Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Cell culture wikipedia , lookup
Cell encapsulation wikipedia , lookup
Cellular differentiation wikipedia , lookup
Cell growth wikipedia , lookup
Mechanosensitive channels wikipedia , lookup
Cell nucleus wikipedia , lookup
Biochemical switches in the cell cycle wikipedia , lookup
Organ-on-a-chip wikipedia , lookup
Extracellular matrix wikipedia , lookup
Cytokinesis wikipedia , lookup
Signal transduction wikipedia , lookup
Cell membrane wikipedia , lookup
Endomembrane system wikipedia , lookup
List of types of proteins wikipedia , lookup
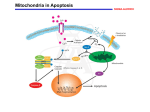
PERSPECTIVES agonists and antagonists. Cell 96, 625–634 (1999). 16. Shimizu, S. & Tsujimoto, Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl Acad. Sci. USA 97, 577–582 (2000). 17. von Ahsen, O. et al. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cytochrome c release. J. Cell Biol. 150, 1027–1036 (2000). 18. Chai, J. C. et al. Structural and biochemical basis of apoptotic activation by Smac/DIABLO. Nature 406, 855–862 (2000). 19. Antonsson, B., Montessuit, S., Lauper, S., Eskes, R. & Martinou, J.-C. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem. J. 345, 271–278 (2000). 20. Gross, A., Jockel, J., Wei, M. C. & Korsmeyer, S. J. Enforced dimerization of BAX results in its translocation, mitochondrial dysfunction and apoptosis. EMBO J. 17, 3878–3885 (1998). 21. Cheng, E. H.-Y. et al. Conversion of Bcl-2 to a Bax-like death effector by caspases. Science 278, 1966–1968 (1997). 22. Saito, M., Korsmeyer, S. J. & Schlesinger, P. H. Baxdependent transport of cytochrome c reconstitued in pure liposomes. Nature Cell Biol. 2, 553–555 (2000). 23. Gilbert, R. J. C. et al. Two structural transitions in membrane pore formation by pneumolysin, the poreforming toxin of Streptococcus pneumoniae. Cell 97, 647–655 (1999). 24. Bernardi, P., Scorrano, L., Colonna, R., Petronilli, V. & Di Lisa, F. Mitochondria and cell death. Eur. J. Biochem. 264, 687–701 (1999). 25. Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341, 233–249 (1999). 26. Eskes, R., Desagher, S., Antonsson, B. & Martinou, J.-C. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 20, 929–935 (2000). 27. Marzo, I. et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281, 2027–2031 (1998). 28. Shimizu, S., Narita, M. & Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487 (1999). 29. Shimizu, S., Ide, T., Yanagida, T. & Tsujimoto, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 275, 12321–12325 (2000). 30. Shimizu, S., Konishi, A., Kodoma, T. & Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl Acad. Sci. USA 97, 3100–3105 (2000). 31. Vander Heiden, M. G., Chandel, N. S., Schumacker, P. T. & Thompson, C. B. Bcl-xL prevents cell death following growth factor withdrawal by facilitating mitochondrial ATP/ADP exchange. Mol. Cell 3, 159–167 (1999). 32. Vander Heiden, M. G. et al. Outer mitochondrial membrane permeability can regulate coupled respiration and cell survival. Proc. Natl Acad. Sci. USA 97, 4666–4671 (2000). 33. Zhuang, J., Dinsdale, D. & Cohen, G. M. Apoptosis, in human monocytic THP.1 cells, results in the release of cytochrome c from mitochondria prior to their ultracondensation, formation of outer membrane discontinuities and reduction in inner membrane potential. Cell Death Differ. 5, 953–962 (1998). 34. Kroemer, G., Dallaporta, B. & Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60, 619–642 (1998). 35. Gross, A. et al. Biochemical and genetic analysis of the mitochondrial response of yeast to Bax and Bcl-xL. Mol. Cell. Biol. 20, 3125–3136 (2000). 36. Priault, M., Chaudhuri, B., Clow, A., Camougrand, N. & Manon, S. Investigation of Bax-induced release of cytochrome c from yeast mitochondria. Eur. J. Biochem. 260, 684–691 (1999). 37. Kissova, I. et al. The cytotoxic action of Bax on yeast cells does not require mitochondrial ADP/ATP carrier but may be related to its import to the mitochondria. FEBS Lett. 471, 113–118 (2000). 38. Priault, M., Camougrand, N., Chaudhuri, B., Schaeffer, J. & Manon, S. Comparison of the effects of Bax-expression in yeast under fermentative and respiratory conditions: investigation of the role of adenine nucleotides carrier and cytochrome c. FEBS Lett. 456, 232–238 (1999). 39. Pastorino, J. G., Chen, S.-T., Tafani, M., Snyder, J. W. & Farber, J. L. The overexpression of Bax produces cell death upon induction of the mitochondrial permeability transition. J. Biol. Chem. 273, 7770–7775 (1998). 40. Bossy-Wetzel, E., Newmeyer, D. D. & Green, D. R. Mitochondrial cytochrome c release in apoptosis occurs upstream of DEVD-specific caspase activation and independently of mitochondrial transmembrane depolarization. EMBO J. 17, 37–49 (1998). 41. Zhivotovsky, B., Orrenius, S., Brustugun, O. T. & Doskeland, S. O. Injected cytochrome c induces apoptosis. Nature 391, 449–450 (1998). 