Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
History of genetic engineering wikipedia , lookup
Group selection wikipedia , lookup
Genetic drift wikipedia , lookup
Protein moonlighting wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Saethre–Chotzen syndrome wikipedia , lookup
Gene desert wikipedia , lookup
Gene therapy wikipedia , lookup
Genome (book) wikipedia , lookup
Fetal origins hypothesis wikipedia , lookup
Gene expression profiling wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Genome evolution wikipedia , lookup
Gene nomenclature wikipedia , lookup
Genome editing wikipedia , lookup
Population genetics wikipedia , lookup
The Selfish Gene wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Frameshift mutation wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Helitron (biology) wikipedia , lookup
Point mutation wikipedia , lookup
Gene expression programming wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Public health genomics wikipedia , lookup
Designer baby wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
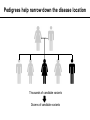
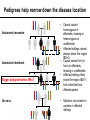
CSE291: Personal genomics for bioinformaticians Class meetings: TR 3:30-4:50 MCGIL 2315 Office hours: M 3:00-5:00, W 4:00-5:00 CSE 4216 Contact: [email protected] Today’s schedule: • 3:30-4:10 Prioritization and Filtering • 4:10-4:15 Break • 4:15-4:50 Time to work on PS5 (+command line tips and info for final presentations) Announcements: • • PS5 due Thursday Reading posted for Thursday Prioritizing and Filtering variants CSE291: Personal Genomics for Bioinformaticians 03/07/17 Outline • The challenge: needles in haystacks • Annotation annotation annotation • Family information • Prior knowledge of gene function • Selection • Remaining challenges The challenge: needles in haystacks The challenge: sifting through piles of variants … From the exome alone, THOUSANDS of candidate varian The challenge: sifting through piles of variants 1. Have I seen this variant before? 6. Is the gene likely relevant to this disease? 2. Is this variant in affected family members? 3. Does this mutation affect gene function? GAAAATATCATGTGGTGTTTCC GAAAATATCATATGGTGTTTCC 5. Gene expression pattern of this gene makes sense? 4. Is this position conserved across species? Common approach: progressively apply filters from highest to least confidence Caveat: some truly pathogenic variants will fail these filters! Annotation annotation annotation Annotating impact of mutations on genes Loss of function variants LOFTEE (https://github.com/konradjk/loftee), VEP plug-in to annotate LoF Assesses suspected LoF variants: • Stop gain (nonsense) • Splice site disrupting • Frameshift variants KEY: filters to identify true vs. false positive LoF annotations, e.g.: • Nonsense variants in last 5% of the gene unlikely to be that damaging (why?) • Nonsense variants in an exon without canonical splice sites around it likely false positive (why?) • Splice sites in very small introns (e.g. <15bp) likely not that critical • If the LoF allele matches the ancestral allele, likely not really LoF (why?) Are missense variants important? Polyphen2: predict impact of an amino acid substitution on gene structure and function • 8 sequence based features, 3 structure based features • Classify variants as: probably damaging, possibly damaging, benign • See also: SIFT, MutationTaster, SNAP, and more. Most people use multiple methods and e.g. require more than one Adzhubei et al. Nature Methods 2010 Ensemble annotations Ensemble: combination of different methods Idea: there’s lots of annotations out there. Some combination of which are probably important. Let’s combine them into a single classifier CADD: Combined Annotation-Dependent Depletion Kircher, et al. Nature Genetics 2014 (Shendure Lab) Features (63 annotations total): • VEP annotations (e.g. nonsense, missense) • SIFT, PolyPhen2 • Mappability • Conservation (PhastCons, PhyloP, GERP) • Segmental duplications • Expression • Histone modifications SVM Classifier Train on simulated data to determine: • Observed (likely benign) vs. • Simulated de novos (likely pathogenic) Family information Pedigrees help narrow down the disease location Thousands of candidate variants Dozens of candidate variants Pedigrees help narrow down the disease location Autosomal recessive Autosomal