Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Neuronal ceroid lipofuscinosis wikipedia , lookup
Oncogenomics wikipedia , lookup
Saethre–Chotzen syndrome wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Genomic imprinting wikipedia , lookup
Epigenomics wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Minimal genome wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Copy-number variation wikipedia , lookup
Public health genomics wikipedia , lookup
Transposable element wikipedia , lookup
Primary transcript wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Genetic engineering wikipedia , lookup
Gene expression programming wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Gene therapy wikipedia , lookup
Genome (book) wikipedia , lookup
Gene nomenclature wikipedia , lookup
Genomic library wikipedia , lookup
Gene expression profiling wikipedia , lookup
Point mutation wikipedia , lookup
Gene desert wikipedia , lookup
Human genome wikipedia , lookup
Non-coding DNA wikipedia , lookup
Pathogenomics wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
History of genetic engineering wikipedia , lookup
Metagenomics wikipedia , lookup
Genome evolution wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Microevolution wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Designer baby wikipedia , lookup
Genome editing wikipedia , lookup
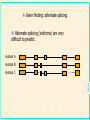
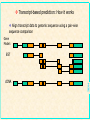
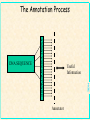
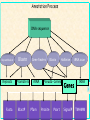
Gene Prediction and Annotation techniques Basics Chuong Huynh NIH/NLM/NCBI Sept 30, 2004 [email protected] NCBI Acknowledgement: Daniel Lawson, Neil Hall What is gene prediction? Detecting meaningful signals in uncharacterised DNA sequences. Knowledge of the interesting information in DNA. Sorting the ‘chaff from the wheat’ GATCGGTCGAGCGTAAGCTAGCTAG ATCGATGATCGATCGGCCATATATC ACTAGAGCTAGAATCGATAATCGAT CGATATAGCTATAGCTATAGCCTAT coding regions in genomic sequence’ NCBI Gene prediction is ‘recognising protein- Basic Gene Prediction Flow Chart Obtain new genomic DNA sequence 1. Translate in all six reading frames and compare to protein sequence databases 2. Perform database similarity search of expressed sequence tag Sites (EST) database of same organism, or cDNA sequences if available Use gene prediction program to locate genes NCBI Analyze regulatory sequences in the gene ACEDB View NCBI Why is gene prediction important? -Increased volume of genome data generated -Paradigm shift from gene by gene sequencing (small scale) to large-scale genome sequencing. -No more one gene at a time. A lot of data. -Foundation for all further investigation. Knowledge of the protein-coding regions underpins functional genomics. NCBI Note: this presentation is for the prediction of genes that encode protein only; Not promoter prediction, sequences regulate activity of protein encoding genes NCBI Map Viewer Genome Scan Models Genes Contig GenBank Mouse EST hits NCBI Human EST hits NCBI Artemis – Free Genome Visualization/ Annotation Workbench NCBI Genome WorkBench NCBI Knowing what to look for What is a gene? Not a full transcript with control regions The coding sequence (ATG -> STOP) Start Middle End NCBI N ORF Finding in Prokaryotes NCBI • Simplest method of finding DNA sequences that encode proteins by searching for open reading frames • An ORF is a DNA sequence that contains a contiguous set of codons that species an amino acid • Six possible reading frames • Good for prokaryotic system (no/little post translation modification) • Runs from Met (AUG) on mRNA stop codon TER (UAA, UAG, UGA) • http://www.ncbi.nlm.nih.gov/gorf/ NCBI ORF Finder ORF Finder (Open Reading Frame Finder) NCBI Annotation of eukaryotic genomes Genomic DNA transcription Unprocessed RNA RNA processing Mature mRNA AAAAAAA Gm3 translation Nascent polypeptide folding ab initio gene prediction (w/o prior knowledge) Comparative gene prediction (use other biological data) Active enzyme Function Reactant A Product B NCBI Functional identification Two Classes of Sequence Information NCBI • Signal Terms – short sequence motifs (such as splice sites, branch points,Polypyrimidine tracts, start codons, and stop codons) • Content Terms – pattern of codon usage that are unique to a species and allow coding sequences to be distinguished from surrounding noncoding sequences by a statistical detection algorithm Problem Using Codon Usage NCBI • Program must be taught what the codon usage patterns look like by presenting the program with a TRAINING SET of known coding sequences. • Different