Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Fischer–Tropsch process wikipedia , lookup
Asymmetric hydrogenation wikipedia , lookup
Marcus theory wikipedia , lookup
Kinetic resolution wikipedia , lookup
Woodward–Hoffmann rules wikipedia , lookup
Elias James Corey wikipedia , lookup
Discodermolide wikipedia , lookup
Stille reaction wikipedia , lookup
Aldol reaction wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
Tiffeneau–Demjanov rearrangement wikipedia , lookup
George S. Hammond wikipedia , lookup
1,3-Dipolar cycloaddition wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Aza-Cope rearrangement wikipedia , lookup
Wolff rearrangement wikipedia , lookup
Ene reaction wikipedia , lookup
Hydroformylation wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
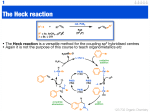
Physical organic chemistry wikipedia , lookup
Petasis reaction wikipedia , lookup
Vinylcyclopropane rearrangement wikipedia , lookup
Asymmetric induction wikipedia , lookup
Strychnine total synthesis wikipedia , lookup
1 The Diels-Alder reaction Me CO2Me + Me Me Δ CO2Me + CO2Me major Me CHO + minor Me Δ Me CHO + CHO toluene, 120°C, no catalyst benzene, 25°C, SnCl4 59 96 : : 41 4 Lewis acid improves selectivity • Diels-Alder (DA) reaction is incredibly valuable method for the synthesis of 6-rings • It is not within the remit of this course to go into detail about this reaction • We are interested in the stereochemical outcome but need a bit of revision... • Normally DA is highly regioselective (as seen above) • It is controlled by the ‘relative sizes’ of the p orbitals in the LUMO & HOMO involved • More accurately referred to as the orbital coefficients • In the presence of a Lewis acid dienophile is polarised giving higher regioselectivity and a faster reaction NMe2 NMe2 CO2Me CO2Me + regioselectivity often follows simple electronic argument (consider which C is δ+ve or δ–ve) HOMO LUMO NMe2 CO2Me Advanced organic 2 Endo vs. exo selectivity A secondary orbital overlap H H A A endo B A B D C H C D A D B C H C B D favoured D B endo H A A C H H ≡ H exo B C C ≡ D D B exo • Endo transition state & adduct is more sterically congested thus thermodynamically • • less stable But it is normally the predominant product The reason is endo transition state is stabilised by π orbital overlap of the group on C or D with the diene HOMO; an effect called ‘secondary orbital overlap’ • The reaction is suprafacial and we observe that the geometry of the diene & dienophile is preserved Advanced organic 3 Diels-Alder reaction A A A H A H B draw a cube add the diene D C B D add dienophile (endo product has substituents directly under diene) C B H H D remember other substituents present C B D do reaction (make new bonds) C B A H H H H A H B H H C D H should be able to see relative stereochemistry • The ‘cube’ method is a nice way to visualise the relative stereochemistry • Finally, remember that the dienophile invariably reacts from the less hindered face • If you are a little rusty on the Diels-Alder reaction either re-read your lecture notes or any standard organic text book MeO OMe H + MeO H NO2 O2N H H NO2 Advanced organic 4 Chiral auxiliaries on the dienophile O O BnOH + + Cl OBn achiral dienophile O + OBn O OBn 1 : 1 mixture of enantiomers achiral diene • One diastereoisomer is formed - the endo product • But mixture of enantiomers • If we add a chiral auxiliary then there are two possible endo diastereoisomers • But one predominates - thus we can prepare a single enantiomer O R O HN R O O O Cl N O Et2AlCl + Me (S)-valine derivative O N O O Me Me Me Me R chiral dienophile achiral diene Me single(ish) diastereoisomer R = H 86% de R = Me 90% de >98% endo BnOH R O OBn single enantiomer Advanced organic 5 Explanation of diastereoselectivity s-cis favoured Et2 O Al O H N O Me O O Me N Et2AlCl2 Et Et Al O O O N Me Me s-trans disfavoured Et2 O Al O H N O Me Me O Et2 O Al O H N O Me Me Me Me lower face blocked • Coordination to the Lewis acid