shedding, this neuroprotective fragment of PrPc could potentially be a biomarker of AD. 78 LIST OF JOURNAL ABBREVIATIONS ACS Chem Biol ACS Chemical Biology Alzheimers Res Ther Alzheimers Research and Therapy Ann Neurol Annals of Neurology Annu Rev Neurosci Annual Review of Neuroscience Arch Intern Med Archives of Internal Medicine Behav Brain Res Behavioural Brain Research Biochem J Biochemical Journal Biol Psychiatry Biological Psychiatry Brain Res Brain Research Brain Res Dev Brain Res Brain Research Developmental Brain Research Curr Opin Cell Biol Current Opinions in Cell Biology Curr Opin Neurobiol Current Opinion in Neurobiology Curr Top Med Chem Current Topics in Medicinal Chemistry Dev Neurosci Developmental Neuroscience Embo J The EMBO Journal Eur J Neurosci European Journal of Neuroscience Expert Rev Neurother Expert Review of Neurotherapeutics J Biol Chem Journal of Biological Chemistry J Cell Biol Journal of Cell Biology J Chem Neuroanat Journal of Chemical Neuroanatomy J Comp Neurol Journal of Comparative Neurology 79 J Geriatr Psychiatry Neurol Journal of Geriatric Psychiatry and Neurology J Neurochem Journal of Neurochemistry J Neurosci Journal of Neuroscience J Neurosci Res Journal of Neuroscience Research J Pharmacol Exp Ther Journal of Pharmacology and Experimental Therapeutics J Physiol Paris Journal of Physiology - Paris Life Sci Life Science Mol Cell Biol Molecular and Cellular Biology Mol Neurobiol Molecular Neurobiology Nat Neurosci Nature Neuroscience Nat Protoc Nature Protocols Neurobiol Aging Neurobiology of Aging Neurobiol Dis Neurobiology of Disease Neurosci Lett Neuroscience Letters Physiol Genomics Physiological Genomics Proc Natl Acad Sci U S A Proceedings of the National Academies of Sciences, United States of America Stem Cell Res Stem Cell Research Trends Cell Biol Trends in Cell Biology Trends Neurosci Trends in Neuroscience 80 REFERENCES Adamec E, Mohan P, Vonsattel JP, Nixon RA (2002) Calpain activation in neurodegenerative diseases: confocal immunofluorescence study with antibodies specifically recognizing the active form of calpain 2. Acta Neuropathol 104:92-104. Aizawa S, Teramoto K, Yamamuro Y (2012) Histone deacetylase 9 as a negative regulator for choline acetyltransferase gene in NG108-15 neuronal cells. Neuroscience 205:63-72. Alier K, Ma L, Yang J, Westaway D, Jhamandas JH (2011) Abeta inhibition of ionic conductance in mouse basal forebrain neurons is dependent upon the cellular prion protein PrPC. J Neurosci 31:16292-16297. Aloe L (2004) Rita Levi-Montalcini: the discovery of nerve growth factor and modern neurobiology. Trends Cell Biol 14:395-399. Auld DS, Mennicken F, Quirion R (2001) Nerve growth factor rapidly induces prolonged acetylcholine release from cultured basal forebrain neurons: differentiation between neuromodulatory and neurotrophic influences. J Neurosci 21:3375-3382. Barrett GL (2000) The p75 neurotrophin receptor and neuronal apoptosis. Prog Neurobiol 61:205-229. Bartus RT, Dean RL, 3rd, Beer B, Lippa AS (1982) The cholinergic hypothesis of geriatric memory dysfunction. Science 217:408-414. Basi GS, Hemphill S, Brigham EF, Liao A, Aubele DL, Baker J, Barbour R, Bova M, Chen XH, Dappen MS, Eichenbaum T, Goldbach E, Hawkinson J, Lawler-Herbold R, Hu K, Hui T, Jagodzinski JJ, Keim PS, Kholodenko D, Latimer LH, Lee M, Marugg J, Mattson MN, McCauley S, Miller JL, Motter R, Mutter L, Neitzel ML, Ni H, Nguyen L, Quinn K, Ruslim L, Semko CM, Shapiro P, Smith J, Soriano F, Szoke B, Tanaka K, Tang P, Tucker JA, Ye XM, Yu M, Wu J, Xu YZ, Garofalo AW, Sauer JM, Konradi AW, Ness D, Shopp G, Pleiss MA, Freedman SB, Schenk D (2010) Amyloid precursor 81 protein selective gamma-secretase inhibitors for treatment of Alzheimer's disease. Alzheimers Res Ther 2:36. Bateman