Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Copy-number variation wikipedia , lookup
Oncogenomics wikipedia , lookup
Genome (book) wikipedia , lookup
History of genetic engineering wikipedia , lookup
DNA barcoding wikipedia , lookup
Point mutation wikipedia , lookup
Designer baby wikipedia , lookup
Gene expression profiling wikipedia , lookup
DNA sequencing wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Transposable element wikipedia , lookup
Public health genomics wikipedia , lookup
Microsatellite wikipedia , lookup
Minimal genome wikipedia , lookup
Sequence alignment wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Microevolution wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Non-coding DNA wikipedia , lookup
Human genome wikipedia , lookup
Genomic library wikipedia , lookup
Whole genome sequencing wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Human Genome Project wikipedia , lookup
Helitron (biology) wikipedia , lookup
Genome editing wikipedia , lookup
Pathogenomics wikipedia , lookup
Genome evolution wikipedia , lookup
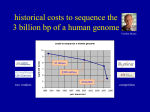
BINF6201/8201: Molecular Sequence Analysis Dr. Zhengchang Su Office: 351 Bioinformatics Building Email: [email protected] Office hours: Tuesday and Thursday: 2:00~3:00pm 08-23-2016 Textbook and reading/reference materials Ø Textbook: Bioinformatics and Molecular Evolution by Paul G. Higgins and Teresa K. Attwood, Blackwell Publishing, 2005. Relevant materials: http://www.wiley.com/WileyCDA/WileyTitle/ productCd-1405106832.html Ø Reference textbooks: 1. Molecular Evolution, Wen-Hsiung Li, Sinauer Association 2. Biological Sequence Analysis, Richard Durbin et. al. Cambridge Univ. Press 3. Introduction to Linear Algebra, 5th Ed. Gilbert Stanger, Wellesley-Cambridge Press 4. Statistical Methods in Bioinformatics, 2th Ed. Warren J. Ewens, Springer 5. Deep Learning, Ian Goodfellow, et. al. The MIT Press Ø Additional readings from the current literature may be assigned as appropriate Ø Lecture slides and lab materials and instructions: http:// bioinfo.uncc.edu/zhx/binf8201/binf8201.html Students Evaluation Ø Weekly or bi-weekly homework assignments, Ph.D students may have additional assignments (20%). Ø Two midterm exams (60%): 10/6(Thursday) and 12/13 (Tuesday) Ø Classroom participation will count for 5% of the grade. Ø Lab participation will count for 5% of the grade. Development of DNA sequencing technologies Ø Chain termination (Sanger) method (1977): read length 800~1000 bps Ø Automation of sequence determination (late 1980s) Ø Shotgun whole genome sequencing strategy (1995) Ø Massively parallel (Next generation (NexGen)) sequencing technologies (2004): read length: 300bps, ~1010 bps/day/machine 1. 454 pyrosequencing: 454 Life Sciences/Roche Diagnostics 2. Solexa sequencing/Illumina, Genome Analyzers, HiSeq2000, HiSeq2500 MiSeq, and HiSeq X 10, … 3. SOLiD sequencing/Life Technologies: 5500 W Series Genetic Analyzer 4. Helico BioSciences: Single molecule sequencing 5. Pacific Biosciences: Single molecule sequencing. Read length: a few kB 6. Oxford Nanopore technologies: Single molecule sequencing 7. Polonator: open source 8. Ion Torrent/Life Technologies: personal sequencer: non optical sequencing 9. ……… Major sequence databases Ø Three almost equivalent biological sequence databases International Sequence Database Collaboration 1. GenBank at NCBI 2. European Molecular Biology Laboratory (EMBL) Nucleotide Sequence Database at European Bioinformatics Institute (EBI) 3. DNA database of Japan (DDBI) Ø Features 1. All published biological sequences are traditionally requested to be deposited in the one of these three databases. 2. Data are exchanged among these three databases on a daily basis. 