Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Genetic engineering wikipedia , lookup
Genetic drift wikipedia , lookup
Oncogenomics wikipedia , lookup
Minimal genome wikipedia , lookup
Population genetics wikipedia , lookup
Transposable element wikipedia , lookup
Gene desert wikipedia , lookup
Dominance (genetics) wikipedia , lookup
History of genetic engineering wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Medical genetics wikipedia , lookup
Genome (book) wikipedia , lookup
Designer baby wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Behavioural genetics wikipedia , lookup
Polymorphism (biology) wikipedia , lookup
Pathogenomics wikipedia , lookup
Metagenomics wikipedia , lookup
Genomic library wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Microevolution wikipedia , lookup
Copy-number variation wikipedia , lookup
Non-coding DNA wikipedia , lookup
Microsatellite wikipedia , lookup
Genealogical DNA test wikipedia , lookup
Genome editing wikipedia , lookup
Genome evolution wikipedia , lookup
Human genome wikipedia , lookup
Public health genomics wikipedia , lookup
Human Genome Project wikipedia , lookup
Molecular Inversion Probe wikipedia , lookup
Human genetic variation wikipedia , lookup
Haplogroup G-M201 wikipedia , lookup
A30-Cw5-B18-DR3-DQ2 (HLA Haplotype) wikipedia , lookup
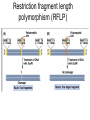
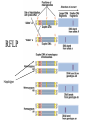
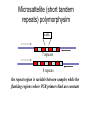
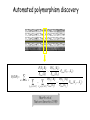
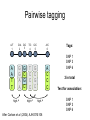
Polymorphism Haixu Tang School of Informatics Genome variations underlie phenotypic differences cause inherited diseases Restriction fragment length polymorphism (RFLP) RFLP Haplotype Microsattelite (short tandem repeats) polymorphysim AATG 7 repeats 8 repeats the repeat region is variable between samples while the flanking regions where PCR primers bind are constant Which Suspect, A or B, cannot be excluded from potential perpetrators of this assault? Single nucleotide polymorphism • The highest possible dense polymorphism • A SNP is defined as a single base change in a DNA sequence that occurs in a significant proportion (more than 1 percent) of a large population. Some Facts • In human beings, 99.9 percent bases are same. • Remaining 0.1 percent makes a person unique. – Different attributes / characteristics / traits • how a person looks, • diseases he or she develops. • These variations can be: – Harmless (change in phenotype) – Harmful (diabetes, cancer, heart disease, Huntington's disease, and hemophilia ) – Latent (variations found in coding and regulatory regions, are not harmful on their own, and the change in each gene only becomes apparent under certain conditions e.g. susceptibility to lung cancer) SNP facts • SNPs are found in – coding and (mostly) noncoding regions. • Occur with a very high frequency – about 1 in 1000 bases to 1 in 100 to 300 bases. • The abundance of SNPs and the ease with which they can be measured make these genetic variations significant. • SNPs close to particular gene can acts as a marker for that gene. SNP maps • Sequence genomes of a large number of people • Compare the base sequences to discover SNPs. • Generate a single map of the human genome containing all possible SNPs => SNP maps How do we find sequence variations? • look at multiple sequences from the same genome region • use base quality values to decide if mismatches are true polymorphisms or sequencing errors Automated polymorphism discovery P( SNP) all var iable P ( S N | RN ) P( S1 | R1 ) ... PPrior ( S1 ,..., S N ) PPrior ( S1 ) PPrior ( S N ) P( SiN | R1 ) P( Si1 | R1 ) S ... ... PPrior ( Si1 ,..., SiN ) P ( S ) P ( S ) S i1 [ A,C ,G ,T ] S iN [ A,C ,G ,T ] Prior i1 Prior iN Marth et al. Nature Genetics 1999 Large SNP mining projects genome reference EST WGS BAC ~ 8 million Sachidanandam et al. Nature 2001 How to use markers to find disease? genome-wide, dense SNP marker map • genotyping: using millions of markers simultaneously for an association study • question: how to select from all available markers a subset that captures most mapping information (marker selection) • depends on the patterns of