Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Transitional fossil wikipedia , lookup
DNA damage theory of aging wikipedia , lookup
DNA vaccination wikipedia , lookup
Genomic library wikipedia , lookup
Oncogenomics wikipedia , lookup
Cancer epigenetics wikipedia , lookup
Primary transcript wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Pathogenomics wikipedia , lookup
United Kingdom National DNA Database wikipedia , lookup
Mitochondrial DNA wikipedia , lookup
Human genome wikipedia , lookup
Genome evolution wikipedia , lookup
Epigenomics wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Nucleic acid double helix wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Molecular cloning wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
DNA supercoil wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Genealogical DNA test wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
History of genetic engineering wikipedia , lookup
Designer baby wikipedia , lookup
Non-coding DNA wikipedia , lookup
DNA barcoding wikipedia , lookup
Koinophilia wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Microsatellite wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
Genome editing wikipedia , lookup
Point mutation wikipedia , lookup
Metagenomics wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Helitron (biology) wikipedia , lookup
Microevolution wikipedia , lookup
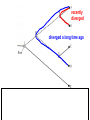
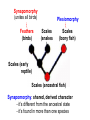
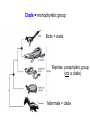
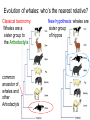
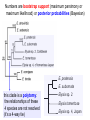
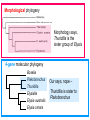
Phylogeny or “tree thinking” The evolutionary history of extant organisms can be understood in terms of their shared inheritance - which extant species evolved from the same ancestor? - how were ancestral traits modified in different lineages? A hypothesis of the evolutionary history of a group is called its phylogeny - often summarized in a branching diagram called a phylogenetic tree Since we can’t travel back in time to identify common ancestors, relationships of existing species must be estimated or inferred from data – therefore, a phylogeny is always a hypothesis Tree thinking and phylogeny Nodes: branching points Branches: lines connecting nodes Phylogenetic tree Topology: branching pattern Taxon = any named group of organisms A+B Two or more = taxa C+D Sister Taxa: two taxa (= named group of organisms) that are more closely related to each other than either is to a 3rd taxon recently diverged diverged a long time ago Branches can be rotated at a node, without changing relationships among the taxa these 3 trees are all equivalent Relationships can be resolved or unresolved A node with more than 2 branches from it is called a polytomy, and means the relationships are not fully resolved - may the data aren’t good enough to figure out the true relationships among descendents of that ancestor - maybe ancestor had multiple direct descendents -- rapid speciation can occur during an adaptive radiation Characters (traits) change across a phylogeny Plesiomorphy: ancestral (or primitive) character state Apomorphy: derived character state, modified from the ancestral (primitive) state Synapomorphy: derived character shared by more than one species or group (shared derived character) Synapomorphy ancestral derived Synapomorphy (unites all birds) Feathers (birds) Plesiomorphy Scales (snakes ) Scales (bony fish) Scales (early reptile) Scales (ancestral fish) Synapomorphy: shared, derived character - it’s different from the ancestral state - it’s found in more than one species Homologous trait: structurally similar, but functionally different - human arm, dolphin flipper, bat wing are homologous: all descended from same ancestral tetrapod appendage, with modification Homoplasy: structurally different (unrelated), but functionally similar - example: streamlined shape of sharks + killer whales - adaptation to similar environment, driven by natural selection flippers = yes arms = no similar coding for these non-relatives Distinguishing between true homology and homoplasy is critical if we are to sort out the phylogeny, or evolutionary relationship, among living things Homoplasy = problem for phylogenetics Homoplasy: non-homologous traits look superficially similar - resemblance is not due to common ancestry, but rather to convergent evolution in 2 different groups, which tricks us - you don’t want to code non-relatives as having the same traits, or they will falsely group together on your tree wings in birds wings in bats Cladistics uses synapomorphies to identify clades, which (a) are the groups we want to name (true groups) (b) tell us something about the evolution of a group For a long time, this was the only way to infer phylogeny, using