Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Viral phylodynamics wikipedia , lookup
Genealogical DNA test wikipedia , lookup
Ridge (biology) wikipedia , lookup
Point mutation wikipedia , lookup
Mitochondrial DNA wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
Genomic imprinting wikipedia , lookup
Transposable element wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Gene expression programming wikipedia , lookup
Genomic library wikipedia , lookup
Short interspersed nuclear elements (SINEs) wikipedia , lookup
Gene expression profiling wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Genome (book) wikipedia , lookup
Designer baby wikipedia , lookup
Quantitative trait locus wikipedia , lookup
History of genetic engineering wikipedia , lookup
Pathogenomics wikipedia , lookup
Microsatellite wikipedia , lookup
Human genome wikipedia , lookup
Minimal genome wikipedia , lookup
Non-coding DNA wikipedia , lookup
Microevolution wikipedia , lookup
Metagenomics wikipedia , lookup
Genome editing wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
Genome evolution wikipedia , lookup
Maximum parsimony (phylogenetics) wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Lecture 7
Difficult problems….and solutions
Platypus (Ornithorhynchus anatinus)
Non-homogenous evolution
Taxon1
Taxon2
Taxon3
Taxon4
1
3
ACGTAAGTCATCGTAGC Mutations at some
ATGGAAATTATCGCGGT
sites are lethal, so
ACATAAATCATCGTAGA
they are invariant
ACGCAAGTCATCGAAGT
2
1
4
Assuming equal
substitution rates
across sites
3
2
4
Allowing some sites to be
invariant – reveals more parallel
evolution among the variant sites
Rates can also differ among the variable sites due to fitness
effects, differential mutability and codon bias - again leading
homogenous models to underestimate parallel change
Such rate variation can
often be accommodated
by assuming a gamma
distribution of rates
across sites in the
likelihood (or distance)
model
Non-homogenous data partitions
Rifleman
Broadbill
Flycatcher
Lyrebird
Indigobird
ZebraFinch
Rook
Codon pos.
Partition 1
GTAACACTAGCC
GTCACACTAGCC
GTTACATTAGCC
GTTACTTTAGCA
GTAACCCTAGCC
GTAACCTTAGCA
GTAACTCTAGCA
123123123123
Partition 2
Kolaczkowski and Thornton
(Nature, 2004)
Rifleman
Red for
variable sites,
most change at
3rd positions
Reconstructed under a
single likelihood model
Competing hypotheses for the interrelations of
the mammalian sub-classes
reptiles
monotremes
marsupials
Marsupionta
placentals
Theria
Janke et al. (PNAS, 1997)
ML analysis of complete
mitochondrial genome
protein-coding sequences
Marsupionta
ppn. constant sites
1.0
0.8
0.6
0.4
0.2
0
0.1
0.2
0.3
0.4
0.5
Purine base frequency
Model
df
TN93+I+ (concatenated) 40
TN93+I+ (partitioned)
480
Grouping of protein - coding
and RNA - coding genes based
on observed constant site
proportions and Purine base
frequency. RNAloops ( );
RNAstems ( ); COI ( );
NADH6; ( ); ATPase8,
NADH2, NADH4L ( );
ATPase6, NADH1, NADH3,
NADH4, NADH5( ); COII,
0.6
COIII, Cytb ( ).
AIC
162260.5
158054.3
Theria
Reptiles
Monotremes
Placentals
KH-test p-value - Phillips et al. (MPE, 2003)
Marsupials
Partitioned ML:
Theria is favoured
Compositional heterogeneity
Stationarity: A standard assumption of most phylogeny
reconstruction methods is that underlying substitution
processes are the same across the tree
When violated, biases arise that provide signals in the data
that can overwhelm the “true” phylogenetic signal
Shifting substitution processes (e.g. AG being favoured
in some branches but G A in others) can result in signals
for relationships arising due to similar DNA or protein
sequence composition, rather than shared ancestry.
Extreme example:
NJ tree - mt 3rd
codon positions,
transitions only
Ostrich
Rook
Brushtail Possum
Fin Whale
Vidua
Wallaroo
Rhea
Armadillo
53
Hippopotamus
61
52
Green Turtle
68
Painted Turtle
Bandicoot
Opossum
Branch thickness
proportional to T:C ratio
Mole
Platypus
Aardvark Elephant
Composition 2 test (stochastic test)
Taxon
A
C
G
T
----------------------------------------------Rifleman
165
154
82
95
Broadbill
203
142
48
103
Flycatcher
195
115
60
126
Lyrebird
138
142
127
89
Indigobird
137
144
128
87
Zebra Finch
141
143
124
88
Rook
145
144
118
89
Expected
160.57
140.57
98.14
96.71
Chi-square = (Exp-Obs)2
Exp*
= 119.211273 df= (n-1)(t-1)= 18
P < 0.0001
Tells only of the presence of a bias and is unreliable when most
of the variation occurs among a small number of character states
Relative compositional variability (magnitude metric)
Allows the magnitude of compositional heterogeneity to
be compared between sequences or coding regimes (for
the same taxa)
n
RCV =
(| Ai - A* | + | Ti - T* | + | Ci- C* | + | Gi - G* |) / n.t
i 1
Where Ai is the observed frequency of adenine for taxon
i, A* is the average frequency of adenine across all taxa,
n is the number of taxa and t is the number of sites
Accounting for compositional heterogeneity
1. LogDet distances - recover additive distances
between sequences when base composition varies
Euglena(y)
A C G T
For each pair of DNA sequences x and y, a 4 4
matrix with each possible pair of sites
Olithodiscus(x)
A
C
G
T
224 5
24 8
Fxy=
3
149 1
16
24 5
230 4
5
19 8
175
0.249
0.003
0.027
0.006
0.006
0.166
0.006
0.021
0.027
0.001
0.256
0.009
Dxy = -ln[det Fxy] = 6.216
0.009
0.018
0.004
0.194
a. Jukes-Cantor distances
Anacystis Chlamydomonas
Lockhart et al.
