Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Point mutation wikipedia , lookup
Short interspersed nuclear elements (SINEs) wikipedia , lookup
Gene desert wikipedia , lookup
Essential gene wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Gene expression programming wikipedia , lookup
Human genetic variation wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
Population genetics wikipedia , lookup
Mitochondrial DNA wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Transposable element wikipedia , lookup
Ridge (biology) wikipedia , lookup
Genetic engineering wikipedia , lookup
Gene expression profiling wikipedia , lookup
Genomic imprinting wikipedia , lookup
Oncogenomics wikipedia , lookup
Designer baby wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Non-coding DNA wikipedia , lookup
Genome (book) wikipedia , lookup
Metagenomics wikipedia , lookup
Human genome wikipedia , lookup
Public health genomics wikipedia , lookup
History of genetic engineering wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Human Genome Project wikipedia , lookup
Genome editing wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Helitron (biology) wikipedia , lookup
Genomic library wikipedia , lookup
Microevolution wikipedia , lookup
Minimal genome wikipedia , lookup
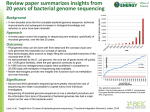
Bioinformatic Insights into the Evolution of Bacterial Genomes H o w a rd O c h m a n Department of Integrative Biology University of Texas Bacterial Genomics (before 1995) • Bacterial chromosomes are (typically) circular, with a single replication origin • Bacterial genomes are tightly packed with coding genes & functional elements, and have little repetitive or non-coding DNA • Bacterial genomes are typically small, ranging in size from 0.5-10 Mb • Bacterial coding genes have no introns, are arranged in operons, and are assorted onto both strands • Gene order is conserved among closely related bacteria • Base composition varies among species (25-75% GC) and is similar among closely related taxa • Base composition is relatively homogeneous over the entire chromosome • Rates & patterns of mutations vary with gene location & transcriptional status The field of Bacterial Genomics is often considered to have begun in Haemophilus influenzae Mycoplasma genitalium What was learned from the first full genome sequences? 1. Possible to assemble full genomes by WGS with paired-end reads 2. Enabled resolution complete gene inventories (& encoded functions) Mycoplasma pneumoniae (816 kb; 736 ORFs) Mycoplasma genitalium (580 kb; 564 ORFs) 1. Start to observe a consistent trend in genome size & gene number 2. Members of the same genus can differ in genome size & contents What is the source of such differences in genome contents? Loss of ancestral genes? Generation of new genes ? Difficult to resolve without knowledge of ancestral (or outgroup) genome genes acquired by lateral gene transfer (LGT) --Geneswithatypicalfeatures(e.g.,G+Ccontent)areconsideredtoarisebyLGT-Strain- & species-specific genes often have atypical base compositions Sequenced genes present in Salmonell a , but not in E. coli (circa 1990) Differences in base composition among species are caused by mutational biases Gene Potential Function Ma p %G+C cbi Cobinamide synthesis 41 59.3 .233 fljA Flagell ar synthesis 56 40.9 .210 fljB Flagell ar synthesis 56 52.3 .216 inv/spa Host recognition/invasion 59 45.5 .261 nanH Sialidase 20.5 40.9 .263 ORF Unknown 98 38.2 .296 pagC Envelope protein 25 43.4 .274 phoN Phosphatase 96 46.5 .248 rfc LPS Synthesis 31 33.5 .175 sinR Tra nscriptional control 7 39.8 .218 tctABCD Tricarboxylate transport 57 55.0 .278 pgtE Phosphoglycerate transport – 45.3 .277 Salmonell a genome is 52% G+C CAI Inferring the Incidence of Lateral Gene Transfer (LGT) from Sequence Heterogeneity along Bacterial Chromosomes Neisseria meningitidis, 52% G+C % G+C (from Tettelin et al. 2000. Science) ⌇ (Mostly) Free-living bacteria ⌇ (Mostly) Pathogens A relationship between between bacterial lifestyle and genome size but with some exceptions (M. tuberculosis, 4.4 mb ; Aquifex, 1.6 Mb) ⌇ Intracellular pathogens & obligate endosymbionts Remarkable relationship between genome size & gene number across >10-fold range in genome size in organisms representing eight phyla Mycobacteriumleprae Then why haven’t pseudogenes accumulated in all of the other sequenced bacterial genomes? Since mutations occur as an on-going process & pseudogenes are continually being generated, what about all those other (big free-living & small symbiont) genomes that fall right on the diagonal? Yersinia pestis Shigella flexneri Mycobacterium leprae The genomes of other recent pathogens of humans possess high numbers of inactivated genes (e.g., Shigella has 400; Yersinia pestis has 150) In bacteria, non-functional regions are removed by a pervasive mutational bias towards deletions This deletional bias was detected computationally and confirmed experimentally Unlike most eukaryotes (in which noncoding DNA accumulates), bacteria show a strong mutational bias towards deletions of all sizes When comparing pseudogenes to functional their counterparts, deletions outnumber insertions Lineages of bacteria with small genomes derive from ancestors with larger genomes < 2 Mb 2–4 Mb > 4 Mb The progression towards a compact genome 1.(Ancestral)free-living(large-genomed)bacteriamovesintoa(nutrient-rich)host 2.Hostassociationrendersmanygenessuperfluous/useless&deadgenesaccumulate 3.Deletionalbiasreducesgenomesizeandremovesinactivatedgenes Sampling multiple strains within species reveals wide variation in genome size (concept of a “core” vs. “pan” genome) 1. Very large sampling of several phyla at various taxonomic levels 2. Minimal size of bacterial genome is ~500 kb (pathogens & symbionts) A new lower limit to the size of a cellular genome The symbiont of a psyllid collected on a hackberry tree in Tucson, Arizona Carsonella ruddii: the smallest bacterial genome (Nakabachi et al., 2005) 159,662 bp 182 ORFs 16.5% GC 98% coding Other smallest cellular genome is 420 kb, in the aphid symbiont Buchnera …with the current record of 112 kb held by Nasuia deltocephalinicola A thousand bacterial genomes sequenced What has the sequencing of 1000s of genomes told us about factors controlling the size & composition of bacterial genomes? The (New) Observations: • Bacterial genomes are typically small, ranging in size from 0.1–13 Mb • Bacterial genomes are USUALLY tightly packed with coding genes, and USUALLY have little repetitive & non-coding sequences (e.g., pseudogenes). Therefore, in bacteria, genome size is tightly linked to gene number • There is a pervasive mutational bias that removes non-functional regions • Small bacterial genomes derive from lineages with large genomes • Base composition varies among species (13-80% GC) and is relatively homogeneous over the entire chromosome • Many bacterial genomes harbor substantial amounts of laterally acquired DNA • Strains within a named bacterial species can vary greatly in genome size But what are the major determinants of bacterial genome size & complexity? First test if Bacterial Genome Size is Adaptive (i.e., shaped by natural selection) or Non-Adaptive (i.e., shaped by genetic drift) The effects of genetic drift (changes in gene frequencies that take place strictly by chance) are more dramatic in species with small effective population sizes (Ne) & will shape the contents of genomes by affecting the fixation of deleterious mutations Due to the link between genome size and gene density in bacteria, evolutionary forces that act on individual genes will affect genome size -- Fate of Bacterial Genes -No Benefit genes whose presence is affected by drift (decay) Essential (preserved) Butassessingthelevelofdriftaffectingbacteriaisproblematic: 1.InBacteria,itisvery(very)difficulttoestimateNe 2.Methodsbasedonlevelsofintraspecificpolymorphismsare confoundedbyproblemsindefiningbacterialspecies Using Ka/Ks values as a proxy for level of drift 1. Because point mutations that cause amino acid replacements are often deleterious, the rate of nonsynonymous changes (Ka) is expected to be less than the rate of synonymous substitutions (Ks) in functional genes. 2. An increased level of drift, produced from reduced Ne (+ genome-wide relaxation of selection) will result in an increased incidence in slightly deleterious mutations and increase Ka/Ks genome-wide. Examined genome-wide Ka/Ks in species pairs from eight phyla Genome size exhibits a strong negative correlation with the level of genetic drift more efficient selection less efficient selection 1. Same significant relationship is observed when considering all genes shared by a genome-pair or only those genes common to the majority of genomes 2. Similarly significant relationship is observed when considering only pairs with low, or only those with intermediate, levels of sequence divergence 3. Those with high genome-wide Ka/Ks have a lifestyle that reduces Ne (obligate endosymbionts, vector-borne pathogens, extremophiles) Gene density is related to the level of genetic drift 1. Genome-pairs with low levels of drift (i.e., more efficient selection) display a relatively narrow range of coding densities (usually 85-90%) 2. Most genome pairs displaying high levels of drift have coding densities that lie outside of the 85-90% range This occurs due to pseudogene formation in recent pathogens (lowering coding density) and to tight gene-packing in the highly reduced genomes (increasing coding density) Genetic Drift as the Major Determinant of Bacterial Genome Size & Complexity But don’t bacteria have small genomes so that they can replicate quickly? 