morphological character traits Cladistics uses synapomorphies alone to establish recent common ancestry - relies on synapomorphies to define monophyletic groups, or clades (same thing) Monophyletic group includes ALL descendants of an ancestral species Types of non-monophyletic groups Paraphyletic: leaves out one or more descendants of an ancestor Polyphyletic: derived from 2 or more ancestral taxa (= crap) Paraphyletic Polyphyletic Clade = monophyletic group Birds = clade Reptiles: paraphyletic group (not a clade) Mammals = clade Why do we name para- and polyphyletic groups? Taxonomists often assigned class or family status to groups that are very morphologically distinctive, such as birds and cetaceans (whales & dolphins) - does not reflect their evolutionary status within other groups what do you think: should scientists call birds and reptiles both classes of vertebrates, if birds are really a subset of reptiles? Another reason is, mistakes can be made in defining relationships depending on the character that is used Cladistics and parsimony Techniques that identify monophyletic groups based on shared derived characters are cladistic methods - based on the idea that the most related groups will have the most traits in common Parsimony is a principle that simpler explanations are more likely to be correct than complicated explanations - when choosing among hypotheses, we should accept the one that explains the data most simply and efficiently - assumes convergence is less likely than shared descent Cladistics and parsimony When a new trait appears, all descendants will share it as a synapomorphy (barring secondary loss of the trait) feathers Bird evolution Cladistics and parsimony Maximum parsimony = method of phylogeny reconstruction, process by which we infer the evolutionary history of a group based on the traits we see today - the best phylogenetic tree is the one which requires the fewest changes in traits (characters) to account for modern character states in surviving lineages - i.e., assumes that the minimum number of changes is what really happened over the course of evolution Using parsimony to infer evolutionary relationships How are the major types of algae-eating sea slugs related? some have shells; seem “primitive” because they retain a plesiomorphy, the ancestral snail shell some groups have frilly flaps on their backs... are they the most derived = different from the ancestor? “code” the different states for each morphological character, for each existing taxon “has a shell” = 0, “no shell” = 1 # of characters you can use is unlimited the characters you are using a coded representation of one genus feed all this into a computer; ask it to give you the tree that requires the fewest number of overall changes, to produce the distribution of traits seen in modern taxa Parsimony analysis of morphological traits gave this answer for how all genera in this group are related Homoplasy = problem for phylogenetics Problem: Morphological traits may be unreliable for inferring evolutionary relationships if they are frequently subject to convergent evolution - homoplasy may fool us if we rely too heavily on morphology can we find traits to use in phylogeny-building that are largely invisible to selection? DNA sequence data & phylogenetics Most modern phylogenetic trees are based partly or entirely on molecular data, usually DNA sequence information - you can combine molecular and morphological data in a tree - you can combine data from different genes Some changes in DNA are largely “invisible” to selection; mutations steadily accrue over time in a clock-like manner - more distant relatives have more changes in their DNA than close relatives - true for non-coding junk DNA, introns, & silent substitutions ATTCGTATTC ATTCGTTTTC ATTCGTTTTC ATTCGTTTTC ATTCGTATTC Changes provide insight into patterns of relatedness (phylogeny) DNA sequence data & phylogenetics Drawback: any given site in a DNA sequence can only occupy one of 4 possible “character states” -- A, T, G, or C - “character” is the nucleotide at a given position in the DNA - “state” is what the character looks like in a particular species (a “G” at position 137 of the actin gene) Mutations can change the nucleotide sequence, but then later mutations can change the sequence back to the original one such reversions can fool us (= homoplasy) ATTCGTATTC ATTCGTTTTC ATTCGTTTTC ATTCGTATTC ATTCGTATTC ATTCGTTTTC ATTCGTATTC DNA sequence data & phylogenetics Mutations can change the nucleotide sequence, but then later mutations can change the sequence back to the original one This leads to a form of homoplasy when comparing DNA sequences, since there are only 4 possible character states - unlike with morphology, at least this kind of homoplasy cannot result from convergent evolution; it’s “accidental” Amino