(MBE, 1994)
Euglena
b. LogDet distances
Olithodiscus
Tobacco
Chlorella Liverwort
Rice
Euglena Chlamydomonas
Anacystis
Rice
Olithodiscus
Chlorella Liverwort
Tobacco
Chlorophyll a/b
Phycobilin
Chlorophyll a/c
uncertain
Rates-across-sites LogDet has yet to be developed, so this
method is often inconsistent due to poor branch-length estimation
2. Non-homogenous base composition Maximum likelihood
Galtier and Gouy
(MBE, 1998)
ω
λ1.Φ
θ1
λ2
θ2
λ3
θ3
λ1.1Φ
θ1
λ5
θ5
λ4
θ4
λ6
θ6
λ7
θ7
Parameters
symbol
root G+C%
ω
branch-length
λ
root location
Φ
Ts/Tv ratio
κ
equilibrium G+C% θ
number
1
2n-3
1
1
2n-2
Limitations
1. restricted to GC vs. AT bias
2. computer time intensive
3. Character state re-coding
• Often much of the compositional heterogeneity arises
within specific classes of character state
e.g. Purine and Pyrimidine transitions
These can be re-coded: RY-coding involves A,G R and
C,T Y
• Similarly, lumping amino acids into functionally similar
groups e.g. Valine, leucine and Isoleucine as single
category of mid-sized aliphatic amino acids.
Nardi et al. (Science, 2003) found Hexapoda to be paraphyletic
Delsuc et al. (Science,
2003)
1st and 3rd codon positions
RY-coded
Hexapoda
RCVnt = 0.1064
RCVry = 0.0413
Mistaking precision for accuracy
106 nuclear genes: Different methods provide conflicting
Yeast topologies, each with 100% bootstrap support
Phillips et al. (MBE, 2004)
The results underline the importance of understanding how nonphylogenetic signals will bias inference under the model used
Not enough phylogentic signal to resolve the tree
Signal erosion with time
Ans. Use high-value (often
slower evolving) characters
Long unbroken branches
make for “noisier” data
Ans. Increase taxon sampling
Branch-length too short
Ans.
Increase gene sequencing
Stemminess (Fiala and Sokal: Evol., 1985) on uncorrected
distance trees indicates the relative extent of phylogenetic
signal erosion among alternative sequemces (or coding
regimes) for the same taxa
Stemminess = Σ external branch-lengths
total tree-length
Greater phylogenetic
signal retention for slower
evolving genes results in
higher stemminess
12 mitochondrial
protein-coding genes
5 nuclear protein-coding
genes
Stemminess =0.086
Stemminess =0.440
Monodelphis
Monodelphis
Wallaroo
Opossum
Opossum
Brushtail
Wallaroo
Spiny Bandicoot
Wombat
Brushtail
Northern Brown
Bandicoot
Spiny Bandicoot
Northern Brown
Bandicoot
Wombat
Dunnart
Tigercat
Tigercat
Dunnart
Saturation – the problem of multiple changes
at the same sites
• Theory, simulations, and practical
experience all indicate that the sequences
must eventually lose information about
events that were long ago.
• Part of the problem with using DNA
sequence alignments to infer deep events is
that the state space is small {A,C,G,T}
Other sorts of characters
• In an idealised situation where each site had
an infinite state space there would be no
parallel changes or reversals and our
character matrices would be homoplasy
free.
• Obviously it is interesting to try and find
characters that are closer to this ideal than
DNA sequences.
SINEs and LINEs
• SINEs (and LINEs) are Short (or Long)
interspersed nuclear elements.
• Retrotransposed DNA elements that are copied
into the genome.
• Low expectations for the same retrotransposon
sequence to insert in exactly the same position
independently (low homoplasy markers)
Insertion event 1 into
chromosome A
The SINE/LINE is copied
from loci 1 on chromosome A
to loci 2 on chromosome B
Taxon3 (present at
loci 1 and 2)
Taxon2 (present at
loci 1 and 2)
Taxon4 (only
present at loci 1)
Taxon1 (not present
at loci 1 or loci 2)
Loci 2 sequence
Taxon1 ATGCT-------//-------GTCTAGT
Taxon2 AGGCTGTTATGT//TCTCTAGGTCAAGT
Taxon3 ATGCTGCTATGT//TCTCTAGGTCTATT
Taxon4 ATACT-------//-------GTATAGT
Competing hypothesis for the position of the whales
SINEs and LINEs provide
homoplasy free support for
the position of the whales as
sister group to the hippos.
