Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Human genetic variation wikipedia , lookup
Gene desert wikipedia , lookup
History of RNA biology wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Heritability of IQ wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Human genome wikipedia , lookup
Ridge (biology) wikipedia , lookup
Short interspersed nuclear elements (SINEs) wikipedia , lookup
Non-coding DNA wikipedia , lookup
Metagenomics wikipedia , lookup
Heritability of autism wikipedia , lookup
History of genetic engineering wikipedia , lookup
Genomic imprinting wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Epitranscriptome wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Long non-coding RNA wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Frameshift mutation wikipedia , lookup
Gene expression profiling wikipedia , lookup
Pathogenomics wikipedia , lookup
Oncogenomics wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Minimal genome wikipedia , lookup
Designer baby wikipedia , lookup
Point mutation wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Genome-wide association study wikipedia , lookup
Genome (book) wikipedia , lookup
Genome evolution wikipedia , lookup
Helitron (biology) wikipedia , lookup
Microevolution wikipedia , lookup
Public health genomics wikipedia , lookup
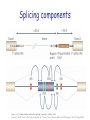
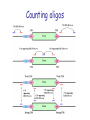
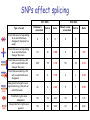
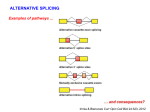
Predicting effect of SNPs and de novo variants on splicing presented by Alexander Tchourbanov Presentation structure Previous work on predicting aberrant splicing events induced by common and de novo genetic variants Proposed plan of action Problem of aberrant splicing Splicing in vertebrate genes is governed by highly degenerate motifs that include donor, acceptor, branch site and repertoire of splicing enhancers and silencers Integrity of human genes is constantly compromised by de novo mutations ~15% disease associated mutations cause aberrant splicing Splicing components Image credit: Understanding alternative splicing: towards a cellular code: Arianne J. Matlin, Francis Clark and Christopher W. J. Smith, Nature Reviews Molecular Cell Biology 6, 386-398 (May 2005) Importance of understanding the aberrant splicing According to Human Gene Mutation Database (HGMD) Professional 2010.4 (http://www.hgmd.cf.ac.uk) 60,489 mutations are missence/nonsense 10,210 mutations have consequences in mRNA splicing Databases DBASS5 and DBASS3 currently contain 900 well-annotated records of disease causing aberrant splicing events (Buratti et. al., Nucleic Acids Research, 2010). Importance of understanding the aberrant splicing Chen R, Davydov E, Sirota M, Butte A: NonSynonymous and Synonymous Coding SNPs Show Similar Likelihood and Effect Size of Human Disease Association. PLoS ONE 2010, 5(10):e13574. Frequently it is difficult to get tissue samples for RNA sequencing (brain samples, retina samples) We need to predict the effect of de novo variants (which includes cancer mutations) and common variants. No association study possible. Existing elements Publication Number of elements predicted Fairbrother, W.G., et al. (Science 2002) 238 hexamers as candidate ESEs Zhang, X.H. and L.A. Chasin (Genes Dev 2004) Putative 2,069 octamers as exonic splicing enhancers and 974 octamers as exonic splicing silencers Wang, Z., et al. (Cell 2004) 133 ESS-containg decanucleotides Yeo, G.W., E.L. Van Nostrand, and T.Y. Liang (PLoS Genet 2007) 133 5’SS ISEs and 299 3’SS ISEs pentamers Goren, A., et al. (Mol Cell 2006) 285 hexamers putative exonic splicing regulatory sequences Zhang, C., et al. (Proc Natl Acad Sci USA 2008) Putative 1131 hexamers Exon-Identity Elements (EIEs) and 708 Intron-Identity Elements (IIEs) Stadler, M.B., et al. (PLoS Genet 2006) 380 hexamers as new candidate ESEs and 132 hexamers as new candidate ESSs Wang, E. T., et. al. (Nature 2008) 187 5’SS ISEs/ISSs and 175 3’SS ISEs/ISSs hexamers supporting the tissue-specific splicing events Orthologos blocks from UCSC GB 2,333,379 extended exons from 23 Tetrapoda organisms were obtained A number of experimental reports showed that genes from distantly related Tetrapoda organisms were correctly expressed and post-transcriptionally modified in transgenic animals (Capetanaki Y et al.: Proc Natl Acad Sci USA 1989, Jacobs GH et al.: Science 2007) The genes encoding well-known RNA binding proteins involved in splicing regulation are enriched with ultraconserved elements (Bejerano G. et al.:Science 2004) Counting oligos Comparing oligo counts Example of 5’SS ISEs found Elements found Using the orthologous exons available for 23 Tetrapoda organisms we have identified 2,546 unique splicing regulatory elements. Among these elements 203 (7.97%) 3’SS and 177 (6.95%) 5’SS supporting motifs are novel and have not been previously reported in systematic screens detecting such elements. Among our predicted elements, 41.08% of sequences were heptamers and 51.81% were octamers and only 6.76% hexamers and 0.35% pentamers Predicting donor splice site Bayesian 5’ splice sites sensor designed during my PhD study has performance better than other sensors, including maximum entropy sensor from MIT. Exonic length distribution Optimal exonic lengths substantially depend on the flanking splicing signals strengths, considering splice site (SS) strengths in the discrete