activates dienophile • The rigid chelate governs reactive conformation (s-cis) as s-trans disfavoured • iso-Propyl group blocks bottom face • Diene’s approach maximises secondary orbital overlap and favours endo product Advanced organic 6 Camphor-derived auxiliary Me Me R O N R + TiCl4 –78°C H S O O Me O N O2S Me Me R = H 99% de R = Me >97% de >98% endo Me R Me Me R N S O O O Ti Ln N SO2 O • A range of auxiliaries can be utilised • Most give good diastereoselectivities Advanced organic 7 Chiral auxiliaries II phenyl group blocks lower face H Me O O BnO Me Me AlCl3 OBn O BnO H Me Me ≡ ≡ + Me O Me O diene approaches from the top H CO2R Me O Me BnO • It is possible to attach the chiral auxiliary to the diene as well O O O O OH MeO O OMe H Ph B(OAc)3 + O H Ph H O OH H O >95% de endo Advanced organic 8 Chiral catalysis and the Diels-Alder reaction O Me MeO + N MeO cat. Br H O Me N Br H O O >97% ee Me Me Me F3CO2S Me N Al N SO2CF3 Me • The fact the Diels-Alder reaction is mediated or catalysed by Lewis acids means enantioselective variants are readily carried out • The aluminium catalyst above has been utilised in enolate chemistry (aldol) reaction and is very effective in this Diels-Alder reaction Advanced organic 9 Chiral catalysis and the Diels-Alder reaction II O O + N lig. (10%) Cu(OTf)2 (9%) H O O O N Cl N N Cl Cl Cl O 92% ee • The oxazolidinone substituent on the dienophile is important • Good selectivities are only achieved when there are two binding points on the dienophile • The two carbonyl groups allow a rigid chelate to be formed & maximise the commincation of chirality O O BH3 / HOAc + OH O Ph H OMe OH OH O H >98% ee OH OMe Ph Advanced organic 10 Organocatalysis and the Diels-Alder reaction OMe cat. (20%) HClO4 O + COEt Et OMe 96% ee endo / exo >200 : 1 Me O Ph O Me N N H O Me Ar N Me O N N N OMe O Et Et Me • Organic secondary amines can catalyse certain Diels-Alder reactions • The reaction proceeds via the formation of an iminium species • This charged species lowers the energy of the LUMO thus catalysing the reaction • In addition one face of dienophile is blocked thus allowing the high selectivity Advanced organic 11 Organocatalysis and the Diels-Alder reaction II OMe O + Ph H O 1. cat. (10%) 2. TFA Ph O O TBSO Ph Tf Ph N N O 87% ee Tf H H Ph TFA H Ph O Me Tf N N MeO H O O H Ph TBSO Tf O TBS Ph O H O • This is an example of a hetero-Diels-Alder reaction • The aldehyde is the dienophile • We have to use a very electron rich diene • The amine catalyst acts as a Lewis acid via two hydrogen bonds Advanced organic 12 Organocatalysis III TBSO H 1. cat. (10%) 2. AcCl Ph + Me N Ph Me Me O Ph >98% ee OH OH O Ph O Ph O O Me H O AcCl Ph TBSO O O H O H O H Ph H O Ph Me N Me • Another hetero-Diels-Alder reaction • It looks very similar to the previous reaction but... • It is believed that only one hydrogen bond activates the aldehyde • The other is used to form a rigid chiral environment for the reaction Advanced organic 13 [3,3]-Sigmatropic rearrangements R2 R2 R2 heat X R1 X R3 R1 X R3 R1 R3 • A class of pericyclic reactions whose stereochemical outcome is governed by • • • geometric requirements of the cyclic transition state Reactions generally proceed via a chair-like transition state in which 1,3-diaxial interactions are minimised General relationship is outlined below... Indicates that geometry of double bonds important to controlling relative stereochemistry R c X a c b d R2 d c a R X b R2 R a X d R X b H the R2 H a b R2 c d Advanced organic 14 Cope rearrangement Ph H Me Me Ph Ph Me H Me 91% Me Me H Me Ph Me Me Ph 1,3-diaxial interactions disfavoured Me H 9% • A very simple example of a substrate controlled [3,3]-sigmatropic rearrangement is • • • the Cope rearrangement To minimise 1,3-diaxial interactions phenyl group is pseudo-equatorial Note: the original stereocentre is destroyed as the new centre is formed This process is often called ‘chirality transfer’ Advanced organic 15 Claisen rearrangements Claisen rearrangement