Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Cracking (chemistry) wikipedia , lookup
Marcus theory wikipedia , lookup
Woodward–Hoffmann rules wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Fischer–Tropsch process wikipedia , lookup
George S. Hammond wikipedia , lookup
Kinetic resolution wikipedia , lookup
Aza-Cope rearrangement wikipedia , lookup
Bottromycin wikipedia , lookup
Stille reaction wikipedia , lookup
Wolff rearrangement wikipedia , lookup
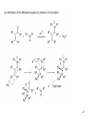
Diels–Alder reaction wikipedia , lookup
Asymmetric hydrogenation wikipedia , lookup
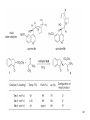
Elias James Corey wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
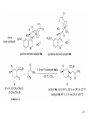
1,3-Dipolar cycloaddition wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
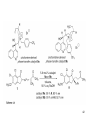
Hydroformylation wikipedia , lookup
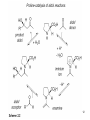
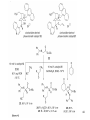
Ene reaction wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
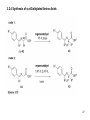
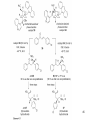
Discodermolide wikipedia , lookup
Aldol reaction wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
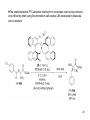
Petasis reaction wikipedia , lookup
Asymmetric induction wikipedia , lookup
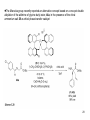
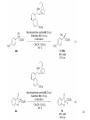
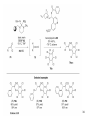
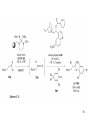
• Organocatalysis • • Albrecht Berkessel, Harald Groeger, Asymmetric Organocatalysis, 2005, Wiley-VCH, p409-435. M.T. Reetz, B. List, S. Jaroch, H. Weinmann (Editors), Ernst Schering Foundation Symposium Proceedings 2007-2 Organocatalysis • http://onlinelibrary.wiley.com/book/10.1002/3527604677 • http://www.springerlink.com/content/x7344h/#section=210299&page=1 • http://www.lib.ntnu.edu.tw/eresource/wiley_ebook.htm • Total Synthesis • • C. Bittner, A. S. Busemann, U. Griesbach, F. Haunert, W.-R. Krahnert, A. Modi, J. Olschimke, P. L. Steck, Organic Synthesis Workbook II, 2001 Wiley-VCH Verlag GmbH http://onlinelibrary.wiley.com/book/10.1002/3527600132 1 Asymmetric Organocatalysis Reference: Albrecht Berkessel, Harald Groeger, Asymmetric Organocatalysis, 2005, Wiley-VCH, p409-435. Tabular Survey of Selected Organocatalysts: Reaction Scope and Availability: 1) Intermolecular Michael addition 2) Mannich reaction 3) Intermolecular aldol reaction 4) Intramolecular aldol reaction 5) Aldol-related reactions (addition of nitrones) 6) Addition to N=N double bonds (a-amination of carbonyl compounds) 7) Addition to N=O double bonds (a-aminoxylation/ hydroxylation of carbonyl compounds L-Proline is commercially available in bulk quantities and represents an economically attractive amino acid organocatalyst. (D-Proline is commercially available, too.) Intramolecular α-alkylation of aldehydes L-Enantiomer commercially available 2 1) Intermolecular Michael addition 2) Intermolecular aldol reaction 3) [3+2]-Cycloadditions 4) Desymmetrization of meso-diols 5) Desymmetrization of meso-epoxides Preparation starting from L-proline in multi-step syntheses Mannich reaction Preparation starting from l-proline in multi-step syntheses 1) Mannich reaction 2) Intermolecular aldol reaction [6.2.1] Readily accessible, using L-penicillamine as starting material 3 Intramolecular aldol reaction Just as L-proline, L-phenylalanine is an economically attractive amino acid organocatalyst, readily available in bulk quantities. 1) Intermolecular Michael addition, including alkylation of heterocyclic aromatics and aniline derivatives 2) [4+2]-Cycloadditions: Diels-Alder reactions 3) [3+2]-Cycloadditions: Nitrone-based reactions Organocatalysts readily prepared from L-phenylalanine, methylamine and acetone or piraldehyde Intermolecular Michael addition Prepared from L-phenylalanine, methylamine and glyoxylic acid in a few steps 4 Tautomerization of enols Prepared from (+)-camphor in a multi-step syntheses Intramolecular Michael addition Commercially available in both enantiomeric forms in bulk quantities; economically attractive organocatalyst 5 1) α-Halogenation of carbonyl compounds 2) Intermolecular Michael addition (including cyclopropanation of enones, enoates etc.) 3) Intramolecular Michael addition 4) β-Lactam synthesis from imines and ketenes 5) β-Lactone synthesis from aldehydes and ketenes 6) Morita-Baylis-Hillman reaction 7) Hydrophosphonylation of aldehydes 8) Diels-Alder reaction 9) Desymmetrization of meso-anhydrides 10) Additions to prochiral ketenes 11) Desymmetrization of meso-diols 12) Desymmetrization of meso-epoxides All four natural cinchona alkaloids (R=H) are commercially available in large quantities. 