Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Genomic imprinting wikipedia , lookup
Genomic library wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Human genetic variation wikipedia , lookup
Gene expression profiling wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Genome (book) wikipedia , lookup
SNP genotyping wikipedia , lookup
Human genome wikipedia , lookup
Frameshift mutation wikipedia , lookup
Non-coding DNA wikipedia , lookup
Designer baby wikipedia , lookup
Pathogenomics wikipedia , lookup
Population genetics wikipedia , lookup
History of genetic engineering wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Genome evolution wikipedia , lookup
DNA barcoding wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Koinophilia wikipedia , lookup
Point mutation wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Genetic code wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Genome editing wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Helitron (biology) wikipedia , lookup
Metagenomics wikipedia , lookup
Microsatellite wikipedia , lookup
Multiple sequence alignment wikipedia , lookup
Sequence alignment wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
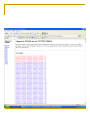
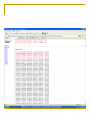
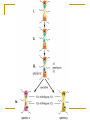
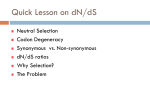
Tutorial #2 Quiz next week Cover everything you’ve seen in the course so far Combination of True/False, definition, short answer, or some similar question from the problem set How to design a PCR primer? Primer length and sequence are of critical importance in designing the parameters of a successful amplification A simple formula for calculating the Tm Tm = 4(G + C) + 2(A + T) When designing a PCR primer, Tm is not the only thing, should also consider; the GC content, any secondary structure or hairpin loop Example Design PCR primer to amplify IFI16 (interferon, gamma-inducible protein 16) NCBI Synonymous Vs Nonsynonymous When studying the evolutionary divergences of DNA sequence Synonymous = silent Nonsynonymous = amino acid altering The rates of these nucleotide substitution maybe used as a molecular clock for dating the evolutionary time of closely related species Calculating Synonymous sites (s) and nonsynonymous sites (n) Each codon has 3 nucleotides, denote by fi (I = 1,2,3) Where s and n for a codon are given by s = ∑3i=1fi and n = (3-s) f1=1/3 (T→C) f2=0 f3=1/3 (A→G) Thus, s = 2/3 and n = 7/3 For DNA sequence of r codons, it will be s = ∑ri=1si and n = (3r-s), where si is the value of s for the ith codon Ex. TTA (Leu) Calculation of s and n for 2 nucleotide differences between 2 codons Ex. GTT (Val) and GTA (Val) 1 synonymous difference Denote sd and nd the number of synonymous and nonsynonymous differences per codon, respectively sd = 1 nd = 0 Con’t Ex. TTT and GTA, 2 pathways to get there Pathway #1: TTT(Phe)↔GTT(Val)↔GTA(Val) Pathway #2: TTT(Phe)↔TTA(Leu)↔GTA(Val) Pathway 1 involve 1 synonymous and 1 nonsynonymous substitution Pathway 2 involve 2 nonsynonymous substitution sd = 1 synonymous substitution / 2 change state = 0.5 nd = 3 nonsysnonymous substitution / 2 change state =1.5 D in the problem set = proportion of synonymous or nonsynonymous differences, therefore, for this nonsynonymous site, the Dn would be 1 / 1.5 = 0.667 Note that sd + nd is equal to the total number of nucleotide differences between the two DNA sequences compared Sequence Alignment Every alignment will have a scoring system Base change cost = 1 Gap cost = 2 Gap extension cost = 1 Ex. ACT GTT GCC AG - C - - GCT Score of this alignment would be 3 + 2x2 + 1 = 8 In this case, a higher score means a worst alignment MLST - Methods Isolate multiple strains of species of interest PCR ~500bp regions of 4-20 housekeeping genes (“loci”) Sequence PCR products Assign “allele numbers” to each locus Arbitrary, each # represents a different sequence 1 2 3 1 2 1 1 1 2 MLST - Methods Collate the information into a table Row = isolate Column = loci Fill in allele numbers 1 2 3 1 2 1 1 1 2 Isolate 1 Isolate 2 Locus A 1 2 Locus B 1 2 Locus C 1 1 Isolate 3 3 1 2 MLST of a Halorubrum Population 36 isolates 4 housekeeping genes atpB ef-2 radA secY 500bp PCR product Allelic profiles vary Few identical pairs All loci polymorphic 8-15 alleles Insights from the MLST Data - 1 How genetically diverse is the saltern Archaeal population? Genetic diversity H = 1-Σxi2 Overall genetic diversity = 0.69 Varied between ponds of different salinity 