42. Deshmukh, M. & Johnson, E. M. Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron 21, 653–655 (1998). 43. Green, D. Apoptotic pathways: papers wraps stone blunts scissors. Cell 102, 1–4 (2000). 44. Desagher, S. et al. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 144, 891–901 (1999). 45. Knudson, C. M. & Korsmeyer, S. J. Bcl-2 and Bax function independently to regulate cell death. Nature Genet. 16, 358–363 (1997). 46. Kane, D. J., Ord, T., Anton, R. & Bredesen, D. E. Expression of Bcl-2 inhibits necrotic neuronal cell death. J. Neurosci. Res. 40, 269–275 (1995). Acknowledgements. We thank B. Antonsson, S. Desagher, M. Koco–Vilbois, K. Maundrell, S. Montessuit, O. Terradillos and X. Roucou for critical reading of the manuscript. OPINION The mitochondrion in apoptosis: how Pandora’s box opens Naoufal Zamzami and Guido Kroemer There is widespread agreement that mitochondria have a function in apoptosis, but the mechanisms behind their involvement remain controversial. Here we suggest that opening of a multiprotein complex called the mitochondrial permeability transition pore complex is sufficient (and, usually, necessary) for triggering apoptosis. As with Pandora’s box, the mitochondrion is full of potentially harmful proteins and biochemical reaction centres. Loss of subcellular and submitochondrial compartmentalization, then, may liberate a flood of toxic compounds, such as reactive oxygen species and proteins that can activate catabolic hydrolases. In most pathways leading to apoptosis, permeabilization of the inner and outer mitochondrial membranes (IMM and OMM, respectively) is critical1,2 (BOX 1). This culminates in the release of certain inactive caspase precursors (procaspases), cytochrome c (a caspase activator), Smac (second mitochondria-derived activator of caspase, also known as DIABLO, which is a caspase co-activator) and apoptosis-inducing factor (AIF; a nuclease activator), as well as a plethora of other proteins from the intermembrane space. All of these proteins must pass through the OMM3, but the IMM is also affected (although its permeabilization may be transient, and is partial for solutes up to 1.5 kDa, allowing proteins in the mitochondrial matrix to be retained). Box 1 | Evidence for the involvement of mitochondria in controlling cell death • Mitochondrial membrane permeabilization (MMP), which can affect both the inner and outer mitochondrial membranes, precedes the signs of necrotic or apoptotic cell death, including the apoptosis-specific activation of caspases. • MMP is a more accurate predictive parameter for cell death than caspase activation (which often is not required for cell death to occur and, in contrast, may also participate in positive signalling). • Many pro-apoptotic proteins and second messengers act on mitochondria to induce MMP. Such signalling molecules include the pro-apoptotic members of the Bcl-2 family, phosphatases and kinases acting on Bcl-2-like proteins, as well as transcription factors (for example, p53 and TR3/Nur-77/NGFI-B). • Bcl-2 and related anti-apoptotic proteins are present in mitochondrial membranes and prevent apoptosis by suppressing MMP. • MMP-mediated release of caspase and nuclease activators is required for full-blown apoptotic cell death. Such activators are normally sequestered in the intermembrane space and include cytochrome c, Smac/DIABLO and apoptosis-inducing factor (AIF). NATURE REVIEWS | MOLECUL AR CELL BIOLOGY VOLUME 2 | JANUARY 2001 | 6 7 © 2001 Macmillan Magazines Ltd PERSPECTIVES Many proteins from the Bcl-2/Bax family regulate apoptosis when locally present in mitochondrial membranes (see BOX 1 in the article by Martinou and Green on page 63). As a rule, close homologues of Bcl-2 (for example, Bcl-xL, Bcl-W, Mcl1 and A1) reside in the mitochondria and stabilize the barrier function of the mitochondrial membranes. Pro-apoptotic proteins can shuttle between a non-mitochondrial localization (the cytosol for Bax, Bad and Bid; microtubules for Bim and mitochondria). Upon induction of apoptosis, these proteins insert into mitochondrial membranes and cause them to be permeabilized. The controversy surrounds the mode of action of these Bcl-2 family members2,4,5. Do they induce permeabilization of mitochondrial membranes in an autonomous fashion, without the need for interactions with sessile mitochondrial proteins? Or do they act on a multiprotein complex called the permeability transition pore complex (PTPC)? The PTPC regulates cell death The PTPC — or mitochondrial megachannel — is formed at the contact site between the IMM and OMM (FIG. 1). Its core components are the adenine nucleotide translocator (ANT, found in the IMM) and the volt- Box 2 | The mitochondrial permeability transition Definition. The