allelic association in the human genome Allelic association • allelic association is the nonrandom assortment between alleles i.e. it measures how well knowledge of the allele state at one site permits prediction at another marker site functional site • significant allelic association between a marker and a functional site permits localization (mapping) even without having the functional site in our collection • by necessity, the strength of allelic association is measured between markers Linkage disequilibrium • LD measures the deviation from random assortment of the alleles at a pair of polymorphic sites D=f( ) – f( ) x f( ) • other measures of LD are derived from D, by e.g. normalizing according to allele frequencies (r2) Haplotype diversity • the most useful multi-marker measures of associations are related to haplotype diversity n markers n 2 possible haplotypes random assortment of alleles at different sites strong association: most chromosomes carry one of a few common haplotypes – reduced haplotype diversity Haplotype blocks Daly et al. Nature Genetics 2001 • experimental evidence for reduced haplotype diversity (mainly in European samples) The promise for medical genetics CACTACCGA CACGACTAT TTGGCGTAT • within blocks a small number of SNPs are sufficient to distinguish the few common haplotypes significant marker reduction is possible • if the block structure is a general feature of human variation structure, whole-genome association studies will be possible at a reduced genotyping cost • this motivated the HapMap project Gibbs et al. Nature 2003 The HapMap initiative • goal: to map out human allele and association structure of at the kilobase scale • deliverables: a set of physical and informational reagents Haplotyping • the problem: the substrate for genotyping is diploid, genomic DNA; phasing of alleles at multiple loci is in general not possible with certainty A C G C T T C A • experimental methods of haplotype determination (single-chromosome isolation followed by whole-genome PCR amplification, radiation hybrids, somatic cell hybrids) are expensive and laborious A example of hyplotyping • Mother GG AT • Father CC AA CA AC TT CT • Children GC AA CC CT • Children GC AT AA TT • Children GC AA AC CT Haplotypes • a • Mother I G A C T • II G T C T b G T A T G A A T • Father I C A A C • II C A A T C A C T C A C C A example of hyplotyping • Mother GG AT • Father CC AA CA AC TT CT • Children GC AA CC CT (M-Ia & F-IIb) • Children GC AT AA TT (M-Ib & F-IIa) • Children GC AA AC CT (M-Ia & F-Ia or M-IIb & F-IIb) ? HapMap Project A freely-available public resource to increase the power and efficiency of genetic association studies to medical traits High-density SNP genotyping across the genome provides information about – SNP validation, frequency, assay conditions – correlation structure of alleles in the genome All data is freely available on the web for application in study design and analyses as researchers see fit HapMap Samples • 90 Yoruba individuals (30 parent-parent-offspring trios) from Ibadan, Nigeria (YRI) • 90 individuals (30 trios) of European descent from Utah (CEU) • 45 Han Chinese individuals from Beijing (CHB) • 45 Japanese individuals from Tokyo (JPT) HapMap progress PHASE I – completed, described in Nature paper * 1,000,000 SNPs successfully typed in all 270 HapMap samples PHASE II – data generation complete, data released * >3,500,000 SNPs typed in total !!! ENCODE-HAPMAP variation project • Ten “typical” 500kb regions • 48 samples sequenced • All discovered SNPs (and any others in dbSNP) typed in all 270 HapMap samples • Current data set – 1 SNP every 279 bp A much more complete variation resource by which the genome-wide map can evaluated Tagging from HapMap • Since HapMap describes the majority of common variation in the genome, choosing non-redundant sets of SNPs from HapMap offers considerable efficiency without power loss in association studies Pairwise tagging A/T 1 A A T T G/A 2 G G A A high r2 G/C 3 G C G C T/C 4 T C C C high r2 G/C 5 A/C 6 A C C C G C G C high r2 After Carlson et al. (2004) AJHG 74:106 Tags: SNP 1 SNP 3 SNP 6 3 in total Test for association: SNP 1 SNP 3 SNP 6