Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Genome evolution wikipedia , lookup
Protein moonlighting wikipedia , lookup
Protein adsorption wikipedia , lookup
List of types of proteins wikipedia , lookup
Epitranscriptome wikipedia , lookup
Eukaryotic transcription wikipedia , lookup
Histone acetylation and deacetylation wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Community fingerprinting wikipedia , lookup
RNA polymerase II holoenzyme wikipedia , lookup
Gene regulatory network wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Transcription factor wikipedia , lookup
Gene expression profiling wikipedia , lookup
Gene expression wikipedia , lookup
Non-coding DNA wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Point mutation wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Interactome wikipedia , lookup
Molecular evolution wikipedia , lookup
Drug design wikipedia , lookup
Promoter (genetics) wikipedia , lookup
Cooperative binding wikipedia , lookup
Ligand binding assay wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
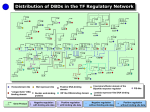
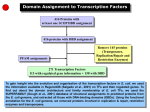
A System Approach to Measuring the Binding Energy Landscapes of Transcription Factors Authors: Sebastian J. et. al Presenter: Hongliang Fei Outline Motivation Terminologies Problem statement and significance Challenges Method Result Conclusion Motivation Quantitatively characterize interactions of network elements; Predict the function of genes in biological networks. Terminologies Affinity: The tendency of a molecule to associate with another; DNA binding domain: any protein motif that binds to double or single-stranded DNA; Terminologies continues Transcription factor: a protein that binds to specific parts of DNA using DNA binding domains. Isoform: A protein that has the same function as another protein but which is encoded by a different gene. Flanking bases: Immediate Neighbors of a mutated base. Problem Statement Given a set of transition factors (TF for short) belonging to a certain basic protein structure family, the problem of measuring Binding Energy Landscapes is to quantify the affinities of molecular interactions. Significance Predict basic function of TF Test basic assumptions of TF (e.g. base additivity ) Test other hypothesis of TF Understand biological network better Challenges A large number of variables in biological interaction lead to so many assays; Many molecular interactions are transient and exhibit nanomolar to micromolar affinities. Low affinity binding events are hard to capture. Method step 1: Use a high-throughput micro fluidic platform to measure affinities of four eukaryotic transcription factors; step 2: results from the platform were used to test hypotheses about transcription factor binding and to predict their in vivo function. Data sources Four eukaryotic TFs belonging to the basic helix-loop-helix (bHLH) family, including Isoforms A and B of Human TF MAX, the yeast TFs Pho4p and Cbf1p. TFs generally bind to a consensus sequence of 5’-CANNTG-3’ 38 genes bound by Pho4p 24 genes bound by Cbf1pb Tool processing Result for binding affinities (N_3 to N_1) From N_3 to N_1, select CAC; From N1 to N3, select GTG (refer to supporting materials) The optimal binding sequence for four TFs is CACGTG for N_3 to N3. Position weight Matrix (PWM) Describes changes in the Gibbs free energy for all 16 possible single-base substitutions. Each isoform has a PWM; Used to test additivity assumption. Comparisons of predicted energy changes with measured values To address the question of how Pho4p and Cbf1p serve distinct biological functions while recognizing seemingly identical consensus motifs, we measured the extent to which these TFs recognize flanking bases. Recognition of flanking bases for pho4p and Cbflp Comparison of A and B Pho4p prefers CC as N_5N_4 and GG as N4N5, extending the motif to 5’-CCCACGTGGG-3’. Cbflp prefers GT as N_5N_4 and AC as N4N5, extending the motif to 5’GTCACGTGAC-3’. Hypothesis testing Whether the sequence-specific binding of bHLH TFs is determined entirely by basic region? Test whether the basic region itself is sufficient to produce the observed flanking base sequence specificity by cloning the basic regions of Pho4p and Cbf1p into the MAX isoform B backbone. Except for a few outliers, the basic region is sufficient to transform original isoform B pattern to patterns resembling Pho4p and Cbflp. Hypothesis testing Whether the binding energy landscapes are sufficient to predict which genes these TFs physically bind. Using a simple model based on calculating a probability of occupancy to generate genes Test these gene’s functions Function distribution related with Pho4p and Cbf1p data sets Prediction Result Comparison Conclusion This platform can measure DNA binding energy very well even in transient and low-affinity interactions; We can successfully predict biological function by pure biophysical measurements. Acknowledgment Thanks for Dr. Huan’s guidance; Thanks to Google, Wiki.