mitochondrial permeability transition involves a sudden (and initially reversible) increase in permeability of the IMM to solutes up to 1.5 kDa. It is commonly defined by its inhibition by cyclosporin A or derivatives of this compound that bind to mitochondrial cyclophilin (peptidyl-prolyl-cis–trans-isomerase) such as N-methyl-Val-4-cyclosporin A. Cyclosporin A-mediated inhibition of the permeability transition is transient (lasting 60 min). Regulation. The permeability transition pore complex (PTPC) functions as a sensor for: • Voltage: The PTPC decodes voltage changes into variations of the probability (the ‘gating potential’) at which pore opening occurs. Pore agonists shift the gating potential to more negative values (physiological = 200 mV, negative inside), favouring pore opening, whereas pore antagonists favour its closure. • Divalent cations: Matrix Ca2+ increases the probability of pore opening. Matrix Mg2+ or Mn2+, and external divalent metal ions including Ca2+ all decrease the probability of pore opening. • Matrix pH: The permeability transition pore is closed at neutral or acidic pH owing to reversible protonation of histidine residues and/or inhibition of the interaction between matrix cyclophilin and the ANT. Alkalinization is permissive for pore opening with a maximum effect at a matrix pH of ~7.3. • Thiol oxidation: Oxidation (disulphide formation) of a critical mitochondrial dithiol (presumably cysteine 56 of the ANT dimer) increases the probability of pore opening. The redox status of this dithiol is in equilibrium with that of matrix glutathione. • Oxidation/reduction state of pyridine nucleotides (NADH/NAD+ and NADPH/NADP+). Oxidation of pyridine nucleotides favours permeability transition. • ANT ligands: The endogenous ANT ligand ADP as well as bongkrekate inhibit permeability transition. Atractyloside, another ANT ligand, induces permeability transition. • Metabolites: Glucose and creatine inhibit permeability transition, presumably through their action on hexokinase and creatine kinase. Ubiquinone O (coenzyme Q) also inhibits permeability transition. Long-chain fatty acids, ceramide and ganglioside GD3 favour permeability transition. • Anti- and pro-apoptotic members of the Bcl-2 family. Metabolic consequences. Full-blown permeability transition causes uncoupling of the respiratory chain with collapse of the electrochemical proton gradient ∆Ψm and cessation of ATP synthesis, matrix Ca2+ outflow, depletion of reduced glutathione, depletion of NADPH, hypergeneration of superoxide anion, and mitochondrial release of intermembrane proteins. Several of the consequences of permeability transition themselves favour opening of the permeability transition pore, implying that permeability transition is a self-amplifying process. Physiological function. Periodic reversible opening of the permeability transition pore allows for the release of Ca2+ from the mitochondrial matrix, thereby participating in Ca2+ homeostasis and/or the generation of Ca2+ waves (Ca2+-induced Ca2+-release). A role in neuronal plasticity has been suggested. The ANT/VDAC couple (and its interacting proteins hexokinase and creatine kinase) may also participate in regulating ATP/ADP transport/synthesis. Irreversible permeability transition triggers mitochondrial autophagy (a process by which cells digest parts of their cytoplasm), apoptosis or necrosis. 68 | JANUARY 2001 | VOLUME 2 age-dependent anion channel (VDAC, located in the OMM). The VDAC is normally permeable to solutes of up to 5 kDa, thereby allowing the free exchange of respiratory-chain substrates such as NADH, FADH and ATP/ADP between the mitochondrial intermembrane space and the cytosol. In contrast, the IMM is almost impermeable — a feature that is essential for generation of the electrochemical proton gradient (∆ψm) used for oxidative phosphorylation. But the permeability of both membranes may suddenly increase in vitro — in isolated mitochondria, for instance, after the addition of atractyloside (which is a ligand of the ANT) — and this ‘permeability transition’6 is mediated by the PTPC (BOX 2). As well as the abundant IMM and OMM proteins — ANT and VDAC respectively — the PTPC contains the peripheral benzodiazepine receptor (located in the OMM), creatine kinase (located in the intermembrane space), hexokinase II (tethered to the VDAC on the cytosolic face of the OMM), cyclophilin D (located in the mitochondrial matrix), as well as Bax/Bcl-2-like proteins (FIG. 1). Direct interactions have been shown for the ANT and cyclophilin D, as well as between the ANT and VDAC6,7. As a consequence of such interactions, changes in the conformation of the ANT (which are indirectly affected by cyclosporin A, which affects the cyclophilin D–ANT interaction) may indirectly impinge on the function of the