Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Gene nomenclature wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
DNA damage theory of aging wikipedia , lookup
Non-coding DNA wikipedia , lookup
Metagenomics wikipedia , lookup
Epigenomics wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Cancer epigenetics wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Genome evolution wikipedia , lookup
Genetic engineering wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Gene expression programming wikipedia , lookup
Gene therapy wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Population genetics wikipedia , lookup
History of genetic engineering wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Oncogenomics wikipedia , lookup
Genome editing wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Genome (book) wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Saethre–Chotzen syndrome wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Microsatellite wikipedia , lookup
Helitron (biology) wikipedia , lookup
Designer baby wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Frameshift mutation wikipedia , lookup
X-inactivation wikipedia , lookup
Skewed X-inactivation wikipedia , lookup
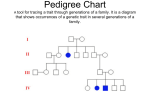
From www.bloodjournal.org by guest on June 17, 2017. For personal use only. Brief report Somatic mosaicism and compound heterozygosity in female hemophilia B Jean-Marc Costa, Dominique Vidaud, Ingrid Laurendeau, Michel Vidaud, Edith Fressinaud, Jean-Pierre Moisan, Albert David, Dominique Meyer, and Jean-Maurice Lavergne Sequencing the complete factor IX gene of 2 sisters with hemophilia B with different phenotypes and no family history of hemorrhagic diathesis revealed a common 5ⴕ splice site mutation in intron 3 (T6704C) in both and an additional missense mutation (I344T) in one. The presence of dysfunctional antigen in the latter strongly suggested that these mutations are in trans. Neither mutation was found in leukocyte DNA from the asymptomatic parents, but the mother was in somatic mosaicism for the shared splice site mutation. This case illustrates the importance of defining the phenotype and considering somatic mosaicism in sporadic cases. It underlines the limitations of complete gene sequencing for the detection of mosaicism and has implication for genetic counseling. (Blood. 2000;96: 1585-1587) © 2000 by The American Society of Hematology Introduction Hemophilia B is an X-linked bleeding disorder resulting from factor IX (F.IX) deficiency,1-3 caused by a wide range of mutations on the F.IX gene.4 Hemophilia B in girls is extremely rare and results from different mechanisms, the most common of which is skewed inactivation of the normal X chromosome in heterozygous girls.5-10 In some cases, the inactivation process does not seem to be random and occurs by either selection in favor of the activity of an X chromosome involved in a balanced X:autosome translocation11 or as a result of genetic differences affecting the X chromosome inactivation itself.12 More rarely, the phenotypic expression of the disease can be related to compound heterozygosity for hemophilic mutations13 or Turner syndrome. We present a family (Figure 1) with no prior bleeding history in whom moderate hemophilia B was diagnosed in 2 sisters having different (II3) and identical (II2) levels of F.IX activity (F.IXC) and antigen (F.IXAg). This discrepancy in the coagulation results implies a different molecular basis for hemophilia. Analysis of the F.IX gene nucleotide sequence confirmed this hypothesis: II2 is heterozygous for a null mutation with skewed inactivation of the normal X chromosome; II3 has an additional mutation in trans and is therefore a compound heterozygote. Neither mutation was detected in DNA from the parents. However, maternal buccal and uroepithelial studies showed somatic mosaicism for the mutation shared by the daughters. The second mutation must have resulted from a de novo mutation in the father’s gametes. parents were asymptomatic. After informed consent, F.IXC was determined by a modified one-stage assay14 and F.IXAg by an immunoenzymatic method using a commercial kit (Asserachrom F.IXAg, Diagnostica