Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Adeno-associated virus wikipedia , lookup
Mitochondrial DNA wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Frameshift mutation wikipedia , lookup
Non-coding RNA wikipedia , lookup
Segmental Duplication on the Human Y Chromosome wikipedia , lookup
Short interspersed nuclear elements (SINEs) wikipedia , lookup
Koinophilia wikipedia , lookup
Gene nomenclature wikipedia , lookup
Oncogenomics wikipedia , lookup
Gene therapy wikipedia , lookup
Human genetic variation wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Gene expression profiling wikipedia , lookup
Genetic engineering wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Gene desert wikipedia , lookup
Bisulfite sequencing wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Transposable element wikipedia , lookup
History of genetic engineering wikipedia , lookup
Non-coding DNA wikipedia , lookup
Copy-number variation wikipedia , lookup
Microsatellite wikipedia , lookup
Public health genomics wikipedia , lookup
Gene expression programming wikipedia , lookup
DNA sequencing wikipedia , lookup
Genome (book) wikipedia , lookup
Minimal genome wikipedia , lookup
Designer baby wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Helitron (biology) wikipedia , lookup
Microevolution wikipedia , lookup
Human genome wikipedia , lookup
Pathogenomics wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genome editing wikipedia , lookup
Genomic library wikipedia , lookup
Metagenomics wikipedia , lookup
Human Genome Project wikipedia , lookup
Genome evolution wikipedia , lookup
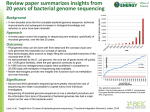
Species tree vs. gene/protein tree Trees can be very different, since genes can have their own histories Very important to know the difference between the trees! a. Gene tree is based a set of orthologous genes (i.e. related by a common ancestor) Often (but certainly not always) the gene tree is similar to the species tree b. Species tree is meant to represent the historical relationship between species. Want to build on characters that reflect time since divergence: In the genomic age, often use as many genes as possible (hundreds to thousands) to generate a species tree: Phylogenomics 1 Phylogenomics: Using Whole-genome information to reconstruct the Tree of Life Several approaches: 1. Concatonate many gene sequences and treat as one Use a ‘super matrix’ of variable sequence characters 2. Construct many separate trees, one for each gene, and then compare Often construct a ‘super tree’ that is built from all single trees 3. Incorporate non-sequence characters like synteny, intron structure, etc. The goal is to use many different # and types of characters to avoid being mislead about the relationship between species. Now recognized that different regions of the genome can have distinct histories. 2 A few other key basic concepts: Selection acts on phenotypes, based on their fitness cost/advantage, to affect the population frequencies of the underlying genotypes. In the case of DNA sequence: • Neutral substitutions = no effect on fitness, no effect on selection Given a ~constant mutation rate, can convert the # of substitutions into time of divergence since speciation = molecular clock theory. • Deleterious substitutions = fitness cost * These are removed by purifying (negative) selection • Advantageous substitutions = fitness advantage * These alleles are enriched for through adaptive (positive) selection 3 Evolutionary genomics relies on one or more quality genome sequences Quality of a genome sequence can dramatically affect evolutionary interpretations Bad genome = bad evolutionary inference Therefore, it’s important to know what makes a good genome sequence 4 Anatomy of a Genome Project A. Sequencing 1. 2. 3. De novo vs. ‘resequencing’ Sanger WGS versus ‘next generation’ sequencing High versus low sequence coverage B. Assembly 1. 2. Draft assembly Gap closure C. Annotation 1. 2. 3. Gene, intron, RNA prediction De novo vs. homology-based prediction Assessing confidence D. Comparison 1. 2. 