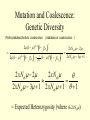Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Maximum parsimony (phylogenetics) wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Public health genomics wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Nutriepigenomics wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Mitochondrial DNA wikipedia , lookup
Genealogical DNA test wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
Oncogenomics wikipedia , lookup
Genetic drift wikipedia , lookup
Non-coding DNA wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Human genetic variation wikipedia , lookup
Genome editing wikipedia , lookup
Genetic engineering wikipedia , lookup
Genome evolution wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Frameshift mutation wikipedia , lookup
Koinophilia wikipedia , lookup
Designer baby wikipedia , lookup
Gene expression programming wikipedia , lookup
Genome (book) wikipedia , lookup
Helitron (biology) wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Computational phylogenetics wikipedia , lookup
History of genetic engineering wikipedia , lookup
Population genetics wikipedia , lookup
A30-Cw5-B18-DR3-DQ2 (HLA Haplotype) wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
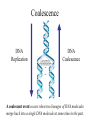
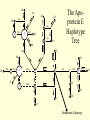
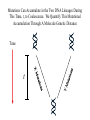
Coalescence DNA Replication DNA Coalescence A coalescent event occurs when two lineages of DNA molecules merge back into a single DNA molecule at some time in the past. COALESCENCE OF n COPIES OF HOMOLOGOUS DNA Gene Tree (all copies of homologous DNA coalesce to a common ancestral molecule) Coalescence in an Ideal Population of N with Ploidy Level x • Each act of reproduction is equally likely to involve any of the N individuals, with each reproductive event being an independent event • Under these conditions, the probability that two gametes are drawn from the same parental individual is 1/N • With ploidy level x, the probability of identity by descent/coalescence from the previous generation is (1/x)(1/N) = 1/(xN) • In practice, real populations are not ideal, so pretend the population is ideal but with an “inbreeding effective size” of an idealized population of size Nef; Therefore, the prob. of coalescence in one generation is 1/(xNef) Sample Two Genes at Random The probability of coalescence exactly t generations ago is the probability of no coalescence for the first t-1 generations in the past followed by a coalescent event at generation t: t 1 1 1 Prob.(Coalesce at t) 1 xN ef xNef Sample Two Genes at Random The average time to coalescence is: t 1 1 1 Expected(Time to Coalesce ) t1 xNef xNef t 1 xNef The variance of time to coalescence of two genes (ct) is the average or expectation of (t-xNef)2 : t 1 1 1 2 2 ct t xNef 1 xNef (xNef 1) x 2 N ef2 xNef xNef xNef t 1 Sample n Genes at Random n n! n(n 1) Number of pairs of genes = 2 2 (n 2)! 2! Prob.(coalescence in the previous gen.) n 1 n(n 1) = 2xN 2 xN Prob.(no coalescence in the previous gen.) n(n 1) =1 2xN Sample n Genes at Random n(n 1) t -1 n(n 1) Prob.(first coalescence in t generations) = 1 2xN 2xN n(n 1) t -1 n(n 1) 2xN E(time to first coalescence) = t1 2xN 2xN n(n 1) t 1 t 1 2 n(n 1) n(n 1) n(n 1) 2xN 2xN 2 1 t 1 1 4 N 2xN 2xN n(n 1) n(n 1) t 1 Sample n Genes at Random Once the first coalescent event has occurred, we now have n-1 gene lineages, and therefore we simply repeat all the calculations with n-1 rather than n. In general, the expected time and variance between the k–1 coalescent event and the kth event is: 2xN E(time between k 1 and k coalescent events) = (n k 1)(n k) 2xN 2xN 1 (n k 1)(n k) (n k 1)(n k) 2 k n1 E(time to coalescence of all n genes) = 2xN 2xN1 k 1 (n k 1)(n k) 1 n Sample n Genes at Random The average times to the first and last coalescence are: 2xNef/[n(n-1)] and 2xNef(1-1/n) •Let n = 10 and x=2, then the time span covered by coalescent events is expected to range from 0.0444Nef to 3.6Nef. •Let n = 100, then the time span