dominant Bigger pedigree=better. Why? De novo • Causal variant homozygous in affecteds, missing or heterozygous in unaffecteds • Affected siblings almost always share the region IBD=2 • Causal variant het (or hom) in affecteds, missing in unaffecteds • Affected siblings likely share the region IBD=1, both inherited from affected parent • Mutation not present in parents or affected siblings Prior knowledge of gene function Databases of clinical consequences of variants Has my candidate gene been previously implicated in a human disease? If yes, is it related to the current disease I’m trying to solve? Gene ontologies Does the annotated function of my gene make sense? e.g. for Marfan Syndrome, FBN1 • Biological process: skeletal/heart/kidney development • Cellular component: basement membrane, extracellular, microfibril • Molecular function: Calcium ion binding, structural, protein binding http://waclawikgen677s10.weebly.com/gene- Incorporating gene expression data - tissue Is this gene expressed in tissues that make sense for this disease? e.g. if disease is primarily liver related, the causal gene is probably expressed in liver We now have resources (e.g. GTeX) reporting expression across tissues CFTR Expression by tissue http://www.gtexportal.org/home/gene/CFTR Incorporating gene expression data from patients • Identified novel exon in COL6A1 formed by dominantly acting splice gain event, causes external collagen-VI-like dystrophy • Overall, diagnosis rate of 35% in patients with undiagnosed neuromuscular diseases • Can incorporate RNA-seq from family members to leverage traditional pedigree approaches Cummings et al. 2016 Selection Types of selection Positive selection: a new mutation confers a selective advantage, and rises to frequency quickly. OR a new environmental factor makes an existing mutation suddenly more advantageous. • Examples: LCT (lactase persistence), EDAR1 • Tests: Long haplotypes, high derived allele frequency Purifying selection: mutations in critical regions of the genome are often deleterious and quickly eliminated • Examples: protein coding sequence vs. introns, ultraconserved regions • Tests: all of these compare observed vs. expected variation • Tajima’s D, Fu and Li Test, many others • Genetic constraint (Tuesday) Selection says disease-causing mutations are rare Effect size severe mild rare e.g. Tay-Sachs Nonexistent Severe Mendelian disorders (removed by selection) (well actually… AD APO e4. why?) Likely many examples, but low power to detect these e.g. high cholesterol, Crohn’s Disease, Type II Diabetes (many common alleles with small effect sizes) common Allele Frequency Metrics of purifying selection Site frequency spectrum: the distribution of allele frequencies of a given set of SNPs in a population or sample Summarizing the SFS: • % Singletons (seen 1 time in the population). Higher=rarer=stronger selection • Mean MAF (higher=more common=weaker selection) • % variable sites (how many positions were never variable. Higher=weaker selection) Purifying selection for variant classes MAPS: mutability adjusted % singletons Generally, the more variants that are singletons, the more deleterious that mutation class is Caveat: these metrics describe categories of variants, and may or may not be useful for predicting impact of a specific mutation Lek et al. 2016 Conservation across species informative • Phastcons: compute conservation based on phylogenetic tree + HMM • phyloP: compute conservation using sequence alignment and model of neutral evolution • GERP: identify constrained elements in multiple sequence alignment by quantifying “substitution deficits” Davydov et al. 2010 Remaining challenges Some examples defy our annotation pipelines • Cystic fibrosis (deltaF508 in CFTR) I I F G V rs113993960 GAAAATATCATCTTTGGTGTTTCC GAAAATATCAT---TGGTGTTTCC I I In-frame deletion! (Usually would prioritize frameshifts) F G V • Synonymous variants can be pathogenic! (e.g. in MDR1, multidrug resistance. UBE1 spinal muscular • atrophy Deep intronic mutation The non-coding genome… • With exome sequencing, we analyze 2% of the genome but still have too many variants. • Helped by the fact that we have a decent idea of how to analyze coding variants. • How will we deal with the overwhelming number of false positives from WGS? • Requires… annotation and prioritization! Final projects + PS5