3. Comparing gene content, lineage specific gene loss, gain, emergence Comparing genome structure (chromosomes, breakpoints, etc) Comparing evolutionary rates of change (rates of amino-acid, nucleotide substitution) 5 Sequencing Approaches Old school: Sanger Whole Genome Shotgun (WGS) 6 Overlapping sequencing ‘reads’ are assembled into a ‘contig’ 10-fold representation at this point 2-fold representation at this point The coverage of a genome = average coverage across all base pairs 8 - >10-fold is typically considered high coverage 1-3-fold is considered low coverage ** Even high average coverage can include ‘gaps’ (i.e. regions with NO coverage) See Lander-Waterman formula (poisson distribution that incorporates the number and length of reads, size of genome, coverage, and amount of overlap between reads) For 500 Mb target, 600bp read length, 5X coverage: ~29k gaps; 10X coverage: 393 gaps 7 Sequencing Approaches Advantages of Sanger Whole Genome Shotgun (WGS) * High quality sequence data * Individual sequence reads are long (~1,000 bp) * WGS is less work than map-based sequencing Disadvantages of Sanger Whole Genome Shotgun (WGS) * Still a lot of processing involved * Sanger sequencing is expensive and slow (gel-based sequencing) * True WGS sequencing requires good sequencing to get assembly to work 8 Sequencing Approaches New technology: ‘Next generation’ sequencing Includes ‘454’, Illumina/Solexa, SOLiD, and other types of sequencing Advantages: * New technology is much cheaper per genome * Generates a huge amount of sequence per run Disadvantages: * Has a higher sequencing error rate per base pair * Generates short reads (100 - 200 bp) - more challenging assembly * Generates a huge amount of sequence (and massive data files) per run 9 Several different ‘next-generation’ sequencing methods 454: emulsion sequencing per well: > 500bp read length SOLiD: emulsion amplification, bead attachment to solid surface, Ligation-based sequencing interrogates each base in 2 ligation reactions 10 Solexa (Illumina) Sequencing >100 bp read length 11 Solexa (Illumina) Sequencing >100 bp read length 12 Solexa (Illumina) Sequencing >100 bp read length 13 Next-generation (deep) sequencing - Very high (>100X) coverage - Much cheaper per bp covered - Rapid improvements in technology (including single-molecule sequencing) But - Much higher error rate (~1%) - Short reads cause assembly challenges - Some require prior amplification - Sequence-specific bias in sequencing efficiency For 500 Mb target, 100bp read length, 5X coverage: ~168k gaps; 10X coverage: 2,250 gaps 14 Anatomy of a Genome Project A. Sequencing 1. 2. 3. De novo vs. ‘resequencing’ Sanger WGS versus ‘next generation’ sequencing High versus low sequence coverage B. Assembly 1. 2. Draft assembly Gap closure C. Annotation 1. 2. 3. Gene, intron, RNA prediction De novo vs. homology-based prediction Assessing confidence D. Comparison 1. 2. 3. Comparing gene content, lineage specific gene loss, gain, emergence Comparing genome structure (chromosomes, breakpoints, etc) Comparing evolutionary rates of change (rates of amino-acid, nucleotide substitution) 15 ‘scaffold’ ‘contigs’ ‘reads’ Sanger WGS de novo assembly Goal is to have no gaps & complete scaffolds (chromosomes) Challenges: Some regions difficult to sequence through (centromeres, heterochromatin, etc) Repetitive regions make assembly difficult/ambiguous 16 ‘Next-generation sequencing’ de novo assembly ** Short read length a real challenge 17 ‘Next-generation sequencing’ de novo assembly ** Short read length a real challenge OR Matching to a ‘reference’ genome * paired-end reads Challenges: Can have lots of gaps, miss any new sequence not in the reference, repetitive regions not sequenced well, can totally miss structural rearrangements 18 Anatomy of a Genome Project A. Sequencing 1. 2. 3. De novo vs. ‘resequencing’ Sanger WGS versus ‘next generation’ sequencing High versus low sequence coverage B. Assembly 1. 2. Draft assembly Gap closure C. Annotation 1. 2. 3. Gene, intron, RNA prediction De novo vs. homology-based prediction Assessing confidence D. Comparison 1. 2. 