RJ, Siemers ER, Mawuenyega KG, Wen G, Browning KR, Sigurdson WC, Yarasheski KE, Friedrich SW, Demattos RB, May PC, Paul SM, Holtzman DM (2009) A gamma-secretase inhibitor decreases amyloidbeta production in the central nervous system. Ann Neurol 66:48-54. Bekris LM, Millard S, Lutz F, Li G, Galasko DR, Farlow MR, Quinn JF, Kaye JA, Leverenz JB, Tsuang DW, Yu CE, Peskind ER (2012) Tau phosphorylation pathway genes and cerebrospinal fluid tau levels in Alzheimer's disease. Am J Med Genet B Neuropsychiatr Genet 159B:874883. Berse B, Blusztajn JK (1995) Coordinated up-regulation of choline acetyltransferase and vesicular acetylcholine transporter gene expression by the retinoic acid receptor alpha, cAMP, and leukemia inhibitory factor/ciliary neurotrophic factor signaling pathways in a murine septal cell line. J Biol Chem 270:22101-22104. Berse B, Lopez-Coviella I, Blusztajn JK (1999) Activation of TrkA by nerve growth factor upregulates expression of the cholinergic gene locus but attenuates the response to ciliary neurotrophic growth factor. Biochem J 342 ( Pt 2):301-308. Biasini E, Turnbaugh JA, Unterberger U, Harris DA (2012) Prion protein at the crossroads of physiology and disease. Trends Neurosci 35:92-103. Bibel M, Hoppe E, Barde YA (1999) Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. Embo J 18:616-622. Bielarczyk H, Szutowicz A (1989) Evidence for the regulatory function of synaptoplasmic acetyl-CoA in acetylcholine synthesis in nerve endings. Biochem J 262:377-380. Bird TD (1993) Alzheimer Disease Overview. 82 Bissonnette CJ, Lyass L, Bhattacharyya BJ, Belmadani A, Miller RJ, Kessler JA (2011) The controlled generation of functional basal forebrain cholinergic neurons from human embryonic stem cells. Stem Cells 29:802-811. Blusztajn JK, Berse B (2000) The cholinergic neuronal phenotype in Alzheimer's disease. Metabolic Brain Disease 15:45-64. Bolognesi B, Kumita JR, Barros TP, Esbjorner EK, Luheshi LM, Crowther DC, Wilson MR, Dobson CM, Favrin G, Yerbury JJ (2010) ANS binding reveals common features of cytotoxic amyloid species. ACS Chem Biol 5:735-740. Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathologica 82:239-259. Brady DR, Phelps PE, Vaughn JE (1989) Neurogenesis of basal forebrain cholinergic neurons in rat. Brain Res Dev Brain Res 47:81-92. Chao MV, Hempstead BL (1995) p75 and Trk: a two-receptor system. Trends Neurosci 18:321-326. Checler F (2012) Two-steps control of cellular prion physiology by the extracellular regulated kinase-1 (ERK1). Prion 6:23-25. Cho G, Lim Y, Zand D, Golden JA (2008) Sizn1 is a novel protein that functions as a transcriptional coactivator of bone morphogenic protein signaling. Mol Cell Biol 28:1565-1572. Cooper JD, Lindholm D, Sofroniew MV (1994) Reduced transport of [125I]nerve growth factor by cholinergic neurons and down-regulated TrkA expression in the medial septum of aged rats. Neuroscience 62:625-629. Costantini C, Weindruch R, Della Valle G, Puglielli L (2005) A TrkA-to-p75NTR molecular switch activates amyloid beta-peptide generation during aging. Biochem J 391:59-67. 83 Dale JK, Vesque C, Lints TJ, Sampath TK, Furley A, Dodd J, Placzek M (1997) Cooperation of BMP7 and SHH in the induction of forebrain ventral midline cells by prechordal mesoderm. Cell 90:257-269. Davies P, Maloney AJ (1976) Selective loss of central cholinergic neurons in Alzheimer's disease. Lancet 2:1403. de Souza LC, Chupin M, Lamari F, Jardel C, Leclercq D, Colliot O, Lehericy S, Dubois B, Sarazin M (2012) CSF tau markers are correlated with hippocampal volume in Alzheimer's disease. Neurobiol Aging 33:12531257. Dickson DW, Crystal HA, Mattiace LA, Masur DM, Blau AD, Davies P, Yen SH, Aronson MK (1992) Identification of normal and pathological aging in prospectively studied