VDAC, or vice versa. So the PTPC may simultaneously control permeability of the OMM and the IMM. Alternatively, initial permeabilization of the IMM may cause the mitochondrial matrix to swell (through osmosis), leading to rupture of the OMM. (The IMM, with its folded christae, does not burst as its surface area is greater than that of the OMM.) Ultrastructural evidence for matrix swelling or rupture of the OMM has been obtained in many models of cell death, including hepatocyte apoptosis induced by injection of an antibody recognizing the CD95 ‘death receptor’ in the plasma membrane8. This particular phenotype is deficient in knockout mice that lack either of the two pro-apoptotic proteins from the Bcl-2 family (Bid or Bak) or overexpress a Bcl-2 transgene in the liver9. However, mitochondrial swelling and membrane rupture is not a universal feature of apoptosis10–13, suggesting alternative possibilities of membrane permeabilization, including the formation of pores in the OMM. Many reports show that apoptosis correlates with signs of a permeability transition, such as loss of the mitochondrial transmembrane potential (∆ψm); that induction of a www.nature.com/reviews/molcellbio © 2001 Macmillan Magazines Ltd PERSPECTIVES permeability transition is enough to trigger cell death; and that, by inhibiting this permeability transition, cell death can be prevented (for reviews see REFS 6,14). For example, cyclosporin A (and N-methyl-4-Valcyclosporin A, which is a non-immunosuppressive derivative that acts on cyclophilin D) can inhibit apoptosis induced in vivo by brain trauma15, by ischaemia reperfusion damage6, or by injection of an antibody against CD95 (which mainly affects hepatocytes)8. Moreover, bongkrekate, an ANT ligand that inhibits the PTPC, prevents apoptosis induced by diverse stimuli including glucocorticoids (in thymocytes)14, excitotoxins (in neurons)16 and tumour necrosis factor (in hepatocytes)17. However, such pharmacological inhibitors are not universally cytoprotective, implying that permeabilization of the mitochondrial membrane may involve individual PTPC components (for example, the VDAC without the ANT/cyclophilin D) or occur in a completely PTPC-independent fashion. Several viral or bacterial proteins that modulate apoptosis also interact with components of the PTPC: for example, Vpr from HIV-1 (which binds the ANT and is pro-apoptotic)18; vMIA/UL37 from cytomegalovirus (which binds the ANT and is anti-apoptotic)19; the hepatitis virus B X-protein (which binds the VDAC and is pro-apoptotic)20; and porin B from Neisseria meningidis (which binds the VDAC and is anti-apoptotic)21. Bcl-2 and Bax interact with the PTPC Co-immunoprecipitation studies indicate that Bcl-2 and Bax can interact — directly or indirectly — with the VDAC in the OMM22. Bcl-2, Bcl-xL, Bax and Bak interact directly with the ANT, as shown by three independent methods: co-purification, coimmunoprecipitation and yeast two-hybrid screening23. The two-hybrid screen revealed that a short stretch of human ANT2 (amino acids 105–156) suffices for the interaction with Bcl-2-related proteins23. The apoptosisregulatory potential of this portion of the ANT has been confirmed by deletion mapping: overexpression of full-length mouse Ant1 (which has 95% amino-acid identity with human ANT2) or its amino-terminal half (amino acids 1–141) induces apoptosis, but expression of an even shorter truncation mutation (amino acids 1–101) does not. So the Bcl-2/Bax-binding site (amino acids 105–156) overlaps with the apoptosis-regulatory region of ANT (amino acids 102–141), as well as with its Vpr-binding consensus motif (WXXF; amino acids 109–113). Interestingly, a substitution in the Glucose-6phosphate + ADP Glucose + ATP Kinases Phosphatases pH change Caspases HK II HVB-X Porin B BH3 peptides Bax Bcl-2 PBR OMM VDAC VDAC Creatine + ATP mtCK Creatine-P + ADP vMIA Vpr Atractyloside/palmitate Bongkrekate/ATP/ADP ANT ANT ∆ψm IMM Cardiolipin Ca2+/oxidation? Cyclosporin A Cyp-D Figure 1 | Hypothetical molecular architecture of the permeability transition pore complex and its regulation. The permeability transition pore complex (PTPC) involves several transmembrane proteins: the adenine nucleotide translocator (ANT), the voltage-dependent anion channel (VDAC) and the peripheral benzodiazepine receptor (PBR). It also involves members of the Bax/Bcl-2 family, as well as associated proteins such as hexokinase II (HK II), mitochondrial creatine kinase (mtCK) and the peptidyl–prolyl isomerase cyclophilin D (Cyp-D). The VDAC functions as a nonspecific pore, allowing diffusion of solutes up to 5 kDa. The ANT is responsible for exchange of ATP and ADP on the inner membrane. The exact function of the PBR is unknown. Agents or metabolites labelled in green facilitate PTPC opening; agents in red inhibit pore