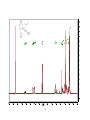
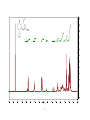
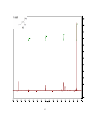
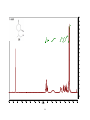
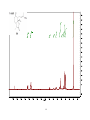
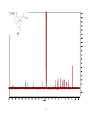
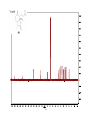
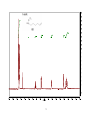
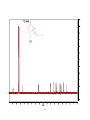
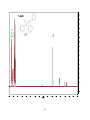
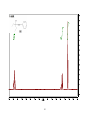
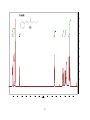
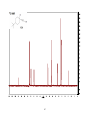
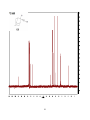
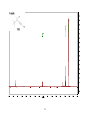
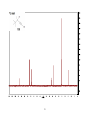
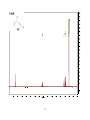
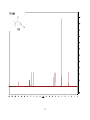
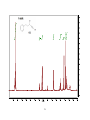
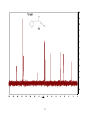
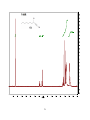
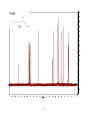
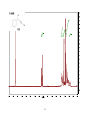
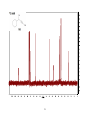
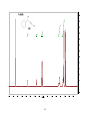
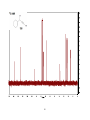
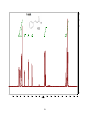
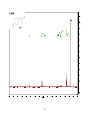
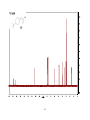
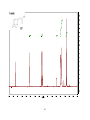
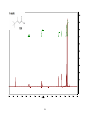
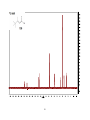
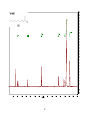
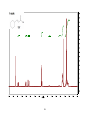
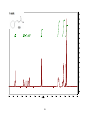
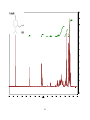
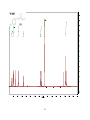
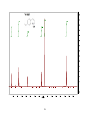
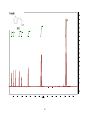
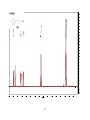
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Woodward–Hoffmann rules wikipedia , lookup
Bottromycin wikipedia , lookup
George S. Hammond wikipedia , lookup
Fischer–Tropsch process wikipedia , lookup
Asymmetric induction wikipedia , lookup
Elias James Corey wikipedia , lookup
Kinetic resolution wikipedia , lookup
Enantioselective synthesis wikipedia , lookup
Diels–Alder reaction wikipedia , lookup
1,3-Dipolar cycloaddition wikipedia , lookup
Stille reaction wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Polythiophene wikipedia , lookup
Tiffeneau–Demjanov rearrangement wikipedia , lookup
Aza-Cope rearrangement wikipedia , lookup
Discodermolide wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
Artemisinin wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
Ene reaction wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Wolff rearrangement wikipedia , lookup
Petasis reaction wikipedia , lookup
Hydroformylation wikipedia , lookup
FLORIDA STATE UNIVERSITY COLLEGE OF ARTS AND SCIENCES ORGANIC SYNTHESIS AND METHODOLOGY RELATED TO THE MALARIA DRUG ARTEMISININ By DOUGLAS A. ENGEL A Dissertation submitted to the Department of Chemistry and Biochemistry in partial fulfillment of the requirements for the degree of Doctor of Philosophy Degree Awarded: Summer Semester, 2009 The members of the committee approve the dissertation of Douglas A. Engel defended on June 30th, 2009. __________________________________ Gregory B. Dudley Professor Directing Dissertation ___________________________________ Thomas Keller Outside Committee Member __________________________________ Marie Krafft Committee Member __________________________________ Lei Zhu Committee Member __________________________________ Geoffery Strouse Committee Member Approved: _____________________________________ Joesph Schlenoff, Chair, Department of Chemistry The Graduate School has verified and approved the above-named committee members. ii This manuscript is dedicated to my family and friends who have supported me through out my life. iii ACKNOWLEDGMENTS Academically, I could not have achieved what I have without the guidance of Dr. Gregory Dudley. It the early years, he was an invaluable source of information and gave me the opportunity to work on some amazing projects. The reactions course he taught my first year in graduate school was probably the most educational course I have ever taken. In the later years, despite my protests, he has forced me to draw my own conclusions and rarely relinquishes an answer to a question I am capable of determining the answer to. I am truly appreciative of this forced evolution from a graduate student to an independent scientist. On the personal level, I have too many thank yous to include in this short acknowledgements page. So thank you to everyone who has bettered my life over the last 5 years. This includes my family who has always supported me, whether they agreed with me or not, and one of the best group of friends one could ask for. So thank you; Mom, Dad, Tiffany, Sean, Van, Shawn, Sam, and everyone else. Financially, I have to thank my mother once again. Her answer was always ―just put it on the credit card.