nondemented elderly humans. Neurobiol Aging 13:179-189. Doucette R, Ball MJ (1987) Left-right symmetry of neuronal cell counts in the nucleus basalis of Meynert of control and of Alzheimer-diseased brains. Brain Res 422:357-360. Eiden LE (1998) The cholinergic gene locus. J Neurochem 70:2227-2240. Fagan AM, Garber M, Barbacid M, Silos-Santiago I, Holtzman DM (1997) A role for TrkA during maturation of striatal and basal forebrain cholinergic neurons in vivo. J Neurosci 17:7644-7654. Flames N, Pla R, Gelman DM, Rubenstein JL, Puelles L, Marin O (2007) Delineation of multiple subpallial progenitor domains by the combinatorial expression of transcriptional codes. J Neurosci 27:9682-9695. Fluharty BR, Biasini E, Stravalaci M, Sclip A, Diomede L, Balducci C, La Vitola P, Messa M, Colombo L, Forloni G, Borsello T, Gobbi M, Harris DA (2013) An N-terminal Fragment of the Prion Protein Binds to Amyloid-beta Oligomers and Inhibits Their Neurotoxicity in Vivo. J Biol Chem 288:78577866. 84 Foletta VC, Lim MA, Soosairajah J, Kelly AP, Stanley EG, Shannon M, He W, Das S, Massague J, Bernard O (2003) Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J Cell Biol 162:1089-1098. Francis PT, Palmer AM, Snape M, Wilcock GK (1999) The cholinergic hypothesis of Alzheimer's disease: a review of progress. Journal of Neurology, Neurosurgery & Psychiatry:137-147. Gelman DM, Martini FJ, Nobrega-Pereira S, Pierani A, Kessaris N, Marin O (2009) The embryonic preoptic area is a novel source of cortical GABAergic interneurons. J Neurosci 29:9380-9389. Gimbel DA, Nygaard HB, Coffey EE, Gunther EC, Lauren J, Gimbel ZA, Strittmatter SM (2010) Memory impairment in transgenic Alzheimer mice requires cellular prion protein. Journal of Neuroscience 30:6367-6374. Gnahn H, Hefti F, Heumann R, Schwab ME, Thoenen H (1983) NGF-mediated increase of choline acetyltransferase (ChAT) in the neonatal rat forebrain: evidence for a physiological role of NGF in the brain? Brain Res 285:4552. Gong CX, Singh TJ, Grundke-Iqbal I, Iqbal K (1993) Phosphoprotein phosphatase activities in Alzheimer disease brain. J Neurochem 61:921927. Greferath U, Trieu J, Barrett GL (2012) The p75 neurotrophin receptor has nonapoptotic antineurotrophic actions in the basal forebrain. J Neurosci Res 90:278-287. Grothe M, Heinsen H, Teipel SJ (2012) Atrophy of the cholinergic Basal forebrain over the adult age range and in early stages of Alzheimer's disease. Biol Psychiatry 71:805-813. Guadagna S, Esiri MM, Williams RJ, Francis PT (2012) Tau phosphorylation in human brain: relationship to behavioral disturbance in dementia. Neurobiol Aging 33:2798-2806. 85 Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, Hazrati L, Collinge J, Pocock J, Lashley T, Williams J, Lambert JC, Amouyel P, Goate A, Rademakers R, Morgan K, Powell J, St George-Hyslop P, Singleton A, Hardy J (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368:117-127. Gunther EC, Strittmatter SM (2010) Beta-amyloid oligomers and cellular prion protein in Alzheimer's disease. J Mol Med (Berl) 88:331-338. Haense C, Kalbe E, Herholz K, Hohmann C, Neumaier B, Krais R, Heiss WD (2012) Cholinergic system function and cognition in mild cognitive impairment. Neurobiol Aging 33:867-877. Heredia L, Helguera P, de Olmos S, Kedikian G, Sola Vigo F, LaFerla F, Staufenbiel M, de Olmos J, Busciglio J, Caceres A, Lorenzo A (2006) Phosphorylation of actin-depolymerizing factor/cofilin by LIM-kinase mediates amyloid beta-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer's disease. J Neurosci 26:6533-6542. Hernandez CM, Kayed R, Zheng H, Sweatt JD, Dineley KT (2010) Loss of alpha7 nicotinic receptors enhances beta-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and septohippocampal pathology in a mouse model of Alzheimer's disease. J Neurosci 30:24422453. Hersh LB, Shimojo M (2003) Regulation of cholinergic gene expression by the neuron restrictive silencer factor/repressor element-1 silencing transcription factor. Life Sci 72:2021-2028. Huber LJ, Chao MV (1995) A potential interaction of p75 and trkA NGF receptors revealed by affinity crosslinking and immunoprecipitation. J Neurosci Res 40:557-563. Imbimbo BP, Giardina GA (2011) gamma-secretase inhibitors and modulators for the treatment of Alzheimer's disease: disappointments and hopes. Curr Top Med Chem 11:1555-1570. 