3. Comparing gene content, lineage specific gene loss, gain, emergence Comparing genome structure (chromosomes, breakpoints, etc) Comparing evolutionary rates of change (rates of amino-acid, nucleotide substitution) 19 Genome Annotation: predicting genetic features ‘Simplest’ predictions: Open Reading Frames (ORFs) - De novo predictions: based on expectation of what ORFs should look like GCGCTTACGTTATCTGCAATATGTCTTCGAACGATTCGAACGATACCG ACAAGCAACATACACGTCTGGATCCTACCGGTGTGGACGACGCCTACA TTCCTCCGGAGCAGCCGGAAACAAAGCACCATCGCTTTAAAATCTCTA AAGACACCCTGAGAAACCACTTTATCGCTGCGGCCGGTGAGTTCTGCG GCACATTCATGTTTTTATGGTGCGCTTACGTTATCTGCAATGTCGCTA ACCATGATGTCGCATAACCGAGATGAGATGATAAAAAA 20 Genome Annotation: predicting genetic features ‘Simplest’ predictions: Open Reading Frames (ORFs) - De novo predictions: based on expectation of what ORFs should look like GCGCTTACGTTATCTGCAATATGTCTTCGAACGATTCGAACGATACCG ACAAGCAACATACACGTCTGGATCCTACCGGTGTGGACGACGCCTACA TTCCTCCGGAGCAGCCGGAAACAAAGCACCATCGCTTTAAAATCTCTA AAGACACCCTGAGAAACCACTTTATCGCTGCGGCCGGTGAGTTCTGCG GCACATTCATGTTTTTATGGTGCGCTTACGTTATCTGCAATGTCGCTA ACCATGATGTCGCATAACCGAGATGAGATGATAAAAAA Features of ORFs used in computational predictions: * Start with ATG * End with stop codon (e.g. TAA) * Should be in one frame (i.e. length divisible by 3 for each codon) * Have a size range (max. size can be >10 kb, min size can be 30 bp; median is probably ~few kb depending on organism) 21 Genome Annotation: predicting genetic features ‘Simplest’ predictions: Open Reading Frames (ORFs) - De novo predictions: based on expectation of what ORFs should look like GCGCTTACGTTATCTGCAATATGTCTTCGAACGATTCGAACGTCGAAT AGACGATGAGACGAGATAGAGCGAGCAAAAGGTAGGATACCGACAA GCAACATACACGTCTGGATCCTACCGGTGTGGACGACGCCTACATTCC TCCGGAGCAGCCGGAAACAAAGCACCATCGCTTTAAAATCTCTAAAGA CACCCTGAGAAACCACTTTATCGCTGCGGCCGGTGAGTTCTGCGGCAC ATTCATGTTTTTATGGTGCGCTTACGTTATCTGCAATGTCGCTAACCA TGATGTCGCATAACCGAGATGAGATGATAAAAAA Many ORFs have introns - splice junction signals are short and variable = difficult to predict. 22 Genome Annotation: predicting genetic features ‘Simplest’ predictions: Open Reading Frames (ORFs) - De novo predictions: based on expectation of what ORFs should look like - Homology-based assignments: find sequences homologous to known ORFs/proteins Met Ser Ser Gln Asp Ser Asn Asp Ser Asp Lys Gln … Met Ser Ser Ans Asp Ser Asn GCGCTTACGTTATCTGCAATATGTCTTCGAACGATTCGAACGTCGAAT Asp Thr Asp Lys Gln .. AGACGATGAGACGAGATAGAGCGAGCAAAAGGTAGGATACCGACAA GCAACATACACGTCTGGATCCTACCGGTGTGGACGACGCCTACATTCC TCCGGAGCAGCCGGAAACAAAGCACCATCGCTTTAAAATCTCTAAAGA CACCCTGAGAAACCACTTTATCGCTGCGGCCGGTGAGTTCTGCGGCAC ATTCATGTTTTTATGGTGCGCTTACGTTATCTGCAATGTCGCTAACCA TGATGTCGCATAACCGAGATGAGATGATAAAAAA 23 Genome Annotation: predicting genetic features ‘Simplest’ predictions: Open Reading Frames (ORFs) - De novo predictions: based on expectation of what ORFs should look like - Homology-based assignments: find sequences homologous to known ORFs/proteins - Matches to cDNA library or RNA transcripts from sequencing RNA transcript GCGCTTACGTTATCTGCAATATGTCTTCGAACGATTCGAACGTCGAAT … AGACGATGAGACGAGATAGAGCGAGCAAAAGGTAGGATACCGACAA GCAACATACACGTCTGGATCCTACCGGTGTGGACGACGCCTACATTCC TCCGGAGCAGCCGGAAACAAAGCACCATCGCTTTAAAATCTCTAAAGA CACCCTGAGAAACCACTTTATCGCTGCGGCCGGTGAGTTCTGCGGCAC ATTCATGTTTTTATGGTGCGCTTACGTTATCTGCAATGTCGCTAACCA TGATGTCGCATAACCGAGATGAGATGATAAAAAA 24 Genome Annotation: predicting genetic features Other Predictions: * Open Reading Frames (ORFs) * Non-coding RNAs (tRNAs, rRNA, other small RNAs, miRNAs, etc * Regulatory elements (ENCODE project) * Transposable elements (TEs) * Origins of DNA replication 25 Anatomy of a Genome Project A. Sequencing 1. 2. 3. De novo vs. ‘resequencing’ Sanger WGS versus ‘next generation’ sequencing High versus low sequence coverage B. Assembly 1. 2. Draft assembly Gap closure C. Annotation 1. 2. 3. Gene, intron, RNA prediction De novo vs. homology-based prediction Assessing confidence D. Comparison 1. 2. 3. Comparing gene content, lineage specific gene loss, gain, emergence Comparing genome structure (chromosomes, breakpoints, etc) Comparing evolutionary rates of change (rates of amino-acid, nucleotide substitution) 26 In what ways can a bad genome sequence affect the following: Comparisons of: * Genome size, organization (chromosomes/plasmids), structure * Gene/ncRNA content: number of genes, duplicates, size of gene families, etc * Sequence differences related to: gene evolution, regulatory evolution * RNA & protein abundance across species, for all RNAs/proteins 27