opening. Proteins or peptides carrying the Bcl-2 homology region-3 (BH3) motif may act on either Bax or Bcl-2 (or their homologues) in the outer mitochondrial membrane. Ca2+ has been postulated to act on ANT-associated cardiolipin molecules; cyclosporin A acts on cyclophilin D. HIV-1 Vpr, the viral mitochondrial inhibitor of apoptosis (vMIA), hepatitis virus B X-protein (HVB-X), and Neisseria meningitidis porin B also act on the PTPC. (OMM, outer mitochondrial membrane; IMM, inner mitochondrial membrane; ∆ψm electrochemical proton gradient.) highly conserved alanine residue 114 in ANT1 causes a dominant human mitochondriopathy24. Whether this pathology is related to a deficient control of apoptosis, however, remains unclear. There is still controversy about where the Bcl-2-like proteins act. Most authors suggest that these proteins are found mainly in the OMM, although a few reports indicate that Bcl-2 (REFS 25,26) and Bax23 are localized to the IMM. Pro-apoptotic stimuli may cause Bax to translocate from the OMM to the IMM23 or cause Bcl-2 to move from the IMM to the OMM26, using as-yetunknown mechanisms. It is also conceivable that protruding domains of Bcl-2 and Bax, which are anchored to the OMM, bind to the IMM within the OMM–IMM contact sites. Indeed, Bcl-2 is particularly enriched at these contact sites. NATURE REVIEWS | MOLECUL AR CELL BIOLOGY Bcl-2 and Bax act on the PTPC The biochemical features of pore-forming proteins can be studied by reconstituting them into synthetic lipid bilayers, either in proteoliposomes or in planar membranes. In response to various pro-apoptotic agents, including atractyloside23, Ca2+, the thiol crosslinker diamide27 and the HIV-1 protein Vpr18, ANT proteoliposomes become permeabilized to hydrophilic compounds with a relative molecular mass of less than 1.5 kDa. Proteoliposomes containing both the ANT and recombinant Bax show higher permeabilization responses to atractyloside23or Vpr18 than do proteoliposomes containing either ANT or Bax alone. In contrast, Bcl-2 prevents the ANT-mediated permeabilization of liposomes responding to atractyloside23 or Vpr18. Analogous results have been obtained for VDAC-containing proteoliposomes (which VOLUME 2 | JANUARY 2001 | 6 9 © 2001 Macmillan Magazines Ltd PERSPECTIVES Bcl-2 Bax Nonspecific Autonomous channel formation sets off local ion imbalances and indirectly stabilizes OMM/IMM Specific Nonspecific Cooperative channel formation within the PTPC or with PTPC components (ANT, VDAC), in OMM and/or IMM Membrane insertion and oligomerization leads to formation of protein-permeable conduits in OMM MMP and cell death Figure 2 | Alternative hypotheses for the modus operandi of Bcl-2/Bax-like proteins. Although Bax and Bcl-2 heterodimerize and inhibit each other, both proteins can act independently in regulating apoptosis. According to one hypothesis (left), Bcl-2 forms protein-impermeable channels, thereby preventing local ion imbalances and non-physiological closure of the voltage-dependent anion channel (VDAC) in the outer mitochondrial membrane (OMM). Bcl-2 and Bax may also interact with the permeability transition pore complex (PTPC) and regulate channel formation by this two-membranespanning protein complex, regulating the permeability of the inner mitochondrial membrane (IMM) and OMM (middle). Bax-like proteins have also been proposed to oligomerize in the OMM, upon interaction with a truncated form of Bid known as tBid, thereby causing the formation of cytochrome c-permeable conduits. (ANT, adenine nucleotide translocator; MMP, mitochondrial membrane permeabilization.) are permeable to sucrose)22. Bcl-2, as well as peptides derived from the BH4 domain of Bcl-2 (where BH4 stands for Bcl-2 homology domain 4, and is a domain that is missing in pro-apoptotic Bcl-2-like proteins), reduces the permeability of such VDAC proteoliposomes to sucrose, whereas Bax enhances their permeability22. Bax/VDAC liposomes (but not liposomes containing VDAC or Bax alone) are permeable to cytochrome c (14.5 kDa), but impermeable to a 50 kDa protein22. nel is qualitatively different from that formed by Bax alone. With a combination of ANT and Bcl-2 (in a molar ratio of 1:1), atractyloside-induced ion movements are no longer seen, indicating the closure of both the ANT and the Bcl-2 channels28. Electrophysiological experiments also indicate cooperative pore formation by the VDAC plus Bax, and inhibition of VDAC channel formation by Bcl-2 (REF. 29). The VDAC and Bax cooperatively create a large pore, with conductance levels fourfold and Bax, ANT and VDAC channels Bax and the ANT (or VDAC) can cooperate to generate channels with higher conductance levels as well as higher opening probabilities than either of the two compounds alone. Moreover, Bcl-2 and the ANT (or VDAC) mutually inhibit the