Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Molecular Inversion Probe wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Gene expression programming wikipedia , lookup
Cancer epigenetics wikipedia , lookup
Ridge (biology) wikipedia , lookup
Point mutation wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Primary transcript wikipedia , lookup
Mitochondrial DNA wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Genomic imprinting wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Copy-number variation wikipedia , lookup
Genetic engineering wikipedia , lookup
Gene expression profiling wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Oncogenomics wikipedia , lookup
Epigenomics wikipedia , lookup
Transposable element wikipedia , lookup
Public health genomics wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Genome (book) wikipedia , lookup
Designer baby wikipedia , lookup
Microevolution wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Non-coding DNA wikipedia , lookup
History of genetic engineering wikipedia , lookup
DNA sequencing wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Minimal genome wikipedia , lookup
Human genome wikipedia , lookup
Helitron (biology) wikipedia , lookup
Pathogenomics wikipedia , lookup
Genome editing wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genome evolution wikipedia , lookup
Human Genome Project wikipedia , lookup
Exome sequencing wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Genomic library wikipedia , lookup
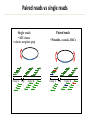
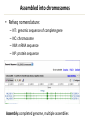
Course Expectations Sequencing technology and (very) large datasets 5/17/2016 Goals for the course • Understand how next-generation sequencing technologies are used in biomedical research • Learn how to use publicly available databases/websites to find specific information about genes • Learn how to analyze gene lists to form hypotheses that can be tested experimentally • Learn to write a results section for a manuscript Logistics • Course website: – http://biochem.slu.edu/bchm628/ • Contact: – Phone: 977-8858 – Email: [email protected] • Office – DRC 611 – Call or email. – Usually at WashU on Thursday afternoons • Lab – DRC 654 Exercise format • There will be 6 exercises, each consisting of 2-4 sections which represent a biological question to be answered with bioinformatics tools/resources from that week or earlier weeks. • You’ll provide the answer in the same format as you would write for the results section of a paper 1. 2. 3. 4. Why did you do this experiment or analysis? What did you actually do? What did you observe? What does it mean? • Include supporting data – Figures with figure legends – Correctly formatted tables of data. Exercises, cont • You will hand in your exercise via email – Exercise in Word or PDF format – Supplemental data in Excel, Word or PDF format. • The exercise should print in portrait orientation. • The exercise should include a header with your name at the top and the file should be named: – Your Name-Ex #. • There is a penalty for turning in your exercises after the deadline. The timestamp on your email is the final determination of whether an exercise is on-time or not. Final project • This will be a project summary of the analyses that you will do over the course of the 4 weeks. • You will be asked to choose 3 genes from your gene lists that you would follow-up on at the bench. – You will be asked to give a rationale for making the choices that you did. • You will analyze the three genes virtually using some of the tools from weeks 1- 4. • You will be asked to propose hypothetical bench experiments for the genes • Final project will be due June 21st at 3:00 pm. Data tables In general, columns describe attributes and rows contain the individual data. The first row contains a header. If you have lots of data, it is generally formatted to have more rows than columns. Table 1: Gene expression for WT cells under conditions X,Y, Z. Gene name Log 2 (Cond. X/untreated) Log 2 (Cond. Y/untreated) Log 2 (Cond. Z/untreated) NM_00522 2.56 3.12 2.75 NM_06588 -1.25 -1.02 -0.98 Table 2: Comparison of clinical parameters for groups 1 and 2. Clinical parameter ALT/AST ratio Leukocyte count 1 Statistical 2 Group 1 (avg ± mean) Group 2 (avg ± mean) P-value 25 ± 1 35 ± 2 0.0021 1200 ± 32 950 ± 65 0.0512 significance was determined by a Mann-Whitney test Statistical significance was determined by 2-tailed t-test Data tables, cont • For the purposes of this class, the tables should be formatted to fit onto a letter size page in portrait orientation. • If your table is so wide that it forces the page into landscape orientation, then it should be