‖ I am guessing she has her eye on some sweet rewards program prize and is just using me to get ever closer to obtaining the coveted pink American Express embroidered teddy bear. Seriously, I am truly appreciative. I also have to thank the Bovay Foundation and the MDS Research Foundation whose financial support made life on the meek graduation stipend a little more bearable. In addition, the completion of this manuscript would have been impossible without the army of editors that so graciously agreed to proofread this document. They include Dr. Mark Kearley, Sami Tlias, Kerry Gilmore, Mike Rosana, and Cece O’Leary. Special thanks go to my sister Tiffany Harris who agreed to proofread a document in a foreign language, and Dave Jones and Dr. Dudley who edited the entire document. iv TABLE OF CONTENTS List of Tables ................................................................................ List of Figures ............................................................................... Abstract .................................................................................... v vi x 1. Total Synthesis of (+)-Dihydro-epi-deoxyarteannuin B and Related Studies ........................................................................ 001 Chapter 1: History of Malaria ..................................................... Chapter 2: Artemisinin: The Last Line of Defense Against Malaria .................................................................... Chapter 3: Synthesis of (+)-Dihydro-epi-deoxyarteannuin B and ()-Dihydroartemisinic Acid…………………….. Chapter 4: Experimental: (+)-Dihydro-epi-deoxyarteannuin B... 001 2. The MeyerSchuster Reaction and Beyond .............................. 094 Chapter 5: Evolution of the MeyerSchuster Reaction .............. Chapter 6: Lewis Acid-Catalyzed MeyerSchuster Reactions: Methodology for the Olefination of Aldehydes and Ketones .................................................................. Chapter 7: MeyerSchuster Experimental……………………… 094 REFERENCES ............................................................................. 224 BIOGRAPHICAL SKETCH ............................................................. 241 v 011 020 038 114 144 LIST OF TABLES Table 1: Allylation of Menthone and Silyloxymenthone 43 ........................ 028 Table 2: Mo-Au Combo Catalysis for Rearrangement of E into α,β-unsaturated Ketones F ......................................................... 101 Table 3: Selected Examples of [(ItBu)AuCl]/AgSbF6 Catalyzed Rearrangement ........................................................................... 106 Table 4: Solvent Screening With AuCl3 Catalyst ....................................... 118 Table 5: Screening of Water Equivalents .................................................. 119 Table 6: Catalyst Loading ......................................................................... 120 Table 7: Initial Screening of Substrates Using Oxygen Activated Alkynes 124 Table 8: Catalyst Loading and Optimization .............................................. 124 Table 9: Olefination of Hindered Ketones ................................................. 126 Table 10: Temperature Effect on (E/Z)-Selectivity .................................... 127 Table 11: MeyerSchuster Reaction of Ethoxyalkynyl Carbinols .............. 128 Table 12: Screening Alternative Catalysts ................................................ 133 Table 13: Effect of Additives ..................................................................... 134 Table 14: Ethoxy Acetylene Addition ........................................................ 135 Table 15: Scandium and Copper Rearrangements ................................... 136 Table 16: Two-Stage Olefination of Ketones and Aldehydes .................... 138 Table 17: Incorporation of External Alcohol Additive ................................. 142 vi LIST OF FIGURES Figure 1: Estimated Incidence of Malaria per 1000 Population, 2006........ 