Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
DNA polymerase wikipedia , lookup
Extrachromosomal DNA wikipedia , lookup
Pathogenomics wikipedia , lookup
DNA sequencing wikipedia , lookup
DNA supercoil wikipedia , lookup
Genome evolution wikipedia , lookup
Transposable element wikipedia , lookup
Epigenomics wikipedia , lookup
Point mutation wikipedia , lookup
Nucleic acid double helix wikipedia , lookup
Human genome wikipedia , lookup
Non-coding DNA wikipedia , lookup
Zinc finger nuclease wikipedia , lookup
History of genetic engineering wikipedia , lookup
Molecular Inversion Probe wikipedia , lookup
Microevolution wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Molecular cloning wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Designer baby wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
Deoxyribozyme wikipedia , lookup
Metagenomics wikipedia , lookup
Genomic library wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Genome editing wikipedia , lookup
Helitron (biology) wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
SNP genotyping wikipedia , lookup
Microsatellite wikipedia , lookup
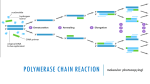
Hivebench Example Created by noelsilvadom BIO 502 Lab (Spring, 2014) 04/10/14 (Techniques in Genomics & Proteomics) DESIGNING PRIMERS EXPERIMENT OBJECTIVE: To design primers for cloning human Rheb1, Rheb2 and TCTP into a mammalian expression vector (pCMV-BICEP4). PRINCIPLE: A primer is a short synthetic oligonucleotide which is used in many molecular techniques from PCR to DNA sequencing. These primers are designed to have a sequence which is the reverse complement of a region of template or target DNA to which we wish the primer to anneal. When designing primers for PCR, sequencing or mutagenesis it is often necessary to make predictions about these primers, for example melting temperature (Tm) and propensity to form dimers with itself or other primers in the reaction. Primer length and sequence are of critical importance in designing the parameters of a successful amplification: the melting temperature of DNA duplex increases both with its length, and with increasing (G+C) content: a simple formula for calculation of the Tm is Tm = 4(G + C) + 2(A + T) oC. Thus, the annealing temperature chosen for a PCR depends directly on length and composition of the primer(s). One should aim at using an annealing temperature (Ta) about 5oC below the lowest Tm of the pair of primers to be used. One consequence of having too low a Ta is that one or both primers will anneal to sequences other than the true target, as internal single-base mismatches or partial annealing may be tolerated: this is fine if one wishes to amplify similar or related targets; however, it can lead to "non-specific" amplification and consequent reduction in yield of the desired product, if the 3'-most base is paired with a target. A consequence of too high a Ta is that too little product will be made, as the likelihood of primer annealing is reduced; another and important consideration is that a pair of primers with very different Tas may never give appreciable yields of a unique product, and may also result in inadvertent "asymmetric" or single-strand amplification of the most efficiently primed product strand. Annealing does not take long: most primers will anneal efficiently in 30 sec or less, unless the Ta is too close to the Tm, or unless they are unusually long. The optimum length of a primer depends upon its (A+T) content, and the Tm of its partner if one runs the risk of having problems such as described above. Apart from the Tm, a prime consideration is that the primers should be complex enough so that the likelihood of annealing to sequences other than the chosen target is very low. For example, there is a ¼ chance (4-1) of finding an A, G, C or T in any given DNA sequence; there is a 1/16 chance (4-2) of finding any dinucleotide sequence (e.g. AG); a 1/256 chance of finding a given 4-base sequence. Thus, a sixteen base sequence will statistically be present only once in every 416 bases (=4 294 967 296 or 4 billion): this is about the size of the human or maize genome, and 1000x greater than the genome size of E. coli. Thus, the association of a greater-than-17-base oligonucleotide with its target sequence is an extremely sequence-specific process, far more so than the specificity of monoclonal antibodies in binding to specific antigenic determinants. Consequently, 17-mer or longer primers are routinely used for amplification from genomic DNA of animals and plants. Too long a primer length may mean that even high annealing temperatures are not enough to prevent mismatch pairing and non-specific priming. 1. Primer Length: It is generally accepted that the optimal length of PCR primers is 18-22 bp. This length is long enough for adequate specificity, and short enough for primers to bind easily to the template at the annealing temperature. 2. Melting Temperature: Melting Temperature (Tm) by definition is the temperature at which one half of the DNA duplex will dissociate to become single stranded and indicates the duplex stability. Primers with melting temperatures in the range of 52-58 oC generally produce the best results. Primers with melting temperatures above 65oC have a tendency for secondary annealing. The GC content of the sequence gives a fair indication of the Tm. 3. Primer annealing temperature: The primer melting temperature is the estimate of the DNA-DNA hybrid stability and critical in determining the annealing temperature. Too high Ta will produce insufficient primer-template hybridization resulting in low PCR product yield. Too low Ta may possibly lead to non-specific products caused by a high number of base pair mismatches. Mismatch tolerance is found to have the strongest influence on PCR specificity. Ta = 0.3 x Tm (primer) + 0.7 Tm (product) – 14.9 where, Tm (primer) = Melting Temperature of the primers Tm (product) = Melting temperature of the product 4. GC Content: The GC content (the number of G's and C's in the primer as a percentage of the total bases) of primer should be 40-60%. 5. GC Clamp : The presence of G or C bases within the lat five bases from the 3' end of primers (GC clamp) helps promote specific binding at the 3' end due to the stronger bonding of G and C bases. More than 3 G's or C's should be avoided in the last 5 bases at the 3' end of the primer. 6. Secondary Structures: Presence of the secondary structures produced by intermolecular or intramolecular interactions can lead to poor or no yield of the product. They adversely affect primer template annealing and thus the amplification. They greatly reduce the availability of primers to the reaction. i) Hairpins: It is formed by intramolecular interaction within the primer and should be avoided. ii) Self Dimer: is formed by intermolecular interactions between the two (same sense) primers, where the primer is homologous to itself. Generally a large amount of primers are used in PCR compared to the amount of target gene. When primers form intermolecular dimers much more readily than hybridizing to target DNA, they reduce the product yield. iii) Cross Dimer: Cross dimers are formed by intermolecular interaction between sense and antisense primers, where they are homologous. 7. Repeats: A repeat is a di-nucleotide occurring many times consecutively and should be avoided because they can misprime. For example: ATATATAT. A maximum number of dinucleotide repeats acceptable are 4 di-nucleotides. 8. Runs: Primers with long runs of a single base should generally be avoided as they can misprime. For example, AGCGGGGGATGGGG has runs of base 'G' of value 5 and 4. A maximum number of runs accepted are 4bp. Target genes: Human Rheb1, Rheb2 and TCTP Cloning Vector: pCMV-BICEP-4 • • • • • • • • Obtain mRNA sequence of the gene listed above (explore NCBI) Determine the coding sequence and highlight it Look at the vector sequences and analyze the multiple cloning sites VERY CAREFULLY Determine suitable sites into which the gene can be cloned (you will have to do a restriction enzyme map of each of the gene using a program like NEB cutter) Find RE sites that do NOT cut the gene- you will use these for cloning into the vector Design the forward (5’) and reverse (3’) primers for the gene (keeping in mind the length and GC content of each primer)- remember to incorporate the RE sites in the primers for cloning; ENSURE THAT THE READING FRAME IS CORRECT Submit primer sequences to me (Primers will be ordered from Invitrogen) Once primers are received, proceed with PCR to amplify the gene