formation of ion channels. Single-channel current measurements involving proteins reconstituted into planar lipid bilayers indicate that the ANT can form large channels with multiple subconductance states (70 to 600 pS) in response to Ca2+. The ANT can also form small channels (30 pS) in response to atractyloside28. A mixture of Bax and the ANT (in a molar ratio of 1:4) has a higher probability of atractyloside-induced pore opening than the ANT alone and shows two conductance levels (30 and 80 pS), as well as cation specificity. At low concentrations (1 nM), Bax does not yield any pronounced macroscopic conductance, unless combined with ANT treated with atractyloside28. So channel formation is more efficient for the combination of the ANT, atractyloside and Bax than for combinations of the ANT plus atractyloside, the ANT and Bax, or atractyloside and Bax. Moreover, the channel formed by Bax at high concentrations is anion specific. So, the ANT/Bax chan- 70 “The intricate relationship between Bcl-2/Bax-like proteins and mitochondrial proteins adds another level of complexity to regulation of cell death.” tenfold greater than those of the VDAC and Bax channels, respectively. Although the VDAC and Bax channels both show ion selectivity and voltage-dependent modulation of their activity, the VDAC–Bax channel has neither of these properties. Cytochrome c reportedly passes through a single VDAC–Bax channel, but not through the VDAC or Bax channels in a planar lipid bilayer29. Anti-apoptotic Bcl-xL and its BH4 oligopeptide reportedly close the VDAC channel30. Studies in isolated mitochondria When incorporated into mitochondrial membranes, Bcl-2 and its anti-apoptotic homologues enhance the resistance of these | JANUARY 2001 | VOLUME 2 mitochondria to a permeability transition. Similarly, microinjection of Bcl-2 into the cytoplasm of intact cells prevents both permeabilization of their mitochondrial membranes and nuclear apoptosis induced by the ANT ligand atractyloside31. If added to purified mitochondria in vitro, recombinant Bax or Bak induce membrane permeabilization, and this is inhibited by various PTPC inhibitors, including Koenig’s polyanion, cyclosporin A, N-methyl-4-Val-cyclosporin A and bongkrekate23,31–33. These are controversial findings, however, as some groups10,11 have not detected any effect of cyclosporin A. Oligomycin, an inhibitor of the F0–F1-ATPase (which, on theoretical grounds, might indirectly affect the ANT), also inhibits mitochondrial membrane permeabilization induced by Bax31–33. At low doses, Bax causes signs of outermembrane permeabilization (that is, release of cytochrome c) but not of inner-membrane permeabilization (for instance, matrix swelling and loss of the ∆ψm). But higher doses of Bax affect both the OMM and the IMM33, and cyclosporin A prevents the effects of Bax on both membranes23,31–33. Studies in intact cells Similar inhibitory profiles to those seen in vitro have been obtained when Bax is microinjected into the cytoplasm of intact cells. In control cells, Bax causes loss of the ∆Ψm and nuclear apoptosis. Our group has found23 that treatment with cyclosporin A, N-methyl-4-Val-cyclosporin A or bongkrekate abolishes both the mitochondrial and the nuclear signs of Bax-induced apoptosis. However, other data10 show that cyclosporin A does not inhibit apoptosis (and the associated mitochondrial membrane permeabilization) induced by transfection with the Bax gene. Proteolytic activation of Bid by caspase-8 (yielding a truncated form of Bid known as tBid) normally leads to the permeabilization of mitochondrial membranes. This permeabilization (and subsequent apoptosis) can be prevented by the ANT-targeted viral mitochondrial inhibitor of apoptosis (vMIA)19. Killing by another pro-apoptotic protein from the Bcl-2 family, BNIP3, is prevented by cyclosporin A and bongkrekate34. Obviously, however, it may be argued that pharmacological inhibition experiments are not truly conclusive — drugs may affect not only components of the PTPC, but also other molecules in the cell — and that genetic interventions on components of the PTPC would be more informative. In yeast cells, several mitochondrial defects www.nature.com/reviews/molcellbio © 2001 Macmillan Magazines Ltd PERSPECTIVES reduce Bax-induced killing, including deletion of the three ANT isoforms23,35, and mutants inactivating the F0–F1-ATPase35,36. These data do not support the idea that Bax kills cells through simple lysis, indicating that Bcl-2-like proteins might modulate cell death through a functional interaction with the PTPC. cell type. Whatever the answer to this conundrum, it seems clear that the intricate relationship between Bcl-2/Bax-like proteins and mitochondrial proteins adds another level of complexity to regulation of cell death. Naoufal Zamzami and Guido