002 Figure 2: Global Distribution of Per Capita GDP ....................................... 003 Figure 3: Overview of Antimalarial Drugs .................................................. 005 Figure 4: Plasmodium falciparum Cycle Within Humans ........................... 006 Figure 5: Model For Formation of the Non-Covalent Heme-Chloroquine Complex .................................................................................... 007 Figure 6: Global Status of Resistance to Chloroquine and Sulphadoxine/ pyrimethamine, the Two Most Widely Used Antimalarial Drugs. 008 Figure 7: The Structure of Artemisinin and its Derivatives ......................... 008 Figure 8: General Reactivity of Artemisinin After Reductive Activation of The Endoperoxide Function ....................................................... 009 Figure 9: Artemisinin and Related Compounds ......................................... 011 Figure 10: Artemisinin is a Natural Product Extracted from Artemisia annua or Sweet Wormwood ................................................................ 012 Figure 11: Schematic Representation of the Engineered Artemisinic Acid Biosynthetic Pathway in S. cerevisiae ...................................... 015 Figure 12: Early Approaches Toward Artemisinin ..................................... 016 Figure 13: Construction of the Schmid and Hofheinz Singlet Oxygen Precursor 17 ............................................................................ 017 Figure 14: Schmid and Hofheinz End Game Strategy ............................... 017 Figure 15: First Example of Dihydroartemisinic acid 21 being converted into Artemisinin 1 ..................................................................... 018 Figure 16: Our Proposal for Generating the Key Endoperoxide ................ 019 Figure 17: Our Synthetic Targets .............................................................. 020 vii Figure 18: Eq (1) Ravindranathan Intramolecular Diels-Alder, Eq (2) Meinwald Intramolecular Diels-Alder ....................................... 021 Figure 19: Avery and Co-Workers’ Strategy to Avoid Epimerization.......... 021 Figure 20: Electrocyclic Stategies to the Decalin Ring System ................. 022 Figure 21: Other Approaches Towards the Decalin Ring System ............. 023 Figure 22: Our Retro-Synthetic Analysis ................................................... 024 Figure 23: The StorkJung Vinylsilane ..................................................... 024 Figure 24: The Original Synthesis of the StorkJung Vinylsilane .............. 025 Figure 25: Early Modifications of the StorkJung Synthesis ...................... 026 Figure 26: Dudley Lab Succint Method for Preparation of StorkJung Vinylsilane 16........................................................................... 026 Figure 27: Hydroboration of Bulk Isopulegol vs. Pure Isopulegol .............. 027 Figure 28: Conversion of Diol 56 to Ketone 43 ......................................... 027 Figure 29: Vinyl Addition to Silyloxymenthone 59 ..................................... 029 Figure 30: Elaboration to First Metathesis Precursor 6322 ..................... 030 Figure 31: Ring Closing Metathesis (RCM) Strategies .............................. 031 Figure 32: Proposed Synthesis of Dihydroartemisinic Acid 21 .................. 032 Figure 33: Olefination Attempts on Menthone and Menthone Derivatives . 033 Figure 34: Proposed Olefination Strategy ................................................. 034 Figure 35: Implementation of Our Olefination Strategy Towards Dihydroartemisinic Acid 21....................................................... 035 Figure 36: Strategy Towards Dihydroartemisinic Acid 21 .......................... 094 Figure 37: Atom-Economical Carbonyl Olefination Strategies ................... 095 Figure 38: Meyer and Schuster’s Rearrangement..................................... 096 Figure 39: Competing Rupe Rearrangement ............................................ 097 viii Figure 40: Hoffmann-La Roche Inc. Application of Vanadate Catalyst ...... 097 Figure 41: Proposed Orthovanadate Mechanism ...................................... 098 Figure 42: Trapping of Oxo-Vanadate Allene R ........................................ 099 Figure 43: Narasaka Lab Perrhenate(VII)/p-Toluenesulfonic Acid System 099 Figure 44: Vidari’s Lab Extension of Toste’s Methodology ........................ 