0.57 in 23% saline pond 0.83 in 36% saline pond Higher than E. coli diversity of 0.47 >5x higher than eukaryotic diversity Insights from the MLST Data - 2 Is recombination occurring in the Archaea? Linkage disequilibrium calculator – mlst.net LD = Alleles are linked and are transferred together during recombination LE = Alleles are not linked and recombination scatters them randomly Halorubrum population is near linkage equilibrium Suggests recombination is occurring Tetraodon Nigroviridis 2X? Nature Reviews Genetics 3; 838849 (2002); Phylogenetic tree Phylogenetics is the field of systematics that focuses on evolutionary relationship between organisms or genes/proteins (phylogeny) A node Human A clade Mouse Fly clade -- A monophyletic taxon taxon -- Any named group of organisms, not necessarily a clade. A phylogenetic tree A node D B A C A+B+C is less than D+B+C Human A clade Mouse Fly So the mouse Sequence is more related to fly than the human sequence is to fly in this example Tetraodon gene evolution Fourfold degenerate (4D) site substitution - a mesure of neutral nucleotide mutations 4D site = 3rd base of codon free to change with no FX on AA # of AA changes at these sites = neutral mutations Fish proteins have diverged faster vs. mammalian homologues Figure 3 Brief generalization of the papers Comparative genomics help identifying region of DNA that are shared between two different species and allows the transfer of information between both species in the common region. It can also detect regions that have gone through chromosomes rearrangement occurring in many different diseases. This information can be of different type. 1) Using one of the species it is possible to transfer annotation information that were not known in the other species, 2) identify region that are under selective pressure, 3) It is also possible to compare for examples regions that have gone through chromosomes rearrangement with annotation genes map to identify genes responsible for a particular disease Homologs Have common origins but may or may not have common activity Orthologs – Homologs produced by speciation. They tend to have similar function Paralogs – Homologs produced by gene duplication. They tend to have differing function Xenologs – Homologs resulting from horizontal gene transfer between two organism BLAST Basic Local Alignment Search Tool Developed in 1990 and 1997 (S. Altschul) A heuristic method (Fast alignment method) for performing local alignments through searches of high scoring segment pairs (HSP’s) 1st to use statistics to predict significance of initial matches - saves on false leads Offers both sensitivity and speed BLAST Looks for clusters of nearby or locally dense “similar or homologous” k-tuples Uses “look-up” tables to shorten search time Uses larger “word size” than FASTA to accelerate the search process Can generate “domain friendly” local alignments Fastest and most frequently used sequence alignment tool – BECAME THE STANDARD Connecting HSP’s Extreme Value Distribution -x P(x) = 1 - e -e = P(x) = 1 – e -(Kmne-lS) Kmne-lS is called Expect or E-value In BLAST, default E cutoff = 10 so P = 0.99995 If E is small then P is small Why does BLAST report an E-value instead of a p value? E-values of 5 and 10 are easier to understand than Pvalues of 0.993 and 0.99995. However, note that when E < 0.01, P-values and Evalue are nearly identical. Expect value Kmne-lS = Expect or E-value What parameters does it depend on? - l and K are two parameters – natural scales for search space size and scoring system, respectively l = lnq/p and K = (q-p)2/q ¨ p = probability of match (i.e. 0.05) ¨ q = probability of not match (i.e. 0.95) Then l = 2.94 and K =0.85 p and q calculated from a “random sequence model” (Altschul, S.F. & Gish, W. (1996) "Local alignment statistics." Meth. Enzymol. 266:460-480.) based on given subst. matrix and gap costs - m = length of sequence - n = length of database - S = score for given HSP Expect value Expect value an intuitive value but… Expect value changes as database changes Expect value becomes zero quickly Alternative: bit score S' (bits) = [lambda * S (raw) - ln K] / ln 2 Independent of scoring system used - normalized Larger value for more similar sequences, therefore useful in analyses of very similar sequences Similarity by chance – the impact of sequence complexity MCDEFGHIKLAN…. High Complexity ACTGTCACTGAT…. Mid Complexity NNNNTTTTTNNN…. Low Complexity Low complexity sequences are more likely to appear similar by chance