Kroemer are at the Centre National de la Recherche Scientifique, UMR1599, Institut Gustave Roussy, 39 rue Camille-Desmoulins, F-94805 Villejuif, France. Correspondence to G.K. e-mail: [email protected] Do Bax/Bcl-2 always act on the PTPC? Recombinant Bax, as well as tBid (but not Bcl-2), has been reported to destabilize lipid bilayers without causing the appearance of ion channels37,38. Alternatively, Bcl-2, Bax and tBid may form channels, with defined levels of conductivity and variable requirements of pH or the local presence of acidic phospholipids39,40. When added to liposomes, Bax can insert into the membranes, oligomerize (presumably to tetramers) and form cytochrome c-permeant conduits with an estimated pore size of around 30 Å (REF. 40). It has been proposed that Bid aids the insertion or oligomerization of Bax12 or Bak13, which would form pores without any interaction with VDAC or ANT. Moreover, recombinant tBid (in contrast to Bax or Bak), added to isolated mitochondria, has been reported41 to cause an exclusive permeabilization of the OMM that is not affected by PTPC inhibitors. All these data may be interpreted to mean that proteins from the Bcl-2/Bax family can permeabilize membranes through a nonspecific effect — that is, without the need for interactions with other proteins from the PTPC. This interpretation is supported by the observation that, during apoptosis, cytochrome c can be released from mitochondria that have an apparently normal ultrastructure and conserve an inner transmembrane potential11,12. How can we reconcile the evidence for these nonspecific effects of Bax/Bcl-2 with the many reports that suggest specific, PTPCdependent effects? One possibility would be to assume that, at physiological concentrations, Bax and Bcl-2 act through these specific effects; for instance, by modulating the probability of channel formation by other proteins. But at higher concentrations, Bax would become able to oligomerize and to exert autonomous, nonspecific effects, involving either channel formation or membrane destabilization. This implies a hierarchic superposition of two levels of regulation. Alternatively, the two pathways could come into action as a function of the local abundance of PTPC components, their isoforms, or PTPC inhibitors/activators (FIG. 2). Indeed, a similar mechanism has been proposed for CD95induced signalling, in which the same primary signal can trigger cell death through completely distinct mechanisms, as a function of the Links DATABASE LINKS Smac | Bcl-2 | Bcl-xL | Bcl-W | Mcl1 | A1 | Bax | Bad | Bid | Bim | creatine kinase | hexokinase II | cyclophilin D | CD95 | ANT2 | mouse Ant1 | BNIP3 ENCYCLOPEDIA OF LIFE SCIENCES Apoptosis: molecular mechanisms 1. 2. 3. 4. 5. 6. 7. 8. 9. 10. 11. 12. 13. 14. 15. 16. 17. 18. 19. Green, D. R. & Reed, J. C. Mitochondria and apoptosis. Science 281, 1309–1312 (1998). Kroemer, G. & Reed, J. C. Mitochondrial control of cell death. Nature Med. 6, 513–519 (2000). Budijardjo, I., Oliver, H., Lutter, M., Luo, X. & Wang, X. Biochemical pathways of caspase activation during apoptosis. Annu. Rev. Cell Dev. Biol. 15, 269–290 (1999). Vander Heiden, M. G. & Thompson, C. B. Bcl-2 proteins: Inhibitors of apoptosis or regulators of mitochondrial homeostasis? Nature Cell Biol. 1, E209–E216 (1999). Gross, A., McDonnell, J. M. & Korsmeyer, S. J. Bcl-2 family members and the mitochondria in apoptosis. Genes Dev. 13, 1899–1911 (1999). Crompton, M. The mitochondrial permeability transition pore and its role in cell death. Biochem. J. 341, 233–249 (1999). Woodfield, K., Ruck, A., Brdiczka, D. & Halestrap, A. P. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem. J. 336, 287–290 (1998). Feldmann, G. et al. Opening of the mitochondrial permeability transition pore causes matrix expansion and outer membrane rupture in Fas-mediated hepatic apoptosis in mice. Hepatology 31, 674–683 (2000). Yin, X.-M. et al. Bid-deficient mice are resistant to Fasinduced hepatocellular apoptosis. Nature 400, 886–891 (1999). Eskes, R. et al. Bax-induced cytochrome c release from mitochondria is independent of the permeability transition pore but highly dependent on Mg2+ ions. J. Cell Biol. 143, 217–224 (1998). von Ahsen, O. et al. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cytochrome c release. J. Cell Biol. 150, 1027–1036 (2000). Eskes, R., Desagher, S., Antonsson, B. & Martinou, J. C. Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane. Mol. Cell. Biol. 20, 929–935 (2000). Wei, M. C. et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 14, 2060–2071 (2000). Kroemer, G., Dallaporta, B. & Resche-Rigon, M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu. Rev. Physiol. 60, 619–642 (1998). Sullivan, P. G., Thompson, M. B. & Scheff, S. W. Cyclosporin A attenuates acute mitochondrial dysfunction following traumatic brain injury. Exp. Neurol. 