OEt OH + Hg+ O O heat H Johnson-Claisen rearrangement OH + MeO OMe Me O H+ OMe O heat OMe OMe Eschenmoser-Claisen rearrangement OH + MeO OMe Me O H+ NMe2 O heat NMe2 NMe2 Ireland-Claisen rearrangement O OH + Me O O Et3N Me R3SiCl base O Me O O heat OSiR3 • One of the most useful sigmatropic rearrangements is the Claisen and all it’s variants O OSiR3 rearrangement Advanced organic 16 ‘Enantioconvergent’ synthesis SET reduction gives most stable alkene OH Na NH3 Me Me OH Me Me NMe2 MeO OMe O NMe2 Me O Me H Me Me Me Me NMe2 Me Me Me ≡ H NMe2 H NMe2 H H i-Pr i-Pr O Me i-Pr H H Me O i-Pr O Me O Me2N H Me H Me2N Me Me NMe2 Me ≡ same configuration H2 Lindlar cat. OH Me OH Me NMe2 MeO OMe Me Me O NMe2 Me Me Me O Me NMe2 O Me H Me Me Me Me heterogeneous hydrogenation leads to syn addition of H2 • Both enantiomers of initial alcohol can be converted into the same enantiomer of • product This process (Eschenmoser-Claisen) shows the importance of alkene geometry Advanced organic 17 Ireland-Claisen reaction H 1. LDA, THF 2. R3SiCl O OSiR3 Me Me Me O O OSiR3 Me H OSiR3 O Me H H O Me Me OSiR3 Me Me O Me H 1. LDA, THF/HMPA 2. R3SiCl H O Me OSiR3 OSiR3 Me Me Me O H O OSiR3 Me Me O H Me OSiR3 Me • Enolate geometry controls relative stereochemistry • Therefore, the enolisation step controls the stereochemistry of the final product Advanced organic 18 Substrate control in Ireland-Claisen rearrangement methyl group is pseudo-equatorial Me Me O O 91% ee OH 1. LHMDS 2. TMSCl Me O Me O H H Me H OTMS OTMS OTMS Me H OTMS OTMS Me HO2C Me 98% syn 91% ee • In a similar fashion to the Cope rearrangement we saw earlier, the Ireland-Claisen • • rearrangement occurs with ‘chirality transfer’ Initial stereogenic centre governs the conformation of the chair-like transition state Largest substituent will adopt the pseudo-equatorial position • Once again, the relative stereochemistry is governed by the geometry of the enolate Advanced organic 19 Auxiliary control in the Ireland-Claisen rearrangement N Ar* Me O N O Ar* Me Me LDA Me O O Me Me Li Me N Ar* Me Li N anti / syn 98:2 94% de for anti Ar* Me NHAr* Me Ar*NH2 = O OMe NH2 • Use of chiral auxiliaries allows the control of absolute stereochemistry • Good news is that it is hard to predict and so will not be examined... Advanced organic 20 Chiral reagent control in the Ireland-Claisen rearrangement i-Pr2NEt CH2Cl2 –78°C R*2B OH O Me O Ph O O Me Me + ArO2S N Ph B N warm Me >97% ee Me SO2Ar R*2B Br OH O warm Et3N Tol / hexane –78°C Me O O O Me Me Me Me 96% ee • Funnily enough, it is possible to carry the reaction out under “reagent” control • Although, it could be argued that this is just a form of temporary auxiliary control! • Enolate formation (enolate geometry) governs relative stereochemistry Advanced organic 21 Chiral catalyst control in the Ireland-Claisen rearrangement Ph MeAl(OR*)2 O Si Me Me Ph Ph SiMe3 O O H Me SiMe3 SiMe2t-Bu MeAl(OR*)2 = O O Al Me SiMe2t-Bu • It is also possible to perform the reactions under chiral catalyst control • Presumably, the Lewis acid coordinates to the oxygen & influences the reactive conformation thus controlling enantioselectivity Advanced organic 22 The Heck reaction R1 X + cat. PdX2 R2 R3N [R33P] R1 = Ar, ArCH2, X = Br, I, OTf R1 R2 • The Heck reaction is a versatile method for the coupling sp2 hybridised centres • Again it is not the purpose of this course to teach organometallics etc Br R3NH Br L Pd L oxidative addition R3N H L L Pd Br L +L H L Pd Br Pd Br Pd(0) (14e) Pd(II) (16e) L Pd(II) (16e) –L L Pd(II) (16e) Pd Br H syn addition β-hydride elimination Br Pd L Advanced organic 23 Alkene isomerisation 0.01% Pd(OAc)2 R3N + O I L Pd I δ+ O δ– O 100°C syn addition Pd(I)Ln H H O β-hydride elimination Ph O L hydroI palladation Pd H Ph O Pd(I)Ln H H Ph O O H Pd L I Ph Ph O O H Pd(I)Ln L Pd I H • β-Hydride elimination is reversible • This alkenes can ‘walk’ or migrate to give the most stable alkene • Only restriction is every step must be syn Advanced organic 24 Enantioselective