Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
X-inactivation wikipedia , lookup
Human genome wikipedia , lookup
DNA sequencing wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Polyadenylation wikipedia , lookup
Gene expression programming wikipedia , lookup
History of genetic engineering wikipedia , lookup
Short interspersed nuclear elements (SINEs) wikipedia , lookup
Pathogenomics wikipedia , lookup
Epigenomics wikipedia , lookup
RNA interference wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Genome evolution wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Non-coding DNA wikipedia , lookup
Designer baby wikipedia , lookup
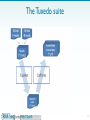
Whole genome sequencing wikipedia , lookup
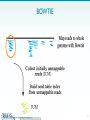
Microevolution wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
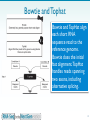
Nucleic acid analogue wikipedia , lookup
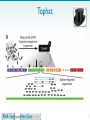
Nucleic acid tertiary structure wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Epigenetics of human development wikipedia , lookup
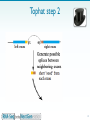
Long non-coding RNA wikipedia , lookup
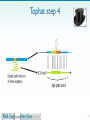
Bisulfite sequencing wikipedia , lookup
Gene expression profiling wikipedia , lookup
Deoxyribozyme wikipedia , lookup
History of RNA biology wikipedia , lookup
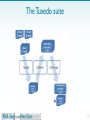
Epitranscriptome wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
RNA silencing wikipedia , lookup
Genomic library wikipedia , lookup
Non-coding RNA wikipedia , lookup
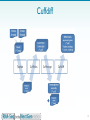
Mir-92 microRNA precursor family wikipedia , lookup
Metagenomics wikipedia , lookup
Methods to analyze RNA expression Is a specific RNA transcribed? In which cells, under which condition is it expressed? How much is there? Is the amount different from other cells/times? RNA analysis techniques 1. Low-to-mid-plex techniques: – Northern blot – Fluorescent in situ hybridization – Reverse transcription PCR (RT-PCR) 2. Higher-plex techniques: – DNA microarray – Tiling array – RNA-Seq 2 RNA sequencing analysis The newest technology for RNA expression analysis • Provides data for all the genes expressed in a particular sample (tissues, conditions, stages, etc.) • Coupled with high throughput sequencing • Quantitative • Highly technical and expensive • Rely heavily on computational statistical analysis 3 Applications of RNA-Seq From raw RNA seq data to transcriptome and differential expression analysis. 1. Abundance estimation 2. Alternative splicing 3. RNA editing 4. Finding novel transcripts And many more….. 4 Experimental design • Control and experimental conditions • Every sample in at least duplicate (triplicate better) • Uniformity of tissue used can be critical to detect small significant differences in transcription levels. 5 Steps in RNA-seq 6 Overall flow chart for RNA-seq 7 Library construction First RNA needs to be converted to cDNA as sequencers are designed for DNA not RNA sequencing. This is done using a special RNA-dependent DNA polymerase known as Reverse transcriptase (RT). The product is known as cDNA. 8 Library construction Each sample (tissue, time point, etc..) is used to prepare RNA. Each RNA then is converted into a library of cDNA fragments. The libraries from several different samples will be sequenced together, so each library has to receive an individual tag (AKA index). 9 Library construction The double-stranded cDNA fragments are attached (ligated) to small ds nucleotide sequences. The SP will be used for sequencing later. The index is a short DNA sequence that is specific to each library. 10 Library construction After size selection and limited amplification by PCR, the library representing short fragments of all the RNAs present in your initial tissues/ embryos/cells is ready for sequencing. When done correctly the number of DNA fragments corresponding to one mRNA is proportional to the initial amount of that specific mRNA. 11 Sequencing • The sequencer • And the “flow cell” where the sequencing takes place. Many libraries can be loaded together on one flow cell. The data from each library will be distinguished later because they each have a different “index”. 12 Data quality Several quality checks are done by the sequencer software to ensure that the sequencing reactions worked correctly. Do not worry about these You will receive individual files where the reads from each of your libraries have been sorted out by their index. 13 Initial data analysis Initially each library is analyzed separately except for the two files of each library if paired ends sequencing was done. 1) The short reads are aligned on the reference genome if available 2) The transcript(s) from each gene are reconstructed. At that point the analysis is done with all the libraries together looking at 3) differential expression and statistical significance. 14 Work flow for data analysis 15 Work flow for data analysis This flow chart also shows when each sample is done separately or compare 16 Work flow for data analysis From RNA-seq reads to differential expression results: Oshlack et al. Genome Biology 2010, 11:220 17 The Tuxedo suite A series of software can be used in succession to perform the series of steps needed for alignment, assembly and expression analysis. It is known as the Tuxedo protocol. Information about software and Tuxedo workflow was first described in: Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, & Pachter L (2012) Nature Protocols 7, 562–578 18 The Tuxedo suite 19 BOWTIE 20 BOWTIE 21 Bowtie and Tophat Bowtie and TopHat align each short RNA sequence read to the reference genome. Bowtie does the initial fast alignment. TopHat handles reads spanning two exons, including alternative splicing. 22 Tophat 23 Tophat step 1 24 Tophat step 2 25 Tophat step 3 26 Tophat step 4 27 Tophat overall 28 Bowtie and Tophat 29 Visualizing TopHat data with IGV 30 The Tuxedo suite 31 Cufflinks Cufflinks identifies splice sites based on TopHat placement of split reads and will merge the short reads to create a gene structure based on RNAseq data. It will also evaluate the proportion of each alternative splice products. The expression of each isoform will be expressed in RPKM: Reads (for that transcript) Per Kilobase (of gene) per Million reads . 32 Cufflinks For figure on previous slide 33 The Tuxedo suite 34 The Tuxedo suite 35 Differential expression DEG = Differentially Expressed Genes 36 Cuffdiff 37 Cuffdiff • Cuffdiff takes all the reads that map to overlapping transcripts (like isoforms) and infers which reads belong to each variant transcript from one gene. • It starts with the reads that map uniquely to each isoform, then iterate to find the most likely distribution of the remaining reads. 38 Cummerbund • Visualization of data generated by Cuffdiff • Graphical representation of over or under expression • Classification of genes by molecular pathways • Rather complicated, but some easy tools are available in DNA subway 39 Cummerbund 40 Differential gene expression 41 Volcano plot 42 Expression Profile of Specific Pathways: HeatMap 43