acids have 20 possible states, which is a bit better - less chance of homoplasy from reverse-mutations to the ancestral amino acid - however, they can also be under selection possibility of convergent evolution exists DNA sequence data & phylogenetics Mutations can change the nucleotide sequence, but then later mutations can change the sequence back to the original one This leads to a form of homoplasy when comparing DNA sequences, since there are only 4 possible character states - unlike with morphology, at least this kind of homoplasy cannot result from convergent evolution; it’s “accidental” Take-home: DNA sequence data & phylogenetics Advantages of DNA-based methods: - DNA sequences do not undergo convergent evolution, so there’s no homoplasy like in morphological characters - you can cheaply acquires 1000’s of nucleotides; each position is potentially information - hopefully, the huge amount of good data will drown out the mis-information present due to “accidental” homoplasy DNA sequence data & phylogenetics Disadvantages of DNA-based methods: - homoplasy arises from reverse-mutations, which makes some nucleotide positions misleading - data can be generated very quickly, but analysis is slow and can require months of computing time (more to follow) Evolution of whales: who’s the nearest relative? Classical taxonomy: Whales are a sister group to the Artiodactyla common ancestor of whales and other Artiodactyls New hypothesis: whales are sister group of hippos 4 kinds of DNA-based analysis There are 4 ways to use DNA info to construct a phylogeny (1) Genetic distance (fast) - “Neighbor-Joining” or NJ tree - pairs up the closest sequences (lowest % difference) as sister taxa, builds a tree from there - discards info about specific changes, focuses on overall similarities and differences - quick and dirty, not very sophisticated Genetic distance Fast, especially when you have a huge amount of sequence data (otherwise many possible trees; slow computation) Branches show how much each of 2 sister taxa has diverged from hypothetical common ancestor 4 kinds of DNA-based analysis (2) Maximum parsimony (medium speed) - uses only informative sites to draw the most parsimonious tree - finds the minimum set of changes that had to occur to produce the data you observe in present-day species - also discards lots of information Parsimony analysis and DNA sequence data Can DNA sequence comparisons tell us who’s related to whales? site 162: C T mutation defines a clade that includes hippos, whales, deer + cows Parsimony analysis and DNA sequence data Can DNA sequence comparisons tell us who’s related to whales? site 166: G C mutation defines hippos + whales as a clade Position 177 shows a reversion: a change from C to T at (1), and change from T back to C at (2) T C (2) C T (1) Every time you posit that a reversion occurred, you make the tree less parsimonious (it costs you) - doesn’t mean reversions don’t happen; just that they are less likely than shared inheritance Tree that requires the smallest number of changes is the most parsimonious, and therefore the “chosen” phylogeny Problem: there can be “ties” where multiple trees require the same number of changes - no way to chose among equally-parsimonious trees 4 kinds of DNA-based analysis (3) Maximum likelihood (slow) - uses a model of DNA sequence evolution to find the most likely tree, given the data - slow; searches all possible trees to find the best one (4) Bayesian inference (medium speed) - also uses model of DNA sequence evolution - rather than looking for best tree, looks at many likely trees (much faster than likelihood) - tells you how often a particular relationship shows up i.e., what % of good trees show the clade [hippo+whale] What is a model of DNA evolution? 0.01 1.0 0.5 0.1 - model may also include a rate multiplier that allows a given site to 10.3 mutate at 10-100 times slower, or faster, than the G T rate 1.0 Model of DNA sequence evolution is a matrix of estimated rates at which all the different mutations occurred, given your data rates are both inferred from your sequence data, and then used to estimate the most likely tree given your data 4 kinds of DNA-based analysis Advantage of likelihood/Bayesian methods: - models of DNA sequence evolution include all sorts of rates of change, which are estimated from the data and used by computer programs to infer the likeliest trees - used to estimate the likelihood of changes at each nucleotide position, based on the overall sequence data A G type of change transition C G transversion likelihood 1.0 some changes are more likely than others 0.1 C A transversion 0.01 DNA-based phylogenetic analysis A G type of change transition C G transversion 0.1 sometimes happens A C transversion 0.01 rarely ever happens A ? C ? ? A G relative likelihood 