Stago, Franconville, France). Polymerase chain reaction (PCR) amplification and sequencing Genomic DNA was prepared from leukocytes using standard procedures.15 All 8 exons, intron-exon boundaries, and 5⬘ and 3⬘ ends of the F.IX gene were amplified by PCR and, after purification on Sephacryl S400 columns (Pharmacia, Orsay, France), fragments were directly sequenced using the ABI PRISM Dye Terminator Cycle Sequencing Reaction Kit (PE Biosystems, Courtaboeuf, France) on an ABI PRISM 377 DNA sequencer according to the manufacturer’s specifications. Competitive oligo priming (COP) PCR We performed COP PCR according to Tada.16 Leukocyte DNA as well as DNA obtained from uroepithelial and buccal cells were studied by COP with allele-specific primers conjugated to different fluorescent dyes, 6-carboxy-fluorescein (FAM), or tetrachloro-6-carboxy-fluorescein (TET), in conjunction with a common primer. The fluorescence of each dye with respect to its amplified DNA locus is scored on a 373 DNA sequencer as previously described.17 Primer sequences for intron 3 splice mutation are as follows: ● ● ● F.IX-FOR1: 5⬘ (TET) TTGGAAGCAGTATGTTGGC 3⬘ F.IX-REV2: 5⬘ (FAM) TTGGAAGCAGTATGTTGGT 3⬘ F.IX-REV: 5⬘ TCAGGTTGGGAGATTGGGTAT 3⬘ Study design Patients and coagulation studies Chromosome X inactivation The proband, a 14-year-old girl with moderate hemophilia B, suffered from hematomas, hemarthrosis, and epistaxis. There was no family history of a bleeding disorder and family studies revealed a milder form of the disease in a sister who suffered only from rare hematomas. The eldest sister and Chromosome X inactivation was analyzed by the method described by Allen18 studying the (CAG) repeat present in the human androgen receptor (HUMARA) gene and determination of the nucleotide sequence of the minimal XIST gene promoter according to Plenge et al.19 From the American Hospital of Paris, Neuilly, France; the UPRESS-JE 2195, Faculté de Pharmacie Paris V, Paris, France; the Centre Hospitalier Régional Universitaire, Nantes, France; and the INSERM U 143 and Assistance Publique Hôpitaux de Paris, Hôpital Bicêtre, Bicêtre, France. Reprints: J. M. Lavergne, INSERM U143, Hôpital Bicêtre, 94275 Bicêtre Cedex, France; e-mail: [email protected]. Submitted October 13, 1999; accepted April 5, 2000. The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked ‘‘advertisement’’ in accordance with 18 U.S.C. section 1734. Supported by a grant from Baxter Laboratories. © 2000 by The American Society of Hematology BLOOD, 15 AUGUST 2000 䡠 VOLUME 96, NUMBER 4 1585 From www.bloodjournal.org by guest on June 17, 2017. For personal use only. 1586 BLOOD, 15 AUGUST 2000 䡠 VOLUME 96, NUMBER 4 COSTA et al Figure 1. Pedigree, haplotypes, and coagulation and mutation studies. (Top) Pedigree and haplotype results of the family with female hemophilia B. Haplotype analysis by restriction fragment length polymorphism using the enzyme Dde I demonstrates that the 2 affected sisters, II2 and II3, share a common allele (2/1) with the mother (I2), whereas the normal sister (II1) inherited the mother’s other allele (2/2). (Bottom) Coagulation and mutation studies. For each member of the family, F.IXC and F.IXAg are indicated as well as the presence (⫹) or the absence (⫺) of each characterized mutation. Results and discussion The female proband (II3) had levels of F.IXC of 1 U/dL and F.IXAg of 28 U/dL, indicating moderately severe hemophilia B, and a karyotype of 46,XX. Her sister (II2) had mild hemophilia B with F.IXC and F.IXAg levels of 7 U/dL. The parents (I1 and I2) and elder sister (II1) showed normal coagulation results. The F.IX gene analysis demonstrated heterozygosity for a unique splice site alteration (T6704C) in the affected sister (II2) as already described in severe hemophiliacs.20 The proband is therefore a carrier for hemophilia B and her low F.IXC and F.IXAg most probably result from a skewed X inactivation.6-8 Inactivation patterns using the HUMARA gene showed that both II1 and II3 had a normal pattern. Unfortunately, X inactivation analysis in the third daughter (II2) was inconclusive because she was not polymorphic for the HUMARA gene (CAG) repeat. But her F.IX level demonstrates that