covered by coalescent events is expected to range from 0.0004Nef to 3.96Nef. •These equations imply that you do not need large samples to cover deep (old) coalescent events, but if you want to sample recent coalescent events, large sample sizes are critical. •For n large, the expected coalescent time for all genes is 2xNef Sample n Genes at Random The variance of time to coalescence of n genes is: n 2xN 2xN 1 2 2 (n k 1)(n k) (n k 1)(n k) 1 4 x N (i)2 (i 1)2 k1 i 2 n1 •Note that in both the 2- and n-sample cases, the mean coalescent times are proportional to Nef and the variances are proportional to Nef2. •The Standard Molecular Clock is a Poisson Clock in Which the Mean = Variance. •The Coalescent is a noisy evolutionary process with much inherent variation that cannot be eliminated by large n’s; it is innate to the evolutionary process itself and is called “evolutionary stochasticity.” Generation Buri’s Experiment on Genetic Drift Number of Populations Fixed for bw Number of Populations Fixed for bw 75 1 0 0 2 0 0 3 0 0 4 0 1 5 0 2 6 1 3 7 3 3 8 9 10 11 12 13 14 15 16 17 18 19 5 5 7 11 12 12 14 18 23 26 27 30 5 6 8 10 17 18 21 23 25 26 28 28 0 2 4 6 8 10 12 14 16 18 20 22 24 26 28 30 32 Number of bw75 Alleles Fixation (Coalescence) Times in 105 Replicates of the Same Evolutionary Process Generation of Fixation Problem: No Replication With Most Real Data Sets. Only 1 Realization. Evolutionary Stochasticity Using the standard molecular clock and an estimator of of 10-8 per year, the time to coalescence of all mtDNA to a common ancestral molecule has been estimated to be 290,000 years ago (Stoneking et al. 1986). This figure of 290,000 however is subject to much error because of evolutionary stochasticity. When evolutionary stochasticity is taken into account (ignoring sampling error, measurement error, and the considerable ambiguity in ), the 95% confidence interval around 290,000 is 152,000 years to 473,000 years (Templeton 1993) -- a span of over 300,000 years! Coalescence of a mtDNA in an Ideal Population of N♀ haploids • Each act of reproduction is equally likely to involve any of the N♀ individuals, with each reproductive event being an independent event • Under these conditions, the probability that two gametes are drawn from the same parental individual is 1/N♀ • Under haploidy, the probability of identity by descent/coalescence from the previous generation is (1)(1/N♀) = 1/(N♀) • In practice, real populations are not ideal, so pretend the population is ideal but with an “inbreeding effective size” of an idealized population of size Nef♀; Therefore, the prob. of coalescence in one generation is 1/(Nef♀) Expected Coalescence Times for a Large Sample of Genes Mitochondrial DNA 2Nef♀=Nef (if Nef♀=1/2Nef) Y-Chromosomal DNA 2Nef♂=Nef (if Nef ♂=1/2Nef) X-Linked DNA 3Nef Autosomal DNA 4Nef 0 Locus MX1 FUT2 CCR5 Lactase FUT6 CYP1A2 Hb-Beta HFE MS205 EDN ECP MC1R PDHA1 RRM2P4 TNFSF5 AMELX APLX HS571B2 5 G6PD 7 Xq13.3 9 MSN/ALAS2 FIX MAO mtDNA Y-DNA TMRCA (In Millions of Years) Estimated Coalescence Times for 24 Human Loci Uniparental Haploid DNA Regions 8 X-Linked Loci 6 Autosomal Loci 4 3 2 1 Coalescence With Mutation Mutation Creates Variation and Destroys Identity by Descent Coalescence Before Mutation Prob.(coalescence before mutation ) Prob.(identity by descent) t 1 … … 1 1 = 1 (1 )2t xNef xNef Prob. of no Prob. of no Prob. of mutation in coalescence coalescence 2t DNA for t -1 gen. at gen. t replications Mutation Before Coalescence … … Mutation Prob.(mutation before coalescence ) 1 2t1 1 2 (1 ) xN ef t Mutation and Coalescence: Genetic Diversity Prob.