86 Iqbal K, Liu F, Gong CX, Alonso Adel C, Grundke-Iqbal I (2009) Mechanisms of tau-induced neurodegeneration. Acta Neuropathol 118:53-69. Johnson EM, Jr., Taniuchi M, Clark HB, Springer JE, Koh S, Tayrien MW, Loy R (1987) Demonstration of the retrograde transport of nerve growth factor receptor in the peripheral and central nervous system. J Neurosci 7:923929. Jope RS, Jenden DJ (1980) The utilization of choline and acetyl coenzyme A for the synthesis of acetylcholine. J Neurochem 35:318-325. Kaplan DR, Miller FD (1997) Signal transduction by the neurotrophin receptors. Curr Opin Cell Biol 9:213-221. Kuner P, Schubenel R, Hertel C (1998) Beta-amyloid binds to p75NTR and activates NFkappaB in human neuroblastoma cells. J Neurosci Res 54:798-804. Larson M, Sherman MA, Amar F, Nuvolone M, Schneider JA, Bennett DA, Aguzzi A, Lesne SE (2012) The complex PrP(c)-Fyn couples human oligomeric Abeta with pathological tau changes in Alzheimer's disease. J Neurosci 32:16857-16871a. Lauren J, Gimbel DA, Nygaard HB, Gilbert JW, Strittmatter SM (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. 457:1128-1132. Li C, Ullrich B, Zhang JZ, Anderson RG, Brose N, Sudhof TC (1995a) Ca(2+)dependent and -independent activities of neural and non-neural synaptotagmins. Nature 375:594-599. Li Y, Holtzman DM, Kromer LF, Kaplan DR, Chua-Couzens J, Clary DO, Knusel B, Mobley WC (1995b) Regulation of TrkA and ChAT expression in developing rat basal forebrain: evidence that both exogenous and endogenous NGF regulate differentiation of cholinergic neurons. J Neurosci 15:2888-2905. 87 Lobello K, Ryan JM, Liu E, Rippon G, Black R (2012) Targeting Beta amyloid: a clinical review of immunotherapeutic approaches in Alzheimer's disease. Int J Alzheimers Dis 2012:628070. Lopez-Coviella I, Berse B, Thies RS, Blusztajn JK (2002) Upregulation of acetylcholine synthesis by bone morphogenetic protein 9 in a murine septal cell line. J Physiol Paris 96:53-59. Lopez-Coviella I, Berse B, Krauss R, Thies RS, Blusztajn JK (2000) Induction and maintenance of the neuronal cholinergic phenotype in the central nervous system by BMP-9. Science 289:313-316. Lopez-Coviella I, Mellott TM, Kovacheva VP, Berse B, Slack BE, Zemelko V, Schnitzler A, Blusztajn JK (2006) Developmental pattern of expression of BMP receptors and Smads and activation of Smad1 and Smad5 by BMP9 in mouse basal forebrain. Brain Res 1088:49-56. Lopez-Coviella I, Follettie MT, Mellott TJ, Kovacheva VP, Slack BE, Diesl V, Berse B, Thies RS, Blusztajn JK (2005) Bone morphogenetic protein 9 induces the transcriptome of basal forebrain cholinergic neurons. Proc Natl Acad Sci U S A 102:6984-6989. Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J (1999) Soluble amyloid beta peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol 155:853-862. Marin O, Anderson SA, Rubenstein JL (2000) Origin and molecular specification of striatal interneurons. J Neurosci 20:6063-6076. Martyn AC, De Jaeger X, Magalhaes AC, Kesarwani R, Goncalves DF, Raulic S, Guzman MS, Jackson MF, Izquierdo I, Macdonald JF, Prado MA, Prado VF (2012) Elimination of the vesicular acetylcholine transporter in the forebrain causes hyperactivity and deficits in spatial memory and longterm potentiation. Proc Natl Acad Sci U S A 109:17651-17656. Massague J, Wotton D (2000) Transcriptional control by the TGF-beta/Smad signaling system. Embo J 19:1745-1754. 