at least a skewed inactivation occurred in the liver. Families have been identified in which unbalanced X inactivation is apparently inherited as an X-linked dominant trait. A mutation involving a single nucleotide in the promoter region of the XIST gene has recently been shown to underlie a skewed pattern of X inactivation in multiple members of 2 unrelated families.19 Sequencing of the minimal promoter showed a normal nucleotide sequence especially the absence of the C43G mutation previously described by Plenge et al.19 The F.IX gene of the proband (II3) showed the same T6704C mutation as her sister’s (II2). However, a second alteration, T31152C, was found in exon 8. This missense mutation, I344T, has not been reported to date but 2 mutations involving the same amino acid residue, I344F and I344S, have been found associated with moderate hemophilia (9th edition of the hemophilia B database; http://www.umds.ac.uk/molgen/haemBdatabase.htm). The different levels of F.IXC and F.IXAg in II3 are only compatible with the presence of each mutation on different alleles and II3 is therefore a compound heterozygote. Analysis of 2 intragenic polymorphic markers within the F.IX gene revealed that the 2 sisters, II2 and II3, inherited the same maternal haplotype, different from the normal eldest sister (II1). Therefore, the T6704C is of maternal origin. To investigate the possibility of somatic mosaicism in the mother (I2), DNA studies were performed on peripheral blood leukocytes and uroepithelial and buccal cells. Sequence analysis showed the unequivocal presence of a normal and mutant sequence at nucleotide 6704 in various amounts in these cells. COP PCR analysis confirmed the sequencing results with less than 5% leukocytes but 10% and 30% of uroepithelial or buccal cells, respectively, carrying the mutation. The second mutation of the proband (II3) probably originated in the father’s gametes. Using the same approach, search for a mosaicism in the father’s buccal and uroepithelial cells for the mutation in exon 8 was negative. Unfortunately, sperm cells were not available and germline mosaicism could not be excluded. Mosaicism has been documented for chromosomal abnormalities, mitochondrial mutations, triplet repeats, and mutations in a growing number of dominant and X-linked single gene disorders.21 For X-linked disorders, the detection of somatic mosaicism implies prior knowledge of the deleterious mutation.Actually, the method of choice for identification of the deleterious mutation relies on DNA sequencing, but the ability of this method to detect somatic mosaicism is poor because the ratio of the mutant to the wild-type allele can be quite small. Somatic mosaicism may be more common than previously thought.22,23 In sporadic cases of hemophilia one must always consider the possibility of maternal somatic/germline mosaicism and therefore systematically examine buccal and uroepithelial DNA. In this family, the mother (I2) must be considered as a carrier even if the germinal mosaicism cannot be documented. Moreover, the implication of the compound heterozygosity in II3 is that any male child has F.IX deficiency. Altogether, these results confirm the initial hypothesis based on coagulation studies that the molecular basis of hemophilia B in these 2 girls is different. Without determination of both low F.IXC and F.IXAg, such a hypothesis could not have been verified and if the entire F.IX gene had not been studied, one of the 2 mutations could have been overlooked with important consequences in genetic counseling. Acknowledgment We wish to thank Dr John F. Davidson for helpful advice. References 1. Thompson AR. Structure, function and molecular defects of the factor IX. Blood.1986;67:565-572. IM, Giannelli F. Molecular pathology of hemophilia B. EMBO J.1989;8:1067-1072. tial inactivation of the parental X chromosome. Eur J Hematol.1991;47:255-261. 2. Kurachi K, Kurachi S, Furukawa M, Yao SN. Biology of factor IX. Blood Coagul Fibrinolysis.1993; 4:953-973. 5. Kitchens CS. Discordance in a pair of identical twins carriers of factor IX deficiency. Am J Hematol.1987; 24:225-228. 