programs search for different patterns. • A NEW training set is needed for each species • Untranslated regions (UTR) at the ends of the genes cannot be detected, but most programs can identify polyadenylation sites • Non-protein coding RNA genes cannot be detected (attempt detection in a few specialized programs) • Non of these program can detect alternatively spliced transcripts Explanation of False Positive/Negative in Gene Prediction Programs NCBI Gene finding: Issues Issues regarding gene finding in general Genome size (larger genome ~ more genes, but …) Genome composition Genome complexity (more complexity -> less coding density; fewer genes per kb) cis-splicing (processing mRNA in Eukaryotics) alternate splicing (e.g. in different tissues; higher organism) Variation of genetic code from the universal code NCBI trans-splicing (in kinetisplastid) Gene finding: genome • Genome composition – Long ORFs tend to be coding – Presence of more putative ORFs in GC rich genomes (Stop codons = UAA, UAG & UGA) • Genome complexity NCBI – Simple repetitive sequences (e.g. dinucleotide) and dispersed repeats tend to be anti-coding – May need to mask sequence prior to gene prediction Gene finding: coding density As the coding/non-coding length ratio decreases, exon prediction becomes more complex Human Fugu worm NCBI E.coli Gene finding: splicing cis-splicing of genes Finding multiple (short) exons is harder than finding a single (long) exon. trans-splicing of genes A trans-splice acceptor is no different to a normal splice acceptor E.coli NCBI worm Gene finding: alternate splicing Alternate splicing (isoforms) are very difficult to predict. Human A Human B Human C NCBI ab initio prediction What is ab initio gene prediction? Prediction from first principles using the raw DNA sequence only. GATCGGTCGAGCGTAAGCTAGCTAG ATCGATGATCGATCGGCCATATATC ACTAGAGCTAGAATCGATAATCGAT CGATATAGCTATAGCTATAGCCTAT NCBI Requires ‘training sets’ of known gene structures to generate statistical tests for the likelihood of a prediction being real. Gene finding: ab initio • What features of an ORF can we use? NCBI – Size - large open reading frames – DNA composition - codon usage / 3rd position codon bias – Kozak sequence CCGCCAUGG – Ribosome binding sites – Termination signal (stops) – Splice junction boundaries (acceptor/donor) Gene finding: features Think of a CDS gene prediction as a linear series of sequence features: Initiation codon Coding sequence (exon) Splice donor (5’) Non-coding sequence (intron) Coding sequence (exon) Termination codon NCBI Splice acceptor (3’) N times A model ab initio predictor Locate and score all sequence features used in gene models dynamic programming to make the high scoring model from available features. e.g. Genefinder (Green) Running a 5’-> 3’ pass the sequence through a Markov model based on a typical gene model e.g. TBparse (Krogh), GENSCAN (Burge) or GLIMMER (Salzberg) e.g. GRAIL (Oak Ridge) NCBI Running a 5’->3’ pass the sequence through a neural net trained with confirmed gene models Ab initio Gene finding programs • Most gene finding software packages use a some variant of Hidden Markov Models (HMM). • Predict coding, intergenic, and intron sequences • Need to be trained on a specific organism. • Never perfect! NCBI What is an HMM? NCBI • A statistical model that represents a gene. • Similar to a “weight matrix” that can recognise gaps and treat them in a systematic way. • Has different “states” that represent introns, exons, and intergenic regions. Malaria Gene Prediction Tool • Hexamer – ftp://ftp.sanger.ac.uk/pub/pathogens/software/hexamer/ • Genefinder – email [email protected] • GlimmerM – http://www.tigr.org/softlab/glimmerm • Phat – http://www.stat.berkeley.edu/users/scawley/Phat • Already Trained for Malaria!!!! The more experimental derived genes used for training the gene prediction tool the more reliable the gene predictor. NCBI GlimmerM Salzberg et al. (1999) genomics 59 24-31 • Adaption of the prokaryotic genefinder Glimmer. Delcher et al. (1999) NAR 2 4363-4641 NCBI • Based on a interpolated HMM (IHMM). • Only used short chains of bases (markov chains) to generate probabilities. • Trained identically to Phat An end to ab initio prediction • • • • • • – Human annotation runs multiple algorithms and scores exon predicted by multiple predictors. – Used as a starting point for refinement/verification Prediction need correction and validation -- Why not just build gene models by comparative means? NCBI • • ab initio gene prediction is inaccurate Have high false positive rates, but also low false negative rates for most