160, 226–234 (1999). Budd, S. L., Tenneti, L., Lishnak, T. & Lipton, S. A. Mitochondrial and extramitochondrial apoptotic signaling pathways in cerebrocortical neurons. Proc. Natl Acad. Sci. USA 97, 6161–6166 (2000). Tafain, M., Schneider, T. G., Pastorino, J. G. & Farber, J. L. Cytochrome c-dependent activation of caspase-3 by tumor necrosis factor requires induction of the mitochondrial permeability transition. Am. J. Pathol. 156, 2111–2121 (2000). Jacotot, E. et al. The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J. Exp. Med. 191, 33–45 (2000). Goldmacher, V. S. et al. A cytomegalovirus-encoded NATURE REVIEWS | MOLECUL AR CELL BIOLOGY 20. 21. 22. 23. 24. 25. 26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 40. 41. mitochondria-localized inhibitor of apoptosis structurally unrelated to Bcl-2. Proc. Natl Acad. Sci. USA 96, 12536–12541 (1999). Rahmani, Z., Huh, K. W., Lasher, R. & Siddiqui, A. Hepatitis B virus X protein colocalizes to mitochondria with a human voltage-dependent anion channel, HVDAC3, and alters its transmembrane potential. J. Virol. 74, 2840–2846 (2000). Massari, P., Ho, Y. & Wetzler, L. M. Neisseria meningitidis porin PorB interacts with mitochondria and protects cells from apoptosis. Proc. Natl Acad. Sci. USA 97, 9070–9075 (2000). Shimizu, S., Narita, M. & Tsujimoto, Y. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399, 483–487 (1999). Marzo, I. et al. Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281, 2027–2031 (1998). Kaukonen, J. et al. Role of adenine nucleotide translocator 1 in mtDNA maintenance. Science 289, 782–785 (2000). Motoyama, S. et al. Bcl-2 is located predominantly in the inner membrane and crista of mitochondria in rat liver. Biochem. Biophys. Res. Commun. 249, 628–636 (1998). Gotow, T. et al. Selective localization of Bcl-2 to the inner mitochondrial and smooth endoplasmic reticulum membranes in mammalian cells. Cell. Death Differ. 7, 666–674 (2000). Costantini, P. et al. Oxidation of a critical thiol residue of the adenine nucleotide translocator enforces Bcl-2independent permeability transition pore opening and apoptosis. Oncogene 19, 307–314 (2000). Brenner, C. et al. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene 19, 329–336 (2000). Shimizu, S., Ide, T., Yanagida, T. & Tsujimoti, Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J. Biol. Chem. 275, 12321–12325 (2000). Shimizu, S., Konishi, A., Kodama, T. & Tsujimoto, Y. BH4 domain of antiapoptotic Bcl-2 family members closes voltage-dependent anion channel and inhibits apoptotic mitochondrial changes and cell death. Proc. Natl Acad. Sci. USA 97, 3100–3105 (2000). Jürgensmeier, J. M. et al. Bax directly induces release of cytochrome c from isolated mitochondria. Proc. Natl Acad. Sci. USA 95, 4997–5002 (1998). Narita, M. et al. Bax interacts with the permeability transition pore to induce permeability transition and cytochrome c release in isolated mitochondria. Proc. Natl Acad. Sci. USA 95, 14681–14686 (1998). Pastorino, J. G. et al. Functional consequences of sustained or transient activation by Bax of the mitochondrial permeability transition pore. J. Biol. Chem. 274, 31734–31739 (1999). Vande Velde, C. et al. BIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol. Cell. Biol. 20, 5454–5468 (2000). Harris, M. H., Vander Heiden, M. G., Kron, S. J. & Thompson, C. B. Role of oxidative phosphorylation in Bax toxicity. Mol. Cell. Biol. 20, 3590–3596 (2000). Matsuyama, S., Xu, Q., Velours, J. & Reed, J. C. Mitochondrial F0F1-ATPase proton pump is required for function of proapoptotic protein Bax in yeast and mammalian cells. Mol. Cell 1, 327–336 (1998). Basañez, G. et al. Bax, but not Bcl-xL, decreases the lifetime of planar phospholipid bilayer membranes at subnanomolar concentrations. Proc. Natl Acad. Sci. USA 96, 5492–5497 (1999). Kudla, G. et al. The destabilization of lipid membrane induced by the C-terminal fragment of caspase 8cleaved Bid is inhibited by the N-terminal fragment. J. Biol. Chem. 275, 22713–22718 (2000). Schendel, S. L. et al. Ion channel activity of the BH3 only bcl-2 family member, BID. J. Biol. Chem. 274, 21932–21936 (1999). Saito, M., Korsmeyer, S. J. & Schlesinger, P. H. Baxdependent transport of cytochrome c reconstituted in pure liposomes. Nature Cell Biol. 2, 553–555 (2000). Shimizu, S. & Tsujimoto, Y. Proapoptotic BH3-only Bcl-2 family members induce cytochrome c release, but not mitochondrial membrane potential loss, and do not directly modulate voltage-dependent anion channel activity. Proc. Natl Acad. Sci. USA 97, 577–582 (2000). Acknowledgements The authors’ work is supported by a special grant from the Ligue Nationale contre le Cancer, as well as grants from ANRS, FRM and EC (to G.K.). VOLUME 2 | JANUARY 2001 | 7 1 © 2001 Macmillan Magazines Ltd