3. Yoshitake S, Schach BG, Foster DC, Davie EW, Kurachi K. Nucleotide sequence of the gene for human factor IX (antihemophilic factor B). Biochemistry. 1985;24:3736-3750. 6. Nisen PD, Waber PG. Nonrandom X chromosome DNA methylation patterns in hemophilic females. J Clin Invest. 1989;83:1400-1403. 8. Wadelius C, Lindstedt M, Pigg M, Egberg N, Pettersson U, Anvret M. Hemophilia B in a 46, XX female probably caused by non-random X inactivation. Clin Genet. 1993;43:1-4. 4. Green PM, Bentley DR, Mibashan RS, Nilsson 7. Kling S, Coffey AJ, Ljung R, et al. Moderate hemophilia B in female carrier caused by preferen- 9. Naumova AK, Plenge RM, Bird LM, et al. Heritability of X chromosome-inactivation phenotype in a large family. Am J Hum Genet. 1996;58:11111119. From www.bloodjournal.org by guest on June 17, 2017. For personal use only. BLOOD, 15 AUGUST 2000 䡠 VOLUME 96, NUMBER 4 10. Lyon MF. Gene action in the X-chromosome of the mouse. Nature. 1961;190:372-373. FEMALE HEMOPHILIA B Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. 11. Zabel BU, Baumann WA, Printke W, GerhardRatschow K. X-inactivation pattern in three cases of X/autosome translocation. Am J Hum Genet. 1978;1:309-317. 16. Tada M, Omata M, Kawai S, et al. Detection of ras gene mutations in pancreatic juice and peripheral blood of patients with pancreatic adenocarcinoma. Cancer Res. 1993;53:2472-2474. 12. Orstavik KH, Orstavik RE, Eiklid K, Tranebjaerg L. Inheritance of skewed X chromosome inactivation. Am J Med Genet. 1996;64:31-34. 17. Lazar V, Diez SG, Laurent A, et al. Expression of human chorionic gonadotropin b subunit in superficial and invasive bladder carcinomas. Cancer Res. 1995;55:3735-3738. 13. Windsor S, Lyung A, Taylor SA, Ewenstein BM, Neufeld EJ, Lillicrap D. Severe hemophilia A in a female resulting from two de novo factor VIII mutations. Br J Haematol. 1995;90:906-909. 14. Langdell RD, Wagner RH, Brinkhous KM. Effect of anti-hemophilic factor IX on one stage clotting test. J Lab Clin Med. 1953;41:637-641. 18. Allen RC, Zoghbi HY, Moseley AB, Rosenblatt HM, Belmont JW. Methylation of HpaII and HhaI sites near the polymorphic CAG repeat in the human androgen-receptor gene correlates with Xchromosome inactivation. Am J Hum Genet. 1992;51:1229-1239. 15. Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual, 2nd ed. Cold 19. Plenge RM, Hendrich BD, Schwartz C. A promoter mutation in XIST gene in two unrelated 20. 21. 22. 23. 1587 families with skewed X-chromosome inactivation. Nat Genet. 1997;17:353-356. Giannelli F, Green PM, Sommer SS, et al. Hemophilia B: a database of point mutations and short additions and deletions, 8th ed. Nucleic Acids Res. 1998;26:265-268. Bernards A, Gusella JF. The importance of genetic mosaicism in human disease. N Engl J Med. 1994;331:1447-1449. Evans DGR, Wallace AJ, Wu CL, Trueman L, Ramsden RT, Strachan T. Somatic mosaicism: a common cause of classic disease in tumorprone syndromes? Lessons from type 2 neurofibromatosis. Am J Hum Genet. 1998;63:727736. Taylor SA, Deugau KV, Lillicrap DP. Somatic mosaicism and female-to-female transmission in a kindred with hemophilia B (factor IX deficiency). Proc Natl Acad Sci U S A. 1991;88:39-42. From www.bloodjournal.org by guest on June 17, 2017. For personal use only. 2000 96: 1585-1587 Somatic mosaicism and compound heterozygosity in female hemophilia B Jean-Marc Costa, Dominique Vidaud, Ingrid Laurendeau, Michel Vidaud, Edith Fressinaud, Jean-Pierre Moisan, Albert David, Dominique Meyer and Jean-Maurice Lavergne Updated information and services can be found at: http://www.bloodjournal.org/content/96/4/1585.full.html Articles on similar topics can be found in the following Blood collections Brief Reports (1936 articles) Hemostasis, Thrombosis, and Vascular Biology (2485 articles) Information about reproducing this article in parts or in its entirety may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requests Information about ordering reprints may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#reprints Information about subscriptions and ASH membership may be found online at: http://www.bloodjournal.org/site/subscriptions/index.xhtml Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Copyright 2011 by The American Society of Hematology; all rights reserved.