88 McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL (1999) Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol 46:860866. Mesulam M (2004) The cholinergic Lesion of Alzheimer's Disease: Pivotal Factor or Side Show? Learning and Memory:43-49. Mineur YS, McLoughlin D, Crusio WE, Sluyter F (2005) Genetic mouse models of Alzheimer's disease. Neural Plast 12:299-310. Moises T, Dreier A, Flohr S, Esser M, Brauers E, Reiss K, Merken D, Weis J, Kruttgen A (2007) Tracking TrkA's trafficking: NGF receptor trafficking controls NGF receptor signaling. Mol Neurobiol 35:151-159. Montagna P, Gambetti P, Cortelli P, Lugaresi E (2003) Familial and sporadic fatal insomnia. Lancet Neurol 2:167-176. Mori T, Yuxing Z, Takaki H, Takeuchi M, Iseki K, Hagino S, Kitanaka J, Takemura M, Misawa H, Ikawa M, Okabe M, Wanaka A (2004) The LIM homeobox gene, L3/Lhx8, is necessary for proper development of basal forebrain cholinergic neurons. Eur J Neurosci 19:3129-3141. Mufson EJ, Li JM, Sobreviela T, Kordower JH (1996) Decreased trkA gene expression within basal forebrain neurons in Alzheimer's disease. Neuroreport 8:25-29. Mufson EJ, Ginsberg SD, Ikonomovic MD, DeKosky ST (2003) Human cholinergic basal forebrain: chemoanatomy and neurologic dysfunction. J Chem Neuroanat 26:233-242. Mufson EJ, Counts SE, Perez SE, Ginsberg SD (2008) Cholinergic system during the progression of Alzheimer's disease: therapeutic implications. Expert Rev Neurother 8:1703-1718. Mufson EJ, Ma SY, Dills J, Cochran EJ, Leurgans S, Wuu J, Bennett DA, Jaffar S, Gilmor ML, Levey AI, Kordower JH (2002) Loss of basal forebrain 89 P75(NTR) immunoreactivity in subjects with mild cognitive impairment and Alzheimer's disease. J Comp Neurol 443:136-153. Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, Gallins PJ, Buxbaum JD, Jarvik GP, Crane PK, Larson EB, Bird TD, Boeve BF, GraffRadford NR, De Jager PL, Evans D, Schneider JA, Carrasquillo MM, Ertekin-Taner N, Younkin SG, Cruchaga C, Kauwe JS, Nowotny P, Kramer P, Hardy J, Huentelman MJ, Myers AJ, Barmada MM, Demirci FY, Baldwin CT, Green RC, Rogaeva E, St George-Hyslop P, Arnold SE, Barber R, Beach T, Bigio EH, Bowen JD, Boxer A, Burke JR, Cairns NJ, Carlson CS, Carney RM, Carroll SL, Chui HC, Clark DG, Corneveaux J, Cotman CW, Cummings JL, DeCarli C, DeKosky ST, Diaz-Arrastia R, Dick M, Dickson DW, Ellis WG, Faber KM, Fallon KB, Farlow MR, Ferris S, Frosch MP, Galasko DR, Ganguli M, Gearing M, Geschwind DH, Ghetti B, Gilbert JR, Gilman S, Giordani B, Glass JD, Growdon JH, Hamilton RL, Harrell LE, Head E, Honig LS, Hulette CM, Hyman BT, Jicha GA, Jin LW, Johnson N, Karlawish J, Karydas A, Kaye JA, Kim R, Koo EH, Kowall NW, Lah JJ, Levey AI, Lieberman AP, Lopez OL, Mack WJ, Marson DC, Martiniuk F, Mash DC, Masliah E, McCormick WC, McCurry SM, McDavid AN, McKee AC, Mesulam M, Miller BL, et al. (2011) Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with lateonset Alzheimer's disease. Nat Genet 43:436-441. Niedowicz DM, Beckett TL, Matveev S, Weidner AM, Baig I, Kryscio RJ, Mendiondo MS, LeVine H, 3rd, Keller JN, Murphy MP (2012) Pittsburgh compound B and the postmortem diagnosis of Alzheimer disease. Ann Neurol 72:564-570. Niewiadomska G, Mietelska-Porowska A, Mazurkiewicz M (2011) The cholinergic system, nerve growth factor and the cytoskeleton. Behav Brain Res 221:515-526. Nobrega-Pereira S, Gelman D, Bartolini G, Pla R, Pierani A, Marin O (2010) Origin and molecular specification of globus pallidus neurons. J Neurosci 30:2824-2834. Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in 90 transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26:10129-10140. Okuda T, Haga T, Kanai Y, Endou H, Ishihara T, Katsura I (2000) Identification and characterization of the high-affinity choline transporter. Nat Neurosci 3:120-125. Olzscha H, Schermann SM, Woerner AC, Pinkert S, Hecht MH, Tartaglia GG, Vendruscolo M, Hayer-Hartl M, Hartl FU, Vabulas RM (2011) Amyloid-like aggregates sequester numerous metastable proteins with essential cellular functions. Cell 144:67-78. Palop JJ, Mucke L (2010) Synaptic depression and aberrant excitatory network activity in Alzheimer's disease: two faces of the same coin? Neuromolecular Med 12:48-55. Perez SE, Dar S, Ikonomovic MD, DeKosky ST, Mufson EJ (2007) Cholinergic forebrain degeneration in the APPswe/PS1DeltaE9 transgenic mouse. Neurobiol Dis 28:3-15. Perry EK, Perry RH, Tomlinson BE, Blessed G, Gibson PH (1980) Coenzyme Aacetylating enzymes in Alzheimer's disease: possible cholinergic 'compartment' of pyruvate dehydrogenase. Neurosci Lett 18:105-110. Picciotto MR, Higley MJ, Mineur YS (2012) Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76:116-129. Piltti KM, Haus DL, Do E, Perez H, Anderson AJ, Cummings BJ (2011) Computer-aided 2D and 3D quantification of human stem cell fate from in vitro samples using Volocity high performance image analysis software. Stem Cell Res 7:256-263. Price M, Lazzaro D, Pohl T, Mattei MG, Ruther U, Olivo JC, Duboule D, Di Lauro R (1992) Regional expression of the homeobox gene Nkx-2.2 in the developing mammalian forebrain. Neuron 8:241-255. 91 Prusiner SB, DeArmond SJ (1990) Prion disease of the central nervous system. Monographs in Pathology:86-122. Ray PG, Meador KJ, Loring DW, Zamrini EW, Yang XH, Buccafusco JJ (1992) Central anticholinergic hypersensitivity in aging. J Geriatr Psychiatry Neurol 5:72-77. Robertson RT, Baratta J, Yu J, Guthrie KM (2006) A role for neurotrophin-3 in targeting developing cholinergic axon projections to cerebral cortex. Neuroscience 143:523-539. Rogers SL, Doody RS, Mohs RC, Friedhoff LT (1998) Donepezil improves cognition and global function in Alzheimer disease: a 15-week, doubleblind, placebo-controlled study. Donepezil Study Group. Arch Intern Med 158:1021-1031. Rubenstein JL, Beachy PA (1998) Patterning of the embryonic forebrain. Curr Opin Neurobiol 8:18-26. Rubenstein JL, Martinez S, Shimamura K, Puelles L (1994) The embryonic vertebrate forebrain: the prosomeric model. Science 266:578-580. Savelieff MG, Lee S, Liu Y, Lim MH (2013) Untangling Amyloid-beta, Tau, and Metals in Alzheimer's Disease. ACS Chem Biol. Savonenko A, Xu GM, Melnikova T, Morton JL, Gonzales V, Wong MP, Price DL, Tang F, Markowska AL, Borchelt DR (2005) Episodic-like memory deficits in the APPswe/PS1dE9 mouse model of Alzheimer's disease: relationships to beta-amyloid deposition and neurotransmitter abnormalities. Neurobiol Dis 18:602-617. Schambra UB, Sulik KK, Petrusz P, Lauder JM (1989) Ontogeny of cholinergic neurons in the mouse forebrain. J Comp Neurol 288:101-122. Schliebs R, Arendt T (2011) The cholinergic system in aging and neuronal degeneration. Behav Brain Res 221:555-563. 92 Schnitzler AC, Lopez-Coviella I, Blusztajn JK (2008a) Purification and culture of nerve growth factor receptor (p75)-expressing basal forebrain cholinergic neurons. Nat Protoc 3:34-40. Schnitzler AC, Lopez-Coviella I, Blusztajn JK (2008b) Differential modulation of nerve growth factor receptor (p75) and cholinergic gene expression in purified p75-expressing and non-expressing basal forebrain neurons by BMP9. Brain Res 1246:19-28. Schnitzler AC, Mellott TJ, Lopez-Coviella I, Tallini YN, Kotlikoff MI, Follettie MT, Blusztajn JK (2010) BMP9 (bone morphogenetic protein 9) induces NGF as an autocrine/paracrine cholinergic trophic factor in developing basal forebrain neurons. J Neurosci 30:8221-8228. Schwab C, Bruckner G, Mares V, Biesold D (1988) A combined method of acetylcholinesterase histochemistry and [3H]thymidine autoradiography: application to neurogenesis of the rat basal forebrain cholinergic system. Neurosci Lett 90:69-74. Selkoe DJ (1997) Alzheimer's disease: genotypes, phenotypes, and treatments. Science 275:630-631. Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298:789-791. Selkoe DJ (2012) Preventing Alzheimer's disease. Science 337:1488-1492. Shimamura K, Hartigan DJ, Martinez S, Puelles L, Rubenstein JL (1995) Longitudinal organization of the anterior neural plate and neural tube. Development 121:3923-3933. Slotkin TA, Seidler FJ, Crain BJ, Bell JM, Bissette G, Nemeroff CB (1990) Regulatory changes in presynaptic cholinergic function assessed in rapid autopsy material from patients with Alzheimer disease: implications for etiology and therapy. Proc Natl Acad Sci U S A 87:2452-2455. Sobreviela T, Clary DO, Reichardt LF, Brandabur MM, Kordower JH, Mufson EJ (1994) TrkA-immunoreactive profiles in the central nervous system: 93 colocalization with neurons containing p75 nerve growth factor receptor, choline acetyltransferase, and serotonin. J Comp Neurol 350:587-611. Sofroniew MV, Howe CL, Mobley WC (2001) Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci 24:1217-1281. Sotthibundhu A, Sykes AM, Fox B, Underwood CK, Thangnipon W, Coulson EJ (2008) Beta-amyloid(1-42) induces neuronal death through the p75 neurotrophin receptor. J Neurosci 28:3941-3946. Sudhof TC (1995) The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature 375:645-653. Sussel L, Marin O, Kimura S, Rubenstein JL (1999) Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development 126:3359-3370. Szutowicz A, Tomaszewicz M, Bielarczyk H (1996) Disturbances of acetyl-CoA, energy and acetylcholine metabolism in some encephalopathies. Acta Neurobiol Exp (Wars) 56:323-339. Takashima A (2010) Tau aggregation is a therapeutic target for Alzheimer's disease. Curr Alzheimer Res 7:665-669. Terry AV, Jr., Buccafusco JJ (2003) The cholinergic hypothesis of age and Alzheimer's disease-related cognitive deficits: recent challenges and their implications for novel drug development. J Pharmacol Exp Ther 306:821827. Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R (1991) Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30:572-580. Thomas MC, Chiang CM (2006) The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol 41:105-178. 94 Um JW, Nygaard HB, Heiss JK, Kostylev MA, Stagi M, Vortmeyer A, Wisniewski T, Gunther EC, Strittmatter SM (2012) Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat Neurosci 15:1227-1235. Walsh DM, Selkoe DJ (2004) Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron 44:181-193. Wanaka A, Matsumoto K, Kashihara Y, Furuyama T, Tanaka T, Mori T, Tanno Y, Yokoya S, Kitanaka J, Takemura M, Tohyama M (1997) LIMhomeodomain gene family in neural development. Dev Neurosci 19:97100. Wang JY, Frenzel KE, Wen D, Falls DL (1998) Transmembrane neuregulins interact with LIM kinase 1, a cytoplasmic protein kinase implicated in development of visuospatial cognition. J Biol Chem 273:20525-20534. Wang JZ, Grundke-Iqbal I, Iqbal K (1996) Restoration of biological activity of Alzheimer abnormally phosphorylated tau by dephosphorylation with protein phosphatase-2A, -2B and -1. Brain Res Mol Brain Res 38:200-208. Wang YJ, Wang X, Lu JJ, Li QX, Gao CY, Liu XH, Sun Y, Yang M, Lim Y, Evin G, Zhong JH, Masters C, Zhou XF (2011) p75NTR regulates Abeta deposition by increasing Abeta production but inhibiting Abeta aggregation with its extracellular domain. J Neurosci 31:2292-2304. Ward SM, Himmelstein DS, Lancia JK, Binder LI (2012) Tau oligomers and tau toxicity in neurodegenerative disease. Biochem Soc Trans 40:667-671. Wedeen VJ, Rosene DL, Wang R, Dai G, Mortazavi F, Hagmann P, Kaas JH, Tseng WY (2012) The geometric structure of the brain fiber pathways. Science 335:1628-1634. Wei B, Huang Z, He S, Sun C, You Y, Liu F, Yang Z (2012) The onion skin-like organization of the septum arises from multiple embryonic origins to form multiple adult neuronal fates. Neuroscience 222:110-123. 