100 Figure 45: Lewis Basic Sites on a Propargyl Alcohol ................................ 102 Figure 46: ―Soft Lewis Acid Activation ....................................................... 102 Figure 47: Two-Step vs. Single-Step Preparation of α,β-Enones via Functinalization of the Propargyl Alcohol ................................. 103 Figure 48: Preparation of β-Monosubstituted (E)-α,β-Unsaturated Ketones ................................................................................... 104 Figure 49: Preparation of β,β-Disubstituted α,β-Unsaturated Ketones ...... 104 Figure 50: Proposed Mechanism for Au(PPh3)NTf2-Catalyzed Rearrangement ........................................................................ 105 Figure 51: SN2’ Type Addition of Au Catalyst ............................................ 107 Figure 52: Chung’s Proposed Cumulene Intermediate ............................. 108 Figure 53: Attempted Regioselective Hydration ........................................ 108 Figure 54: Optimized Hg(OTf)2 Rearrangement ........................................ 109 Figure 55: Hg(OTf)2-Catalyzed Hydration of Ethoxypropargyl Acetates .... 110 Figure 56: α,β-Unsaturated Amide Formation ........................................... 110 Figure 57: Proposed Mechanism for the Ru-Catalyzed Isomerization of Propargyl Alcohols into α,β-Unsaturated Aldehydes ................ 111 Figure 58: Application of the MeyerSchuster Reaction in Total Synthesis ................................................................................. 112 Figure 59: Two-Step Olefination Strategy ................................................. 114 Figure 60: Cr(VI) Rearrangements............................................................ 115 ix Figure 61: Initial Oxo-philic Approaches to the MeyerSchuster Rearrangement ........................................................................ 116 Figure 62: Switch to Triphenyl Propargyl Alcohol 117 ............................... 117 Figure 63: Initial Gold(III) Experiment ....................................................... 117 Figure 64: Initial Substrate Screening with MeCN ..................................... 121 Figure 65: Initial Substrate Screening with CH2Cl2 .................................... 122 Figure 66: Use of Electron-Rich Ethoxyacetylene ..................................... 123 Figure 67: Qualitative Screening for Optimal Conditions ........................... 127 Figure 68: Gold Salts vs. Acid Catalysts ................................................... 130 Figure 69: Hypothesis on the MeyerSchuster Reaction Pathway ............ 131 Figure 70: Incorporation of External Alcohol Additive ................................ 131 Figure 71: Functional Group Compatability ............................................... 137 Figure 72: Test for trans-Esterification ...................................................... 140 Figure 73: Revised Mechanistic Hypothesis of Scandium(III) Salts ........... 141 x ABSTRACT Malaria is a global epidemic, resulting in the deaths of nearly one million people every year. Part 1 of this dissertation will focus on the history of Malaria and ways to combat this devastating disease. Artemisinin has emerged as the drug of choice for treatment of malaria due to its effectiveness against all strains of the malaria parasite. Access to artemisinin through isolation, bio-engineering, and chemical synthesis will be described. Our attempts to access the artemisinin family of anti-malarials through the total synthesis of dihydro-epi-deoxyarteannuin B and dihydroartemisinic acid will be discussed fully. Key features of the syntheses will include alkylation of menthone derivatives using Noyori’s zincate enolate method and nucleophilic addition to a hindered ketone using either organocerium or acetylide nucleophiles. In addition, two alternative olefin metathesis approaches are described for the final cyclization. Problems associated with the olefination of a key intermediate in our efforts toward dihydroartemisinic acid led us to develop a two-step olefination of ketones and aldehydes. Part II will discuss this olefination strategy which consists of acetylide addition to generate a propargyl alcohol followed by a MeyerSchuster rearrangement to the corresponding α,β-enone. A complete history of the MeyerSchuster rearrangement will be presented, highlighting the short comings of the method prior to our work. A complete overview of our research pertaining to the MeyerSchuster reaction will be given. Key topics will include development of a Au(III)-catalyzed rearrangement of propargyl ethynyl ethers into α,β-unsaturated esters and its use in the olefination of hindered ketones, efforts to control the (E/Z)-selectivity of the MeyerSchuster rearrangement, and the search for more affordable catalysts. xi PART 1: TOTAL SYNTHESIS OF (+)-DIHYDRO-EPI-DEOXYARTEANNUIN B AND RELATED STUDIES CHAPTER 1 HISTORY OF MALARIA Malaria is the most devastating tropical parasitic disease in the world and is ranked in the top three of communicable diseases it terms of deaths. 