1a. Bacterial genome size is not caused by selection for replication efficiency Comparisons across species Genetic Drift as the Major Determinant of Bacterial Genome Size & Complexity 2. The effect of drift on bacterial genomes is opposite to the pattern proposed by Lynch (Lynch & Conery 2003) Eukaryotes Bacteria Smaller Ne –> More drift More drift The difference arises from the fact that bacterial genomes comprise sequences that are maintained by selection, or otherwise deleted 1. What is the basis of the variation in bacterial genome size? ✓ 2. What is the basis of the variation in genomic base composition? Proposed that differences in base composition among species are caused by mutational biases (again, a non-adaptive process) In bacteria, variation in genomic base composition has long been thought to be due to mutational biases but this has not slowed the search for an adaptive basis for the observed differences What might be a reason why some bacteria have G+C-rich genomes whereas others have A+T-rich genomes? PE! Thermal tolerance?? GC basepairs (with 3 H-bonds) are stronger than are AT basepairs, so high GC genomes would seem to be less prone to denaturing at higher temperatures O N -- What might be a reason why some bacteria have G+C-rich genomes whereas others have A+T-rich genomes? Wait, there are more..... (Feil&Rocha.2010.PLoSGenetics) Why are there GC-rich bacterial genomes? Experimental analysis of mutations in E. coli and Salmonella (both of which are >50% G+C) Given these mutational patterns, why are E. coli & Salmonella GC-rich? of its mRNA. The folding energy of the entire between folding energy and expression did not mRNA was not significantly correlated with flu- overlap with the Shine-Dalgarno (SD) sequence, Due to the link between genome size and gene density in bacteria, orescence (r = 0.16, P = 0.051), but the folding which suggested that SD occlusion by secondary forces that act on basestructure composition of individual genes energy of the first third of the mRNA wasthe strongly (22, 23) did not play a major role will in affect overall composition correlated: mRNAs with stronger structure pro-genomic inhibiting base expression, probably because our conduced lower fluorescence (r = 0.60, P < 10–15). structs contained no noncoding mutations. By A moving window analysis identified a region, contrast, the region of strongest effect overlapped Gene-level selection on base composition: A simple experiment GFP constructs with synonymous mutations in 3rd positions (Kudla G, et al. 2009. Science. 324: 255) Selected clones of low (40-42%), medium (46-48%) and high (51-54%) G+C content All selected genes had similar codon usage biases: CAIE.coli (0.58-0.68) Strains expressing GC-rich genes have higher growth rates!?! (timepoints are hours after induction with IPTG) The effect is observed with either of two anonymous genes Conclusions: Observations • Bacterial genomes (and those of archaea & probably many eukarya) display a mutational bias towards deletions among small indels • There is a strong negative association between bacterial genome size and the level of drift, such that species with small Ne have small genomes • Bacterial genomes are usually packed with functional genes, but almost every genome contains some pseudogenes • Bacteria with small genomes (pathogens, symbionts) derive from large-genomed ancestors; and during the transition to a host-associated lifestyle, functional redundancy and lower efficacy of selection causes accumulation of pseudogenes Conclusions: Findings • Variation in bacterial genome size, which has usually been attributed to selection for replication efficiency, is actually caused by non-adaptive processes • Variation in the base composition of bacterial genomes, long thought be determined by a strictly neutral mutational process, is now known to be caused by selection.