95 Wurtman RJ (1992) Choline metabolism as a basis for the selective vulnerability of cholinergic neurons. Trends Neurosci 15:117-122. Yeo TT, Chua-Couzens J, Butcher LL, Bredesen DE, Cooper JD, Valletta JS, Mobley WC, Longo FM (1997) Absence of p75NTR causes increased basal forebrain cholinergic neuron size, choline acetyltransferase activity, and target innervation. J Neurosci 17:7594-7605. Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ, Lee VM (2007) Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron 53:337-351. You H, Tsutsui S, Hameed S, Kannanayakal TJ, Zamponi GW (2011) A-beta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. PNAS-Proceedings of the National Academy of Sciences of the United States of America 109:1737-1742. Zampieri N, Xu CF, Neubert TA, Chao MV (2005) Cleavage of p75 neurotrophin receptor by alpha-secretase and gamma-secretase requires specific receptor domains. J Biol Chem 280:14563-14571. Zhang Y, Hong Y, Bounhar Y, Blacker M, Roucou X, Tounekti O, Vereker E, Bowers WJ, Federoff HJ, Goodyer CG, LeBlanc A (2003) p75 neurotrophin receptor protects primary cultures of human neurons against extracellular amyloid beta peptide cytotoxicity. J Neurosci 23:7385-7394. Zhao Y, Marin O, Hermesz E, Powell A, Flames N, Palkovits M, Rubenstein JL, Westphal H (2003) The LIM-homeobox gene Lhx8 is required for the development of many cholinergic neurons in the mouse forebrain. Proc Natl Acad Sci U S A 100:9005-9010. 96 CURRICULUM VITAE Timothy Alfred Norman Jr. Year of Birth: 1983 [email protected] EDUCATION University of North Carolina at Wilmington, Wilmington, NC Bachelor of Science in Biology – May 2005 Boston University School of Medicine, Boston, MA Master of Arts – May 2013 Department of Pathology and Laboratory Medicine EXPERIENCE BOSTON UNIVERSITY SCHOOL OF MEDICINE 9/2011-5/2013 Graduate Student Project 1: Analysis of differential protein expression in neurons using immunohistochemistry and confocal microscopy. Discovered the cellular prion protein was upregulated in cholinergic neurons of the APPswe/PS1dE9 mouse model of AD. Project 2: Confocal imaging, immunofluorescence and quantification of cholinergic innervation in the hippocampus of APPswe/PS1dE9 mice after BMP9 infusion. Teaching Assistant, BT432: Basic Mechanisms of Disease Crafted and delivered powerpoint lectures for cardiovascular th diseases and traumatic brain injury from Robbins and Cotran 8 edition with Dr. Chris Andry MEDCO HEALTH SOLUTIONS 11/2007-4/2011 Lead Pharmacy Technician Communicated assignments and departmental production goals to pharmacists and technicians. Ensured product safety and accuracy while processing 750K prescriptions per week. Performed new hire training and minor supervisory and administrative functions including schedule preparation and goal targets. UNIVERSITY OF UTAH MEDICAL CENTER 3/2006-8/2006 Psychiatric Technician Assessed patient mental status and activities of daily living. Application of crisis prevention and intervention techniques to ensure patient and staff safety. Obtained vital signs, documented patient behavior and assisted with group therapy. PUBLICATIONS Rebecca M. Burke, Timothy A. Norman, Tarik F. Haydar, Jan Krzystof Bluszajn, Tiffany J. Mellot. BMP9 ameliorates the amyloidosis and the cholinergic defect in the APPswe/PS1dE9 mouse model of Alzheimer’s disease. (manuscript in preparation) 97