6 dimeric cinchona alkaloid derivatives 1) α-Halogenation of carbonyl compounds 2) Carboethyoxycyanation of ketones 3) Desymmetrization of meso-anhydrides 4) (Dynamic) kinetic resolution of racemic Anhydrides Commercially available 1) Kinetic resolution of racemic alcohols by acylation 2) Desymmetrization of meso-diols by acylation Preparation starting from L-proline in multi-step syntheses L-proline-derived diamines 7 Chapter 1. Introduction: Organocatalysis – From Biomimetic Concepts to Powerful Methods for Asymmetric Synthesis Chapter 2. On the Structure of the Book, and a Few General Mechanistic Considerations 8 Reference: Albrecht Berkessel, Harald Groeger, Asymmetric Organocatalysis, 2005, Wiley-VCH, P10-11. 9 10 11 12 Pioneering work by Pracejus et al. in 1960, again using alkaloids as catalysts, afforded quite remarkable 74% ee in the addition of methanol to phenylmethylketene. In this particular reaction 1 mol% O-acetylquinine (10, Scheme 1.2) served as the catalyst 13 1971 saw the discovery of the Hajos–Parrish–Eder–Sauer–Wiechert reaction, i.e. the proline (1)-catalyzed intramolecular asymmetric aldol cyclodehydration of the achiral trione 11 to the unsaturated Wieland–Miescher ketone 12 (Scheme 1.3) [12, 13]. Ketone 12 is an important intermediate in steroid synthesis. 14 Surprisingly, the catalytic potential of proline (1) in asymmetric aldol reactions was not explored further until recently. List et al. reported pioneering studies in 2000 on intermolecular aldol reactions. For example, acetone can be added to a variety of aldehydes, affording the corresponding aldols in excellent yields and enantiomeric purity. 15 In the same year, MacMillan et al. reported that the phenylalanine-derived secondary amine 5 catalyzes the Diels–Alder reaction of a,b-unsaturated aldehydes with enantioselectivity up to 94% (Scheme 1.4). 16 A similarly remarkable event was the discovery of the cyclic peptide 14 shown in Scheme 1.5. In 1981 this cyclic dipeptide – readily available from l-histidine and l-phenylalanine – was reported, by Inoue et al., to catalyze the addition of HCN to benzaldehyde with up to 90% ee (Scheme 1.5). Again, this observation sparked intensive research in the field of peptide-catalyzed addition of nucleophiles to aldehydes and imines. 17 Also striking was the discovery, by Julia’, Colonna et al. in the early 1980s, of the poly-amino acid (15)-catalyzed epoxidation of chalcones by alkaline hydrogen peroxide. In this experimentally most convenient reaction, enantiomeric excesses > 90% are readily achieved (Scheme 1.6). 18 An example is the finding by Rawal et al. that hetero-Diels–Alder reactions – a classical domain of metal-based Lewis acids – can be effected with very high enantioselectivity by hydrogen bonding to chiral diols such as TADDOL (16, Scheme 1.7). 19 Chapter 3. Nucleophilic Substitution at Aliphatic Carbon 20 Phase-transfer catalysts are used and form a chiral ion pair of type 4 as an key intermediate. In a first step, an anion, 2, is formed via deprotonation with an achiral base; this is followed by extraction in the organic phase via formation of a salt complex of type 4 with the phase-transfer organocatalyst, 3. Subsequently, a nucleophilic substitution reaction furnishes the optically active alkylated products of type 6, with recovery of the catalyst 3. 21 3.1 α-Alkylation of Cyclic Ketones and Related Compounds The first example of the use of an alkaloid-based chiral phase-transfer catalyst as an efficient organocatalyst for enantioselective alkylation reactions was reported in 1984. Researchers from Merck used a cinchoninium bromide, 8, as a catalyst in the methylation of the 2-substituted indanone 7. The desired product, 9, a key intermediate in the synthesis of (+)-indacrinone was formed in 95% yield and with 92% ee (Scheme 3.2). 22 3.2 α-Alkylation of α-Amino Acid Derivatives Attachment of the 9-anthracenylmethyl group to a bridgehead nitrogen gave high enantioselectivity in the biscinchona-alkaloid-catalyzed dihydroxylation of olefins by osmium tetroxide, Corey and co-workers designed the structurally rigidified chiral quaternary ammonium salt 25 (Scheme 3.6). 23 The development of dimeric cinchona alkaloids as very efficient and practical catalysts for asymmetric alkylation of the N-protected glycine ester 18 was reported by the Park and Jew group. 24 3.2.2 Improving Enantioselectivity During Work-up Because of the high potential of alkaloid-based alkylations for synthesis of amino acids, several groups focused on the further enantiomeric enrichment of the products. In addition to product isolation issues, a specific goal of those contributions was improvement of enantioselectivity to ee values of at least 99% ee during downstream-processing (e.g. by crystallization). For pharmaceutical applications high enantioselectivity of >99% ee is required for optically active α-amino acid products. 