(mutation before coalescence | mutation or coalescence ) 2 (1 ) 2 (1 )2t 1 1 1 xN ef t 1 2t 1 1 xN ef 1 xN ef t (1 )2t 1 1 xN ef t 1 2xNef 2 2xNef 3 1 2xNef 2 2xNef 2xNef 3 1 2xNef 1 1 = Expected Heterozygosity (where xNef) Gene Vs. Allele (Haplotype) Tree Gene Trees vs. Haplotype Trees Gene trees are genealogies of genes. They describe how different copies at a homologous gene locus are “related” by ordering coalescent events. The only branches in the gene tree that we can observe from sequence data are those marked by a mutation. All branches in the gene tree that are caused by DNA replication without mutation are not observable. Therefore, the tree observable from sequence data retains only those branches in the gene tree associated with a mutational change. This lower resolution tree is called an allele or haplotype tree. The allele or haplotype tree is the gene tree in which all branches not marked by a mutational event are collapsed together. Unrooted Haplotype Tree Haplotype trees are not new in population genetics; they have been around in the form of inversion trees since the 1930’s. The Inversion Tree Is Not Always The Same As A Tree of Species Or Populations, In This Case Because of: Transpecific Polymorphism Haplotype Trees Can Coalesce Both Within And Between Species The human MHC region fits this pattern; it takes 35 million years to coalesce, so humans and monkeys share polymorphic clades. Ebersberger et al. (2007) Estimated Trees From 23,210 DNA Sequences In Apes & Rhesus Monkey: Below Are The Numbers That Significantly Resolved the “Species Tree” Haplotype Trees ≠Species or Population Trees It is dangerous to equate a haplotype tree to a species tree. It is NEVER justified to equate a haplotype tree to a tree of populations within a species because the problem of lineage sorting is greater and the time between events is shorter. Moreover, a population tree need not exist at all. Homoplasy & The Infinite Sites Model • Homoplasy is the phenomenon of independent mutations (& many gene conversion events) yielding the same genetic state. • Homoplasy represents a major difficulty when trying to reconstruct evolutionary trees, whether they are haplotype trees or the more traditional species trees of evolutionary biology. • It is common in coalescent theory (and molecular evolution in general) to assume the infinite sites model in which each mutation occurs at a new nucleotide site. • Under this model, there is no homoplasy because no nucleotide site can ever mutate more than once. Each mutation creates a new haplotype. Homoplasy & The Infinite Sites Model QuickTime™ and a TIFF (Uncompressed) decompressor are needed to see this picture. QuickTime™ and a TIFF (LZW) decompressor are needed to see this picture. Homoplasy & The Infinite Sites Model The distributio n of polymorphi c nucleotide sites i n a 9.7 kb region of the human Lipoprotein Lipase gene over nucleotides associated with thre e known mutagenic motifs and all remainin g nucleotide positions. E. g., Apoprotein E Gene Region Exon 4 Exon 3 Exon 2 Exon 1 5361 5229B 5229 A 4951 4075 4036 3937 3701* 3673 3106 2907 2440 1998 1575 1522 1163 832 624 560 545 471 30 8 73 No recombination has been detected in this region. 5.5 5. 4.5 4. 3.5 3. 2.5 2. 1.5 1. 0.5 0. The Apoprotein E Haplotype Tree 21 14 24 560 26 22 0 0 9 4 560 1 2907 560 0 30 10 560 16 29 17 19 0 28 15 0 25 1998 7 5361 2 2440 3 2440 11 12 3937 6 3937 8 832 0 1998 23 0 3106 5 0 18 0 13 20 27 Chimpanzee (Outgroup) 31 A. Maximum Parsimony The Apo-protein E Haplotype Tree Use a Finite Sites mutation model that allows homoplasy. Can show that probability of homoplasy between two nodes increasing with increasing number of observed mutational differences. Therefore, allocate homoplasies to longer branches. Called “Statistical Parsimony” because you can use models to calculate the probability of violating parsimony for a given branch length. TCC 624 C T 560 624 1575 AC T T 560 624 1575 1575 C TTC 560 624 1575 T T A A O OR C 624 OR ATC T 560 624 1575 OR B. Statistical Parsimony TCC C 624 T 560 624 1575 ACT 560 624 1575 T 1575 C TTC 560 624 1575 T T A A O C OR 624 T ATC 560 624 1575 Homoplasy is still common, as shown by circled mutations. In this case, most of the homoplasy is associated with Alu sequences, a common repeat type in the human genome that is known to cause local gene conversion, which mimics the effects of parallel mutations. The