included as a supplemental attachment to the exercise. If the table extends past 1 page, then include it as a supplemental attachment. • Refer to supplemental tables in your write-up and number then and the file as Name_SuppTable1, ect. • Supplemental tables can be in Excel format. Figures • If you can export the figure from whatever program in jpeg or png format, those can be inserted into a Word document easily. • PDFs can be converted to other formats using Illustrator • There are some online converters – http://www.wikihow.com/Convert-PDF-to-JPEG • Screen capture and placement may also work. • Talk to me if you have issues. • I won’t be very picky about high resolution. Figures, cont. • Figures should have figure legends. The figure legends should describe the experiment that lead to the data in the figure and include an explanation for any symbols used. • Figures should be numbered consecutively and should not take up more than ¼ of the page. If larger than that, include as supplemental data. • Create a text box in Word, write the figure legend and then insert the figure above the figure legend. This will allow you to resize as necessary. • Again, talk to me if you have issues. Grading • Grading: – Exercises – Final exam – Class attendance 65 % 25 % 10 % • Grading policy handout – Details about late assignment and tests Lecture outline • Overview of sequencing a genome • Next generation sequencing • High-throughput experiments by sequencing • Genome browsers Genome sequencing Approach depends on the source, size, complexity and goal for the data for a given organism Goal? – De novo sequencing – Re-sequencing for annotation – Sequencing to identify variations • Size and complexity – Virus, bacterial, single-celled eukaryote, mammal, plant – Quasi-species or repetitive sequences • Sample prep – Can it be cultured? – Tissue source: unlimited or limited quantities? – Virus levels, RNA or DNA Genome sizes Genome size (base pairs) Number of genes Hepatitis C virus 0.01 x 106 10 Epstein-Barr virus 0.172 x 106 37 Bacterium (E. coli) 4.6 x 106 4406 Yeast (S. cerevisiae) 12.5 x 106 6172 Nematode worm (C. elegans) 100.3 x 106 19,099 Thale cress (A. thaliana) 115.4 x 106 25,498 Fruit fly (D. melanogaster) 128.3 x 106 13,601 Corn (Z. mays) 2500 x 106 39,469 Human (H. sapiens) 3223 x 106 20,500 Wheat (T. aestivium) 5500 x 106 (x 3) ~95,000 Organism Types of questions • How many genes? – How many functional genetic elements – miRNAs, ncRNAs • What’s different about this genome compared to another one? – Virulence differences in pathogenic organisms – What is the cause of this particular phenotype? • What taxonomic groups are represented in this population of bacteria, viruses or fungi? • How do the gene expression patterns change between samples (and across time)? • Where does this transcription factor bind in the genome? DNA sequencing – Overview • Gel electrophoresis – Predominant in 1980s • Whole genome strategies Cost/base for DNA sequence 1.0E+02 1.0E+01 Physical mapping (BAC clones) Walking Shotgun sequencing Capillary sequencing machines 1.0E+00 • Computational fragment assembly • Next generation technologies 1.0E-05 – – – – – Polony based sequencing – Novel assembly techniques 1.0E-01 1.0E-02 1.0E-03 1.0E-04 1.0E-06 1.0E-07 Traditional approach 1. Shear the very large genome into smaller chunks 2. Clone in vectors that can support large inserts 3. Digest and separate on high resolution gel to determine the clone overlap 4. Pick minimum number of clones 5. Shotgun sequence each clone 6. Read the traces and assemble 7. Make the gene calls 8. Load it into a genome viewer BAC library in DNA sequencing Shotgun sequencing D Sequence each clone Individual sequence reads Contig assembly E Contig A Gap Contig B Paired reads vs single reads Single reads • M13 clones • robotic template prep Contig A Gap Contig B Paired reads • Plasmids, cosmids, BACs Contig A Gap Contig B Gap closure!! Prefer 3-10 mate pairs per gap Inserts of different, but known sizes Steps to Assemble a Genome Some Terminology read a 500-900 long word that comes 1. Find reads outoverlapping of sequencer mate pair a pair of reads from two ends of the same insert fragment 2. Merge some “good” pairs of reads into contigssequence formed contig longer a contiguous by several overlapping reads with no gaps 3. Link contigs to formand supercontigs supercontig an ordered oriented set (scaffold) of contigs, usually by mate pairs consensus sequence derived from the 4. Derive multiple consensus sequence sequence alignment of reads in contig ..ACGATTACAATAGGTT.. Target: 30X coverage or >30 high quality reads per base Assembled into chromosomes • Refseq nomenclature: – – – – NT: genomic