3. However, with the inundation of short read DNA sequences generated by next-generation sequencing technologies, storing these sequences is a huge challenge. Available sequences grow faster than CPU speed Ø Even before the genomic era, both the number and length of sequences increased exponentially with the time. Ø Clearly, effective analysis of these DNA sequences requires using computers and effective algorithms. Ø Although the number of transistors in a computer also increased exponentially in the same time period, the number/length of sequences increases even faster than the number of transistors in a CPU. rt ln N (t ) = rt + ln N 0 . lnN(t) N (t ) = N 0e ; (t) An update of the analysis https://www.nlm.nih.gov/about/2014CJ.html An update of the analysis (continued) https://www.genome.gov/images/content/costpergenome2015_4.jpg Sequence data explosion Ø Since 1995, the number of sequenced genomes has also increased exponentially. Complete and Permanent Draft Genome Total in the GOLD Database Sequence data explosion Genome Total in the GOLD Database in each year Sequence data explosion Project Totals in GOLD (Domain Group) Sequence data explosion Ø Phylogenetic distribution of Bacterial Genome Projects October 2014, 36,959 projects: http://www.genomesonline.org Sequence data explosion Ø Major Sequencing Centers October 2014, 56,020 projects: http:// www.genomesonline.org Sequence data explosion Ø Major Sequencing Centers for Archaeal and Bacterial Genomes October 2014, 37,885 projects: http://www.genomesonline.org Sequence data explosion Ø Major Sequencing Centers for Archaeal and Bacterial Genomes October 2014, 37,885 projects: http://www.genomesonline.org Sequence data explosion Ø Since 2006, the number of meta-genome sequences has increased exponentially thanks to the advent of NGS technologies. Ø 8/20/2014, about 467 meta-genomes are sequenced or are in the process of sequencing. http://www.genomesonline.org/cgi-bin/GOLD/metagenomic_classification.cgi https://www.genome.gov/images/content/costpermb2015_4.jpg Sequence data explosion Ø The speed of computers also increase exponentially with the time. Ø However, how can we use the ever powerful computers to solve biological problems is a very challenging task for both computer science and biology research communities. Sequence-related data explosions Ø More and more biological research depends on computational analyses. Human genome sequence explosion Ø The initial human genome projects (the public and Celera projects, drafts finished in 2001) produced consensus sequences from several individuals. Ø The 1000 Genome Project (http://www.1000genomes.org/) aims to sequence thousands of carefully selected individual genomes, including parents and child trios from different human populations. Ø The UK 10K Genome Project (http://www.uk10k.org/goals.html): funded by Welcome Trust to sequence tens of thousands UK individual genomes. Ø The US 1M Genome Project: funded by the Obama Precision Medicine Initiative to sequence 1M genomes of Americans ( https://www.whitehouse.gov/precision-medicine) Ø The GenomeDemark project (http://www.genomedenmark.dk/english/) will sequence the entire Demark population. Ø So far, it is estimated that more than 100,000 individual human genomes have been sequenced, mainly by NGS technologies, majority of them are not released due to privacy concerns. Ø We are soon entering the precision/personalized medicine era, and how to properly use the data to interpret the variations of phenotypes caused by the variations in individual genomes is a great challenge. What is genomics? Ø The availability of whole genome sequences of organisms has led to the birth of Genomics that studies the organisms based on the genetic information encoded in the genomes. Ø According to the subjects of the study, genomics can be divided into: 1. Functional genomics, which is coupled with the development of relevant high-throughput technologies, such as, • Microarray/RNA sequencing(RNA-Seq): transcriptomics • Chromosome precipitation followed by microarray hybridization (ChIP-Chip) or sequencing (ChIP-seq): epigenomics to characterize transcription factor binding sites and chromatin states or markers. • DNAse-seq: to determine the accessibility of chromatin (nucleosome free region) • Chromatin conformation capture (3C) and its derivatives: to determine the physical proximity of linearly distant DNA segments • Mass spectrometry: Proteomics • Nucleus magnetic resonance (MR) and mass spectrometry: metabolomics 2. Comparative/evolutionary genomics: studies organisms by identifying the similarity