predictors Incorporating similarity info is meant to reduce false positive rate, but at the same also increase false negative rate. Biggest determinant of false positive/negative is gene size. Exon prediction sensitivity can be good Rarely used as a final product Annotation of eukaryotic genomes Genomic DNA ab initio gene prediction (w/o prior knowledge) transcription Unprocessed RNA RNA processing Mature mRNA AAAAAAA Gm3 Nascent polypeptide translation Comparative gene prediction folding (use other biological data) Active enzyme Function Reactant A Product B NCBI Functional identification If a cell was human? The cell ‘knows’ how to splice a gene together. We know some of these signals but not all and not all of the time So compare with known examples from the species and others Central dogma for molecular biology DNA Transcriptome RNA Proteome Protein NCBI Genome When a human looks at a cell Compare with the rest of the genome/transcriptome/proteome data DNA Extract DNA and sequence genome RNA Extract RNA, reverse transcribe and sequence cDNA Peptide sequence inferred from gene prediction NCBI Protein comparative gene prediction Use knowledge of known coding sequences to identify region of genomic DNA by similarity transcriptome - transcribed DNA sequence proteome - peptide sequence genome - related genomic sequence NCBI Transcript-based prediction: datasets Generation of large numbers of Expressed Sequence Tags (ESTs) Quick, cheap but random Subtractive hybridisation to find rare transcripts Use multiple libraries for different life-stages/conditions Single-pass sequence prone to errors Generation of small number of full length cDNA sequences Slow and laborious but focused Systematic, multiplexed cloning/sequencing of CDS Expensive and only viable if part of bigger project NCBI Large-scale sequencing of (presumed) full length cDNAs Gene Prediction in Eukaryotes – Simplified • For highly conserved proteins: – Translate DNA sequence in all 6 reading frames – BLASTX or FASTAX to compare the sequence to a protein sequence database – Or – Protein compared against nucleic acid database including genomic sequence that is translated in all six possible reading frame sby TBLASTN, TFASTAX/TFASTY programs. NCBI • Note: Approximation of the gene structure only. Transcript-based prediction: How it works Align transcript data to genomic sequence using a pair-wise sequence comparison Gene Model: EST NCBI cDNA Transcript-based gene prediction: algorithm BLAST (Altshul) (36 hours) Widely used and understood HSPs often have ‘ragged’ ends so extends to the end of the introns EST_GENOME (Mott) (3 days) Dynamic programming post-process of BLAST Slow and sometimes cryptic Next generation of alignment algorithm Design for looking at nearly identical sequences Faster and more accurate than BLAST NCBI BLAT (Kent) (1/2 hour) Peptide-based gene prediction: algorithm BLAST (Altshul) Widely used and understood Smith-Waterman Preliminary to further processing NCBI Used in preference to DNA-based similarities for evolutionary diverged species as peptide conservation is significantly higher than nucleotide Genomic-based gene prediction: algorithm BLAST (Altshul) Can be used in TBLASTX mode BLAT (Kent) Can be used in a translated DNA vs translated DNA mode Significantly faster than BLAST WABA (Kent) Designed to allow for 3rd position codon wobble Only really used in C.elegans v C.briggsae analysis NCBI Slow with some outstanding problems Comparative gene predictors This can be viewed as an extension of the ab initio prediction tools – where coding exons are defined by similarities and not codon bias GAZE (Howe) is an extension of Phil Green’s Genefinder in which transcript data is used to define coding exons. Other features are scored as in the original Genefinder implementation. This is being evaluated and used in the C.elegans project. NCBI GENEWISE (Birney) is a HMM based gene predictor which attempts to predict the closest CDS to a supplied peptide sequence. This is the workhorse predictor for the ENSEMBL project. Comparative gene predictors A new generation of comparative gene prediction tools is being developed to utilise the large amount of genomic sequence available. Twinscan (WashU) attempts to predict genes using related genomic sequences. Doublescan (Sanger) is a HMM based gene predictor which attempts to predict 2 orthologous CDS’s from genomic regions pre-defined as matching. NCBI Both of these predictors are in development and will be used for the C.elegans v C.briggsae match and the Mouse v Human match later this year. Summary Genes are complex