Apoprotein E Statistical Parsimony Haplotype Tree Estimated Times To Common Ancestor (Method of Takahata et al. 2001) Dhc Nuc.Diff. Between Humans & Chimps Dh Nuc.Diff. Within Humans TMRCA = 12Dh/Dhc 6 Million Years Ago 3.2 The Apoprotein E Haplotype Coalescent 2.4 3937 1.6 4075 2440 1163 73 1998 5229B 308 4036 4951 3673 624 545 1522 471 2907 3106 3701 9 16 6 27 2 28 1 14 29 30 12 13 17 20 5 31 2 0.8 3 4 0 Years (x 105) Estimate the distribution of the age of the haplotype or clade as a Gamma Distribution (Kimura, 1970) with mean T=4N (or N for mtDNA) and Variance T2/(1+k) (Tajima, 1983) where k is the average pairwise divergence among present day haplotypes derived from the haplotype being aged, measured as the number of nucleotide differences. NOTE: VARIANCE INCREASES WITH INCREASING T AND DECREASING k! The Apo-protein E Haplotype Coalescent 3.2 2.4 3937 1.6 Years (x 105) 4075 f(t) 1163 2440 73 1998 0.8 5229B 308 4036 4951 3673 624 9 16 2 545 6 27 2 28 471 1522 2907 3701 1 3 Years (x 105) 14 29 30 3106 12 13 17 4 20 5 31 0 Because of Deviations From The Infinite Sites Model, Corrections Must Also be Made in How We Count the Number of Mutations That Occurred in The Coalescent Process. The Basic Idea of Coalescence Is That Any Two Copies of Homologous DNA Will Coalesce Back To An Ancestral Molecule Either Within Or Between Species Time t Mutations Can Accumulate in the Two DNA Lineages During This Time, t, to Coalescence. We Quantify This Mutational Accumulation Through A Molecule Genetic Distance Time t Molecule Genetic Distance = X + Y. If = the neutral substitution rate, then the Expected Value of X = t and the Expected Value of Y = t, So the Expected Value of the Genetic Distance = 2t Time t Complication: Only Under The Infinite Sites Model Are X+Y Directly Observable; Otherwise X+Y ≥ The Observed Number of Differences. Use Models of DNA Mutation To Correct For Undercounting Molecule Genetic Distance = X + Y = 2 t THE JUKES-CANTOR GENETIC DISTANCE Consider a single nucleotide site that has a probability of mutating per unit time (only neutral mutations are allowed). This model assumes that when a nucleotide site mutates it is equally likely to mutate to any of the three other nucleotide states. Suppose further that mutation is such a rare occurrence that in any time unit it is only likely for at most one DNA lineage to mutate and not both. Finally, let pt be the probability that the nucleotide site is in the same state in the two DNA molecules being compared given they coalesced t time units ago. Note that pt refers to identity by state and is observable from the current sequences. Then, pt+1 pt (1 )2 (1 pt )2 / 3 (1 2 )pt 2 (1 pt )/ 3 Molecule Genetic Distance = X + Y = 2 t THE JUKES-CANTOR GENETIC DISTANCE pt +1 (1 2)pt 2(1 pt )/3 p pt1 pt 2pt 2 (1 pt )/ 3 83 pt 23 Approximating the above by a differential equation yields: dpt 83 pt 23 dt extract 2t from the equation given above: pt 1 3e8t / 3/ 4 pt 14 43 e8 t / 3 3 4 e8 t / 3 pt 14 83 t n 43 pt 13 2t 43 n 43 pt 13 DJC Molecule Genetic Distance = X + Y = 2 t THE JUKES-CANTOR GENETIC DISTANCE DJC 43 n43 pt 13 The above equation refers to only a single nucleotide, so pt is either 0 and 1. Hence, this equation will not yield biologically meaningful results when applied to just a single nucleotide. Therefore, Jukes and Cantor (1969) assumed that the same set of assumptions is valid for all the nucleotides in the sequenced portion of the two molecules being compared. Defining as the observed number of nucleotides that are different divided by the total number of nucleotides being compared, Jukes and Cantor noted that pt is estimated by 1-. Hence, substituting 1- for pt yields: 2t 34 n1 43 DJC Molecule Genetic Distance = X + Y = 2 t THE KIMURA 2-PARAMETER GENETIC DISTANCE The Jukes and Cantor genetic distance model assumes neutrality and that mutations occur with equal probability to all 3 alternative nucleotide states. However, for some DNA, there can be a strong transition bias (e.g., mtDNA): Pyrimidines