sequence of complete gene NC: chromosome NM: mRNA sequence NP: protein sequence Assembly: completed genome, multiple assemblies Calling the genes • De novo computer algorithms – Identify coding sequences by GC content – Start and stop sites – Intron/exon boundaries • Comparison with other known genes • EST libraries Sanger method Sanger sequencing reached its technical limits • Only modestly parallel (394 lanes/machine) • Long read lengths (500-900 bp) & >99.9% correct • Need to clone the DNA to obtain enough for sequencing reaction • At SLU: cost for typical Sanger sequencing is $56/sample with reliable 500 bp of sequence DNA sequencing timeline How many sequenced genomes? NCBI: >16,000 genomes deposited JGI (Joint Genome Institute): >8000 complete >28,000 draft genomes NGS sequencing • Polony: discrete clonal amplifications of a single DNA molecule, grown in a gel matrix. The clusters can then be individually sequenced, producing short reads • Polony-based or cluster-based sequencing is the basis of most second generation sequencers Typical NGS workflow: 1. Library construction to add adapters to sequence 2. Template CLONAL amplification (on a bead or chip) 3. Massively PARALLEL sequencing Library Prep: ~ 6 hours Illumina NGS A) Fragment DNA B) Repair ends/Add A overhang DNA C) Ligate adapters D) Select ligated DNA Cluster generation ~ 6 hours E) Attach DNA to flow cell F) Bridge amplification G) Generate clusters H) Anneal sequencing primer Sequencing 2-6 days I) Extend 1st base, read & deblock K) Generate base calls J) Repeat to extend strand Illumina HiSeq and miSeq • 100 – 200 bp read lengths • Available locally with MoGene and Cofactor Genomics • GTAC (Wash U) has HiSeq 2500, HiSeq 3000 and MiSeq. They offer read lengths from 50bp to 250 bp (single- and paired-end) • Why not use this for all sequencing? – – – – Cost is ~300-400/library and ~$1100/lane of sequencing Generate Tb of data per run Gb per lane Sample prep limitations Ion Torrent – measures pH changes Done on a semiconductor chip Ion Torrent workflow Illumina vs Ion Torrent • • • • Illumina has greater capacity but longer run times Latest versions of both have read lengths ~200 bp SLU has an Ion Torrent machine Cost is ~$270/sample, including the sequencing • Can do single- or pair-end reads • Paired end are 2X cost for library construction, but necessary for de novo genome assembly Bioinformatics challenges • Each flow cell in the Illumina Hiseq 2500 can generate a billion bases of sequence – Raw read files are Tb in size – Processed read files are several 700-800 Mb – Alignment files 150-300 Mb • Assembly of millions of short (75-100 bp) reads into vertebrate genome – Need high-performance compute (HPC) cluster for vertebrate sized genomes* • What biomolecular species to interrogate – 25,000 genes – 160,000 transcripts – miRNA, non-coding RNA Sequencing has become a standard technique • • • • RNA sequencing for expression ChIP sequencing for TF site identification DNA sequencing for variants Identification of populations/genetic changes in highly variable viruses and bacteria • Metagenomics – Identification of unknown/non-culturable communities of bacteria/viruses/fungi Where is all this data deposited? • NCBI: National Center for Biotechnology Information • Databases are well integrated • Well integrated with literature (PubMed) • EBI: European Bioinformatics Institute • • • • • Same base data as NCBI, but offers different front-end Much better list-based searching More protein-based information (domains, complexes & interactions) Not as well integrated with literature Transcript variants differ from NCBI because of different annotation pipelines NCBI Ensembl main page Genome viewers • Provides chromosomal context to the gene(s) of interest • See transcript variants in graphical view • Have “tracks” of additional information: – – – – – Variants (SNPs) Expression data Repetitive sequences Comparative data (with other species) Download genomic sequence • Ensembl genome viewer (useast.ensembl.org) • UCSC genome viewer (genome.ucsc.edu) Genetic maps • Chromosomal banding patterns – Stain with Giemsa (G-banding pattern) Chromosomes are numbered based on size Giemsa binds to phosphate groups & attaches to regions that are AT rich Dark regions heterchromatic, late replicating and AT rich Lighter regions euchromatic, early replicating and GC rich Chromosome nomenclature p (petite) = short arm q (queue) = long arm Bands are numbered going away from centromere 4q21.1 represents chromosome 4, long arm 2nd band, 1st sub-band and 1st sub-sub-band Today in computer lab • Finding genes and transcripts using NCBI and EBI • Visualization of genes and transcripts with genome browsers