structure which are difficult to predict with the required level of accuracy/confidence We can predict stops better than starts We can only give gross confidence levels to predictions (i.e. confirmed, partially confirmed or predicted) Gene prediction is only part of the annotation procedure Curation of gene models is an active process – the set of gene models for a genome is fluid and WILL change over time. NCBI Movement from ab initio to comparative methodology as sequence data becomes available/affordable The Annotation Process ANNALYSIS SOFTWARE DNA SEQUENCE Useful Information NCBI Annotator Annotation Process DNA sequence Blastn Repeats Promoters Fasta BlastP Gene finders rRNA Pfam Blastx Halfwise Pseudo-Genes Prosite Psort tRNA scan Genes SignalP tRNA TMHMM NCBI RepeatMasker Artemis • Artemis is a free DNA sequence viewer and annotation tool that allows visualization of sequence features and the results of analyses within the context of the sequence, and its six-frame translation. NCBI • http://www.sanger.ac.uk/Software/Artemis/ NCBI atcttttacttttttcatcatctatacaaaaaatcatagaatattcatcatgttgtttaaaataatgtattccattatgaactttattacaaccctcgtt tttaattaattcacattttatatctttaagtataatatcatttaacattatgttatcttcctcagtgtttttcattattatttgcatgtacagtttatca tttttatgtaccaaactatatcttatattaaatggatctctacttataaagttaaaatctttttttaattttttcttttcacttccaattttatattccg cagtacatcgaattctaaaaaaaaaaataaataatatataatatataataaataatatataataaataatatataatatataataaataatatataatat ataatatataataaataatatataatatataatatataataaataatatataataaataatatataatatataatatataatactttggaaagattattt atatgaatatatacacctttaataggatacacacatcatatttatatatatacatataaatattccataaatatttatacaacctcaaataaaataaaca tacatatatatatataaatatatacatatatgtatcattacgtaaaaacatcaaagaaatatactggaaaacatgtcacaaaactaaaaaaggtattagg agatatatttactgattcctcatttttataaatgttaaaattattatccctagtccaaatatccacatttattaaattcacttgaatattgttttttaaa ttgctagatatattaatttgagatttaaaattctgacctatataaacctttcgagaatttataggtagacttaaacttatttcatttgataaactaatat tatcatttatgtccttatcaaaatttattttctccatttcagttattttaaacatattccaaatattgttattaaacaagggcggacttaaacgaagtaa ttcaatcttaactccctccttcacttcactcattttatatattccttaatttttactatgtttattaaattaacatatatataaacaaatatgtcactaa taatatatatatatatatatatatatatatatattataaatgttttactctattttcacatcttgtccttttttttttaaaaatcccaattcttattcat taaataataatgtattttttttttttttttttttttttattaattattatgttactgttttattatatacactcttaatcatatatatatatttatatat atatatatatatatatatatattattcccttttcatgttttaaacaagaaaaaaaactaaaaaaaaaaaaaataataaaatatatttttataacatatgt attattaaaatgtatatataaaaatatatattccatttattattatttttttatatacattgttataagagtatcttctcccttctggtttatattacta ccatttcactttgaacttttcataaaaattaatagaatatcaaatatgtataatatataacaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaata tatatatatatatatatacatataatatatatttcatctaatcatttaaaattattattatatattttttaaaaaatatatttatgataacataaaaaga atttaattttaattaaatatatataattacatacatctaatattattatatatatataataagttttccaaatagaatacttatatattatatatatata tatatatatatatattcttccataaaaagaataaaataaaataaaaacaccttaaaagtatttgtaaaaaattccccacattgaatatatagttgtattt ataaaattaaagaaaaagcataaagttaccatttaatagtggagattagtaacattttcttcattatcaaaaatatttatttcctaattttttttttttg taaaatatatttaaaaatgtaatagattatgtattaaataatataaatatagcaaaatgttcaattttagaaatttgcctctttttgacaaggataattc aaaagatacaggtaaaaaaaaaaaaataaagtaaaacaaaacaaaacaaaaaacaaaaaaaaaaaaaaaaaaaaaaatgacatgttataatataatataa taaataaaaattatgtaatatatcataatcgaagaaacatatatgaaaccaaaaagaaacagatcttgatttattaatacatatataactaacattcata tctttatttttgtagatgatataaaaaattttataaactcttatgaagggatatatttttcatcatccaataaatttataaatgtatttctagacaaaat tctgatcattgatccgtcttccttaaatgttattacaataaatacagatctgtatgtagttgatttcctttttaatgagaaaaataagaatcttattgtt ttagggtaatgaaatatatatagatttatatttttatttatttattatatattattttttaatttttcttttatatatttattttatttagtgtataaaa tgatatcctttatatttatatttacatgggatattcaaataataacaaaaatgagtatacacatatatatatatatatatatatatatgtatattttttt tttttttttatgttcctataggaaagggaagaattcactgatttgtagtgtttacaatattagggaatgcaactttacacttttgaaaaaaattcagtta agcaaaaatattaataacattaaaaagacactgatagcaaaatgtaatgaatatataataacattagaaaataagaaaattactttttatttcttaaata aagattatagtataaatcaaagtgaattaatagaagacggaaaagaacttattgaaaatatctatttgtcaaaaaatcatatcttgttagtaataaaaaa ttcatatgtatatatataccaattagatattaaaaattcccatattagttatacacttattgatagtttcaatttaaatttatcctacctcagagaatct ataaataataaaaaaaagcatataaataaaataaatgatgtatcaaataatgacccaaaaaaggataataatgaaaaaaatacttcatctaataatataa cacataacaattataatgacatatcaaataataataataataataataatattaatggggtgaaagaccatataaataataacactctggaaaataatga tgaaccaatcttatctatatataatgaagatcttaatgttttatatatatgccaaaatatgtataacgtcctttttgttttgaatttaaataacctaagt DNA in Artemis Black bar = stop codon GC content Forward translations NCBI Reverse Translations DNA and amino acids