T C Purines A G where is the rate of transition substitutions, and 2is the rate of transversion substitutions. The total rate of substitution (mutation) Molecule Genetic Distance = X + Y = 2 t THE KIMURA 2-PARAMETER GENETIC DISTANCE Kimura (J. Mol. Evol. 16: 111-120, 1980) showed that GENETIC DISTANCE = Dt = 2()t = -1/2ln(1-2P-Q) - 1/4ln(1-2Q) where P is the observed proportion of homologous nucleotide sites that differ by a transition, and Q is the observed proportion of homologous nucleotide sites that differ by a transversion. Note that if (no transition bias), then we expect P = Q/2, so = P+Q = 3/ Q, or Q = 2/ . This yields the Jukes and Cantor distance, which is therefore a 2 3 special case of the Kimura Distance. If (large transition bias), as t gets large, P converges to 1/4 regardless of time, while Q is still sensitive to time. Therefore, for large times and with molecules showing an extreme transition bias, the distances depend increasingly only on the transversions. Therefore, you can get a big discrepancy between these two distances when a transition bias exists and when t is large enough. Molecule Genetic Distance = X + Y = 2 t Pyrimidines T C Purines A G You can have up to a 12 parameter model for just a single nucleotide (a parameter for each arrowhead). You can add many more parameters if you consider more than 1 nucleotide at a time. If distances are small (Dt ≤ 0.05), most alternatives give about the same value, so people mostly use Jukes and Cantor, the simplest distance. Above 0.05, you need to investigate the properties of your data set more carefully. ModelTest can help you do this (I emphasize help because ModelTest gives some statistical criteria for evaluating 56 different models -- but conflicts frequently arise across criteria, so judgment is still needed). LOOK AT YOUR DATA! Recombination Can Create Complex Networks Which Destroy the “Treeness” of the Relationships Among Haplotypes. Recombination is not Uniformly distributed in the human genome, but rather is Concentrated into “hotspots” that Separate regions of low to no Recombination. Region of Overlap of the Inferred Intervals Of All 26 Recombination and Gene Conversion Events Not Likely to Be Artifacts. Number of Recombination Events 18 16 LD in the human LPL gene Haplotype Trees can be Estimated for these Two regions, but not For the entire LPL region. 14 12 10 8 6 Significant |D’| 4 2 Non-significant |D’| 0 0 1000 2000 3000 4000 5000 (Templeton et al.,AMJHG 66: 69-83, 2000) 6000 7000 8000 9000 10000 Too Few Observations for any |D’| to be significant Because of the random mating equation: Dt=D0(1-r)t Linkage Disequilibrium Is Often Interpreted As An Indicator of the Amount of Recombination. This Is Justifiable When Recombination Is Common Relative To Mutation However, in regions of little to no recombination, the pattern of disequilibrium is determined primarily by the historical conditions that existed at the time of mutation, that is the Haplotype Tree. Note, AfricanAmericans Have More D Than Europeans & EA Because of Admixture: Not All D Reflects Linkage 0. 0.5 Apoprotein E Gene Region 1. 1.5 2. 2.5 3. 3.5 4. 4.5 5. 5.5 Exon 4 Exon 3 Exon 2 Exon 1 These Two Sites Show No Significant Disequilibrium in Any Sample 5361 5229B 5229 A 4951 4075 4036 3937 3701* 3673 3106 2907 2440 1998 1575 1522 1163 832 624 560 545 471 30 8 73 These Two Sites Are in Strong Disequilibrium in All Samples All Four Gametes Exist Because of Homoplasy, Not Recombination These mutations are Well separated in time And show little D 21 2907 560 545 1998 7 5361 2440 2 6 4036 3937 4951 471 5361 832 1998 560 5229B 3673 5 624 15 3106 4951 13 31 4075 560 10 624 4951 560 12 8 308 24 18 23 560 3 20 73 1163 560 27 17 4 560 1 29 3701 832 11 19 624 28 25 5361 624 624 The Apoprotein E Haplotype Tree 1522 30 1575 26 These haplotypes Are T at Site 832 & C At Site 3937 14 624 9 560 These haplotypes Are G at Site 832 & T At Site 3937 1575 22 16 These mutations are close in time And show much Disequilibrium