Heck reaction Pd[(R)-BINAP]2 proton sponge OTf + O CO2Et NMe2 NMe2 O EtO2C 62% >96% ee PPh2 PPh2 proton sponge (R)-BINAP Pd(dba)2 (3%), lig (6%) i-Pr2NEt + O TfO O O PPh2 N 92% >99% ee t-Bu lig amino acid derivative • With the use of chiral ligands the Heck reaction can be enantioselective • Remember that we often see alkene migration Advanced organic 25 Enantioselective Heck reaction II TBSO TBSO Pd[(R)-BINAP]Cl2 AgPO4, CaCO3 I N Me O H 78% 82% ee PPh2 PPh2 O N O Pd2(dba)3 (R)-BINAP Me I O O Ag3PO4 N,N-dimethylaniline Me N (R)-BINAP O O 71% ee • Intramolecular variant allows the construction of ring systems • The silver salt accelerates the reaction and prevents alkene isomerisation Advanced organic 26 Suzuki-Miyuara reaction L Pd0 L –L R2 reductive elimination L Pd0 oxidative addition X R2 R1 L Pd X R1 L Pd R2 R2 transmetallation R1 B(OH)2 • The Suzuki-Miyuara reaction is (normally) the palladium catalysed coupling of an • • alkenyl or aryl halide with an alkenyl or aryl boronic acid Normally the components should be sp2 hybridised to avoid β-eliminations Mechanism etc is (surprise surprise) outside the scope of this course but the wonderful enantioselective examples are not... Advanced organic 27 Enantioselective biaryl formation Me O B (PdClC3H5)2 lig1 CsF + Me Me O Me I PPh2 NMe2 Fe H Me lig1 60% 85% ee Br P(O)(OMe)2 + Pd2(dba)3 (0.2%) lig2 Me Me NMe2 P(O)(OMe)2 PCy2 B(OH)2 95% 86% ee lig2 • Virtually every (if not every...) reaction we have covered in this course has formed a • • stereogenic centre (central chirality) These two examples form axially chiral compounds Please note: both ligands are thought to be mono-dentate (in the active species at least, although they may be bidentate in ‘resting state’) via the phosphine Advanced organic 28 Other catalytic enantioselective reactions Br O Ph Me N + O Pd2(dba)3 (1%) lig1 Ph NaOt-Bu Me Me O N i-Pr2P Me 80% 93% ee lig1 • Pd(0) chemitry has been utilised in the enantioselective arylation of enolates • The reaction is related to much of Pd chemistry you have covered • Below is an example of a chiral variant of the Schrock metathesis catalyst • The reaction involves desymmetrisation by selective reaction if one disubstituted alkene O O L2 (10mol%), PhH, 22°C, 48h N Me N i-Pr i-Pr Ar Me Me Me 91% 98% ee N O Mo THF Me O Ar Ph Me L2 Advanced organic 29 Enantioselective Negishi reactions NiCl2•glyme (10mol%), L1 (13mol%), DMI:THF (7:1), 0°C O Bn Et N Ph + hex ZnBr O Bn Et N Ph hex 90% 95% ee Br NiBr2•diglyme (10mol%), L1 (13mol%), DMA, 0°C Br O + BrZn O O O Cl Cl 82% 91% ee O N i-Pr O N N L1 i-Pr • Last year (2005) saw the first examples of catalytic enantioselective Negishi couplings • The system still has some limitations but is an exciting development • On a practical note, many of the reactions above were run in air!!! Advanced organic 30 Summary of methods for stereoselective synthesis Method Advantages Disadvantages resolution both enantiomers available maximum 50% yield synthesis of (–)-propranolol chiral pool 100% ee guaranteed synthesis of (R)-sulcatol often only 1 enantiomer available Examples chiral auxiliary often excellent ee’s; built in extra steps to introduce resolving agent and remove auxiliary oxazolidinones chiral reagent alpine-borane®, Brown allylation reagents often excellent ee’s; stereoselectivity can be independent of substrate control chiral catalyst economical; only small amounts of recyclable material used only a few reagents are successful and often only for a few substrates only a few reactions are asymmetric hydrogenation; really successful; frequently Sharpless epoxidation a lack of substrate generality • Hopefully this course has shown that the area of stereoselective synthesis (or more particularly, methodology for stereoselective synthesis) is a vast & fascinating topic • There are many reactions we have not covered (there is already far too much material in the course) • I hope you found the course as interesting as I did... Advanced organic