25 3.2.3 Specific Application in the Synthesis of Non-natural Amino Acids The Maruoka group used their highly enantioselective, structurally rigid, chiral spiro catalysts of type 29 in the synthesis of L-Dopa ester (S)-40 and an analog thereof. Initial asymmetric alkylation in the presence of 1 mol% (R,R)-29 gave the intermediate (S)-20q in 81% yield and 98% ee (Scheme 3.16). Subsequent debenzylation provided the desired L-Dopa ester (S)-40 in 94% yield and 98% ee. This reaction has also already been performed on a gram-scale. 26 3.2.4 Synthesis of α,α-Dialkylated Amino Acids 27 The enantioselective PTC-alkylation starting from racemates can be also achieved very efficiently when using the ammonium salt catalyst, 29, developed by Maruoka and co-workers. 28 The Maruoka group recently reported an alternative concept based on a one-pot double alkylation of the aldimine of glycine butyl ester, 44a, in the presence of the chiral ammonium salt 29 as chiral phase-transfer catalyst 29 3.2.6 Solid-phase Syntheses The solid-phase synthesis of α-amino acids via alkaloid-catalyzed alkylation has been investigated by the O’Donnell group. The solid-phase based synthetic approach is particularly useful for rapid preparation of a-amino acids for combinatorial application. 30 3.4 Fluorination, Chlorination, and Bromination Reactions 3.4.1 Fluorination Reactions An enantioselective fluorination method with catalytic potential has not been realized until recently, when Takeuchi and Shibata and co-workers and the Cahard group independently demonstrated that asymmetric organocatalysis might be a suitable tool for catalytic enantioselective construction of C-F bonds. 31 32 3.4.2 Chlorination and Bromination Reactions A similar catalytic procedure for enantioselective formation of C-Br and C-Cl bonds has been reported recently by the Lectka group. 33 34 35 4. Nucleophilic Addition to Electron-deficient C=C Double Bonds 4.1 Intermolecular Michael Addition 36 37 One of these approaches consists in activating the acceptors – mostly α,β-unsaturated aldehydes (R4 = H) and ketones (R4 = alkyl) – by reversible conversion to a chiral iminium ion. As shown in Scheme 4.2a, reversible condensation of an α,β-unsaturated carbonyl compound with a chiral secondary amine provides a chiral α,β-unsaturated iminium ion. Face-selective reaction with the nucleophile provides an enamine which can either be reacted with an electrophile then hydrolyzed or just hydrolyzed to α,β-chiral carbonyl compound. The second approach is the enamine pathway. If the nucleophile is an enolate anion, it can be replaced by a chiral enamine, formed reversibly from the original carbonyl compound and a chiral secondary amine (Scheme 4.2b). 38 4.1.1. Intermolecular Michael Addition of C-nucleophiles 4.1.1.1 Chiral Bases and Phase-transfer Catalysis The first examples of asymmetric Michael additions of C-nucleophiles to enones appeared in the middle to late 1970s. In 1975 Wynberg and Helder demonstrated in a preliminary publication that the quinine-catalyzed addition of several acidic, doubly activated Michael donors to methyl vinyl ketone (MVK) proceeds asymmetrically. Enantiomeric excesses were determined for addition of a-tosylnitroethane to MVK (56%) and for 2-carbomethoxyindanone as the pre-nucleophile (68%). Later Hermann and Wynberg reported in more detail that 2-carbomethoxyindanone (1, Scheme 4.3) can be added to methyl vinyl ketone with ca 1 mol% quinine (3a) or quinidine (3b) as catalyst to afford the Michael-adduct 2 in excellent yields and with up to 76% ee. Because of their relatively low basicity, the amine bases 3a,b do not effect the Michael addition of less acidic pre-nucleophiles such as 4 (Scheme 4.3). However, the corresponding ammonium hydroxides 6a,b do promote the addition of the substrates 4 to methyl vinyl ketone under the same mild conditions, albeit with enantioselectivity not exceeding ca 20%. 39 40 41 42 43 44 45 4.1.1.2 Activation of Michael Acceptors by Iminium Ion Formation, Activation of Carbonyl Donors by Enamine Formation Cheap and readily available Lproline has been used numerous times for the intermediate and reversible generation of chiral iminium ions from a,b-unsaturated carbonyl compounds. For example, Yamaguchi et al. reported in 1993 that the rubidium salt of L-proline catalyzes the addition of di-iso-propyl malonate to the acyclic Michael acceptors 40a–c (Scheme 4.13), with enantiomeric excesses as high as 77%. 46