and difference between their genomes. What is Bioinformatics? Ø For a short answer: “Bioinformatics is the use of computational methods to study biological data and problems”. Ø For a more detailed answer: Bioinformatics is 1. “The development and use of computational methods for studying the structure, function, and evolution of genes, proteins and whole genomes;” 2. “The development and use of methods for the management and analysis of biological information arising from genomics and high-throughput experiments.” Population genetics, molecular evolution and sequence analysis Ø According to the evolutionary theory, biological sequences are related to one another through heredity and variation; Ø Sequence analysis methods are thus based on the principles of evolution of sequences. Ø Therefore, to analyze sequences correctly, we must understand 1. The dynamics changes of genes (loci) in a population of the same species — population genetics; and 2. How gene sequences change during the course of evolution among different species — molecular evolution. Sequence Similarity Ø The similarity of two sequences can be identified by aligning the sequences using an alignment method/algorithm, such as the BLAST or Smith-Waterman method/algorithm. Ø Two parameters to describe the similarity of two sequences 1. Identity 2. Similarity Identities = 38/139 (27%), Similarity = 66/139 (47%), Gaps = 9/139 (6.5%) LELTYIVNFGSELAVVSMLPTFFETTFDLPKATAGILASCFAFVNLVARPAGGLISDSVG + Y + FG +A + LPT+ T + AG + FA ++ARP GG +SD + MSFLYAIVFGGFVAFSNYLPTYITTIYGFSTVDAGARTAGFALAAVLARPVGGWLSDRIA SRKNTMGFLTAGLGVGYLVMSMIKPGTFTGTTGIAVAVVITMLASFFVQSGEGATFALVP R + L + + P ++ T I +AV + + G G FA V PRHVVLASLAGTALLAFAAALQPPPEVWSAATFITLAVCLGV--------GTGGVFAWVA -LVKRRVTGQVAGLVGAYGNVG G V G+V A G +G RRAPAASVGSVTGIVAAAGGLG Homologous Sequence Ø Homology: If the two sequences have a high enough similarity, it is highly likely that they have evolved from a common ancestral sequence, and we say that they are homologous to each other. For example, if two sequences of 100 amino acids have 80% of identity, the probability by chance that the two sequences share this level of identity is very small (1/20)80. Ø Homology of two sequences can only be inferred computationally, but is difficult to be tested experimentally. Orthologs and Paralogs Ø There are two distinct types of homologous relationships, which differ in their evolutionary history and functional implications. Ø Orthologs: Evolutional counterparts derived from a single ancestral gene in the last common ancestor (LCA) of the given two species. Therefore, orthologous genes are related due to vertical evolution. Orthologous genes typically have the same function. Ø Paralogs: homologous genes evolved through duplication within the same or ancestral genome. Therefore, paralogous genes are related due to duplication events. Paralogous genes do not necessary have the same function. duplication speciation speciation Divergence evolution Ø When the similarity between two sequences is very low, say, 8% identity, then they could be still homologous due to divergent evolution; Speciation or duplication homologues Ø Divergently evolved genes usually have similar biochemical functions. Convergence evolution Ø When the similarity between two sequences are very low, say, 8%, they could be of different origin, and the observed sequence similarity is due to convergent evolution under functional selection during the course of evolution. These two sequences are called analogues. analogues Ø Analogues may have similar biochemical functions, and they usually only share several amino acids in the active site of enzymes, called motifs. Horizontal gene transfer (HGT) Ø During evolution, a progeny obtains its genes from its ancestor (vertical gene transfer), however, it also can obtain genes from other species, genera, or even taxa. This phenomenon is called horizontal gene transfer (HGT) or lateral gene transfer (LGT). Ø HGT is very pervasive, in particular, in prokaryote, and is believed to be a major driving force for evolution. LCA (Last common ancestor) • Bacteria Archaea Eukaryota Vertical gene transfer Horizontal gene transfer