Genome-order based phylogeny
Large state-space
• DNA sequences : 4 states per site
• Signed circular genomes with n genes:
2n-1(n1)! states, 1 site
• Circular genomes (1 site)
– with 37 genes:
2.56×1052
states
– with 120 genes:
3.70×10232
states
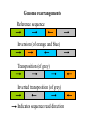
Genome rearrangements
Reference sequence
Inversion (of orange and blue)
Transposition (of grey)
Inverted transposition (of grey)
Indicates sequence read direction
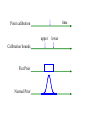
Breakpoint Distance
• Breakpoint distance=5
1
2
3
4
5
6
7
8
9
10
1 –3 –2
4
5
9
6
7
8
10
Minimum Inversion Distance
• Inversion distance=3
1
2
3
4
8
9
10
1
2
3 –8 –7 –6 –5 –4
9
10
1
8 –3 –2 –7 –6 –5 –4
9
10
1
8 –3
9
10
7
5
6
7
2 –6 –5 –4
Distance-based methods
Tandy Warnow, UT-Austin
Maximum Parsimony on Rearranged
Genomes (MPRG)
• The leaves are rearranged genomes.
• Find the tree that minimizes the total number of
rearrangement events
A
A
B
3
6
E
C
2
B
D
Tandy Warnow, UT-Austin
C
3
4
Total length
= 18
F
D
Mitochondrial genome rearrangement
maximum parsimony
Fritzsch et al. (J.Theor. Biol., 2006)
Data choice and analytical
methods are in their infancy
Note non-monophyly of
Nematoda and Mollusca;
Well resolved sequence and
morphology clades
?
An additional possibility is that there are multiple signals:
1. Biases in the data (e.g. compositional heterogeneity),
2. genes have different histories (e.g. lineage sorting or
hybridization)
If a gene has a long coalescent
time, then its relationships
among taxa may differ from the
species tree
Gene tree
Species tree
A
B
C
D
Molecular dating
Genetic change
Genetic divergence
The molecular clock e.g. Zukerkandl and Pauling (J. Theor Biol., 1965)
Time since divergence
Human – Chimpanzee
Human – Mouse
Human – Bird
corrected for
saturation
observed
Time since divergence
Is the data clock-like?
Can the deviation from an ultrametric tree be
explained by the stochastic nature of substitution
(sampling error), or do substitution rates differ
across the tree?
Relative rates tests
HO: Two sister taxa are evolving at the same rate (by
comparison with an outgroup)
Hebsgaard et al. (TIM, 2005)
Molecular clock likelihood ratio test
HO: That a clock model explains the data as well as a
non-clock model
1. Optimize the likelihood of the (unrooted) tree under a
non-clock model (lnLn)
2. Optimise the likelihood of the (rooted) tree under a
clock model (lnLc)
3. Calculate the test statistic = 2(lnLc minus lnLn)
4. This is compared to a 2 distribution critical value
(where the degrees of freedom are the difference in the
number of free parameters being estimated between
the two models = n2)
Linearized trees: Takezaki et al. (MBE, 1995)
Prune the taxa that are the most non-clock-like until the
molecular clock likelihood ratio test is passed
Concerns: 1. removing any branches reduces the power of the
test (so increases the probability of passing) and 2. remaining
branches may hide complementary rate shifts that cancel out
Relaxing the molecular clock
1. Local clocks
2. Autocorrelated rate evolution
r3
r6
r1
r5
r3
r2
Relies on the
identification of
rate classes with
respect to clades
r9
r4
r1
r7
r10
r8
r2
Each rate ri is a function of the rate of its
parent branch. Many different models of
rate change have been applied including:
quadratic, lognormal, exponential,
gamma, Ornstein-Uhlenbeck
3. Uncorrelated rate evolution
Method of Drummond et al. (PLoS Biol., 2006)
r6
r5
r3
r9
r4
r1
r7
r10
r8
r2
Rates ri do not depend on the rate of their parent branch,
but are drawn from a lognormal or exponential distribution
that maximises the posterior probability of the tree
Performance of correlated rates methods on trees simulated
under uncorrelated rates among branches
Ducks
Albatross
Penguins
Calibrating molecular clocks
61 Ma
calibration
90 Ma
Slack et al., (MBE, 2006) estimate
Biogeographical divergences
e.g. New Zealand split from Gondwana
about 80 million years ago and so did
some of New Zealand’s endemic fauna
Fossils that post-date
divergences
time
Point calibration
upper
Calibration bounds
Flat Prior
Normal Prior
lower
Using a lognormal (19Ma-25Ma upper 95%,
mean=21Ma) calibration for cats/hyaenas
25
20
15
Barnett et al. (Curr. Biol., 2005)
10
5
Millions of
0 years ago