range from 1 (weakest) to 5 (strongest). Example of LOD profiles (5’SS ISE) Exon scoring method LOD scores associated with 5’SS,3’SS, exonic length, competing SSs and Enhancer/Silencer signals are combined towards an exon strength Existing splicing prediction software • • • • • • • http://www.umd.be/HSF http://esrsearch.tau.ac.il/ http://genes.mit.edu/burgelab/rescue-ese/ http://genes.mit.edu/exonscan/ http://cryp-skip.img.cas.cz/ http://cubweb.biology.columbia.edu/pesx/ Strongest exonic silencers are the splice sites themselves!!! SpliceScan II performance on mutations Database Prediction method DBASS 5 (Buratti E et al: Nucleic Acids Res 2007) Correct Wrong Accuracy DBASS 3 (Vorechovsky I: Nucleic Acids Res 2006) Correct Wrong Accuracy ExonScan (Wang Z et al: Cell 2004 ) 42 320 11.6% 8 117 6.4% GenScan (Burge C:J Mol Biol 1997) 52 310 14.36% 21 104 16.8% SpliceScan II 100 262 27.62% 40 85 32% Disturbing circadian pacemaker For example, the circadian pacemaker period homolog 1 (Per1) gene locus has intronic non-coding variant rs885747 that has been previously associated with Autism (Nicholas et. al., Molecular Psychiatry, 2007). Haplotype analysis within per1 gave a single significant result: a global P=0.027 for the markers rs2253820-rs885747 We predicted creation of intronic splicing enhancer GCGGGGT as one of the possible causative mechanisms behind rs885747 that promotes aberrant exonic isoform. Disturbing circadian pacemaker Disturbing circadian pacemaker Per1 is a member of the Period family of genes and is expressed in a circadian pattern in the suprachiasmatic nucleus, the primary circadian pacemaker in the mammalian brain. Genes in this family encode components of the circadian rhythms of locomotor activity, metabolism, and behavior. SNPs affect splicing 997 SNPs Type of event NEW! NEW! Alzheimer’s associated 539 SNPs Control Ratio Breast cancer associated Control Ratio Predicted exon corresponding to an annotated exon disappears (becomes too weak) 0 2 0 0 0 - Predicted exon corresponding to an annotated exon changes the score 43 12 3.58 11 2 5.5 Predicted exon sharing a SS with an annotated exon changes the score 242 78 3.10 59 29 2.03 Predicted exon sharing a SS with an annotated exon disappears 23 4 5.75 6 1 6.00 New predicted cryptic exon appears sharing a SS with an annotated exon 26 9 2.89 5 1 5.00 Predicted cryptic exon disappears 50 49 1.02 30 17 1.76 New predicted cryptic exon appears 50 46 1.08 24 25 0.96 rs849563 variant Am J Med Genet B Neuropsychiatr Genet. 2007 Jun 5;144B(4):492-5. Association of the neuropilin-2 (NRP2) gene polymorphisms with autism in Chinese Han population. Wu S, Yue W, Jia M, Ruan Y, Lu T, Gong X, Shuang M, Liu J, Yang X, Zhang D. Institute of Mental Health, Peking University, Beijing, China. Significant genetic association found between autism and two of the SNPs of the NRP2 gene (rs849578: P = 0.017, rs849563: P = 0.027), as well as specific haplotypes, especially those formed by rs849563. rs849563 is synonymous rs849563 predicted mechanism The neuropilin-2 (NRP2) gene is localized to 2q34, an autism susceptibility locus. NRP2 has been demonstrated to both guide axons and to control neuronal migration in the central nervous system. It has been reported that NRP2 may be required in vivo for sorting migrating cortical and striatal interneurons to their correct destination. SpliceScan II tool SpliceScan II tool http://www.wyomingbioinformatics.org/~achu rban/docs/SpliceScanII.tar.gz Is more sensitive than existing splicing simulators (NetUTR, ExonScan) Uses novel 5’ GC SS Bayesian sensor Method allows predicting aberrant splicing events associated with genomic variants ACGMAP companion database http://www.stritch.luc.edu/node/375 Proposed system architecture Shotgun Mate pairs Transcriptome Trios Online submission Phased reference genomes of healthy individuals Haplotype trees Variants calling (GMAP/gsNap) Use PolyPhen (Ramensky et.al., NAR, 2002), SIFT (Kumar et.al., Nature Protocols, 2009) or Panther (Thomas et.al., Genomic research, 2003) to predict destabilizing effects of non-synonymous genetic variants Use SpliceScanII to predict effect of synonymous mutations on splicing Visualize information in the context of existing information (HGMD, UCSC genome browser, dbSNP, PFAM, ASTD) Variants analysis and visualization Chromosome testing at BGI DNA swap Craven et al. Nature, 1-4 (2010) Mito and Thacker Tachibana et.al., Nature, 2009 Wellderly study The Wellderly Study is headed by Scripps Health Chief Academic Officer Dr. Eric J. Topol, who has spent the past four years recruiting healthy elderly individuals youngest participant is at least 80 years old, the median age of this study group is 87 with oldest participant 108 years old free from major diseases and long-term medications This fall Complete Genomics announced that it will sequence, at its own cost, the whole human genomes of 1,000 participants in the Wellderly Study Following announcement NASDAQ: GNOM stocks dropped 7%. The genomic sequences obtained in this study will be a private property of Complete Genomics Archon Genomics X PRIZE http://genomics.xprize.org/life-at-100-plus Third generation platforms 3rd generation platforms (such as Oxford nanopore http://www.nanop oretech.com/) will revolutionize the field soon. Clarke, J. et al. “Continuous base identification for single-molecule nanopore DNA sequencing.” Nature Nanotech. 2009. Thanks!