1.0 often happens Can we figure out what the ancestral nucleotide was at a certain position, if we know the nucleotide at that position in four related species today? DNA-based phylogenetic analysis A G type of change transition C G transversion 0.1 sometimes happens A C transversion 0.01 rarely ever happens A A C A A A A G A G relative likelihood 1.0 often happens C Parsimony Likelihood 2 changes A G often A C rarely Only two changes required, but one is a very rare change (likelihood doesn’t like to let rare things happen) DNA-based phylogenetic analysis A G type of change transition C G transversion 0.1 sometimes happens A C transversion 0.01 rarely ever happens G A A G C G G G relative likelihood 1.0 often happens A G G A C Parsimony 3 changes, don’t like as much Likelihood G A often G A often G C sometimes Likelihood may favor more changes if they are likely changes Tree parameters In addition to estimaitng the model of DNA sequence evolution, the computer program will draw the phylogeny that maximizes the likelihood of seeing your sequence data, by altering 2 things: 1) the topology itself (the tree, or branching relationships) C B C A likelihood: 1.2 0.04 B A B C A 37.1 Tree parameters In addition to estimaitng the model of DNA sequence evolution, the computer program will draw the phylogeny that maximizes the likelihood of seeing your sequence data, by altering 2 things: 1) the topology itself (the tree, or branching relationships) 2) branch lengths (estimate of evolutionary time, or amount of change, since a split in the tree) C B C A C A B likelihood: 10.2 B B A C A B 7.1 C A C A 12.5 B Genes evolve at different rates Some genes evolve faster than others, so you can pick a gene appropriate to the problem you are tackling If comparing closely related species, use fast-evolving mitochondrial genes to show differences mitochondrial lack sophisticated proof-reading enzymes of the nucleus; accumulate mutations faster If comparing distantly related taxa, use a slow-evolving nuclear gene, like 18S ribosomal RNA or histone 3 gene Numbers are bootstrap support (maximum parsimony or maximum likelihood) or posterior probabilities (Bayesian) Statistical measure of how confident we are that a given node is real (that relationships really are how they appear in the tree) - significant bootstrap levels are >70% - significant posterior probabilities are >90% Numbers are bootstrap support (maximum parsimony or maximum likelihood) or posterior probabilities (Bayesian) E. pratensis E. subornata this clade is a polytomy: the relationships of these 4 species are not resolved (it’s a 4-way tie) Elysia sp. 2 Elysia tomentosa Elysia sp. 4, Japan mitochondrial COI gene every slug has a slightly different sequence nuclear histone H3 gene Alderia modesta all alleles differ by silent substitutions the species are 20% different at this gene within a species, every slug has the same allele the two species are 1% different at this conserved gene Alderia willowi Thuridilla Boselia basal, or “primitive” Plakobranchus Elysia - most species-rich genus My phylogeny of sea slug family Elysiidae, based on 4 genes: 2 fast mitochondrial genes 2 slow-evolving nuclear genes Sequences of the fast-evolving mitochondrial COI gene help to distinguish individuals within a species from different populations - Guam versus Australia - different Caribbean islands Also resolve the placement of closely related (recently diverged) species – i.e., near the tips of tree Sequences of conserved, slow-evolving nuclear genes help to resolve ancient events (relationships among genera) - things that happened near the root of the tree, farther back in time For instance, can help answer group-level questions: Who is the sister group of Elysia? Sequences of conserved, slow-evolving nuclear genes help to resolve ancient events (relationships among genera) - things that happened near the root of the tree, farther back in time For instance, can help answer group-level questions: Who is the sister group of Elysia? … a clade of Thuridilla + Plako Does DNA give you the same “answer” (= phylogeny) as morphological traits? Why or why not, and which is better? Morphological phylogeny Morphology says, Thuridilla is the sister group of Elysia 4-gene molecular phylogeny Boselia Plakobranchus Thuridilla Elysiella Elysia australis Elysia ornata Our says, nope – Thuridilla is sister to Plakobranchus DNA sequence data & phylogenetics Many kinds of molecular data are used in evolutionary biology: a) DNA sequence comparisons b) amino acid sequences of protein-coding genes c) secondary structure of RNA (rRNA, tRNA genes) d) presence/absence of introns, transposable elements e) microsatellites f) gene order on the chromosome g) gene duplication events (presence/absence of genes) h) expression patterns of genes (evo-devo)