1 The size and the scope of the malaria pandemic are astounding with the World Health Organization (WHO) reporting an estimated 247 million cases in 2006. Of those cases, nearly 1 million resulted in death, mostly in children under the age of 5.2 It is estimated that a child dies from malaria every 40 seconds, resulting in the loss of thousands of adolescents every day.1 Malaria is so deadly that many experts contend it has contributed significantly to the proliferation of the sickle-cell trait. When inherited from both parents the sickle-cell mutation is fatal, yet when inherited from only one parent the trait is protective against malaria. Natural selection would tend to remove the sickle cell trait from the gene pool, but the risk of death from malaria is so severe that the sickle cell trait remains a prominent and needed mutation.3,4 With the staggering number of malaria cases every year, malaria may be perceived as a global problem. In reality, malaria is centered in tropical regions and extends into subtropical regions across five continents putting 3.3 billion people at risk. Currently, 109 countries are considered to have malaria epidemics with nearly half of those countries being located in Africa2 (Figure 1). Unlike tropical areas, temperate regions possess distinct seasons, and their cold winters are a key element in the fight against malaria. The life cycle of the malaria parasite is highly temperature dependant. In order to become infectious to other individuals, the parasite inside the mosquito must undergo a highly specific life cycle change. The amount of time necessary for this transformation is inversely proportional to the ambient temperature. As 1 average temperatures decline, the time necessary for the life cycle change approaches the life span of the mosquito, leaving a much smaller window for transmission. At temperatures of 16 o C and below malaria parasite development ceases completely. 1,5,6 Seasonal temperatures have the most pronounced affect on malaria, but other climate factors such as humidity and rainfall must also be considered when assessing transmission rates.1 Figure 12: Estimated Incidence of Malaria per 1000 Population, 2006 The correlation between malaria and poverty may also be responsible for the malaria epidemic in the tropical regions. Areas of the world where malaria is prevalent have lower percapita gross domestic products (GDP) than areas that are unaffected (Figure 2). From 1965 to 1990, countries with major segments of their populations living in regions at high risk for malarial transmittance had an average growth of per-capita GDP of 1.9 % less than that of countries with little malaria risk.7 2 Figure 21: Global Distribution of Per Capita GDP. The correlation between poverty and malaria is undisputable; however, there is debate as to whether malaria causes poverty or poverty results in the proliferation of malaria. Most likely it is a combination of both.1,2 Wealthier nations possess the resources to initiate government control programs such as the draining of swamplands and other breeding grounds, along with large scale spraying of pesticides. General urban development including improved housing significantly reduces individuals’ exposure to mosquitoes and the malaria parasite. These factors combined with personal expenditures on bednets and household insecticides led to the elimination of malaria in wealthier nations with temperate climates by the 1950’s. Although economic development and wealth are important factors in controlling malaria, they are not enough by themselves. Wealthy countries with high year-round temperatures still suffer from malaria and are unable to eliminate the disease. 1 Malaria has much broader social and economic impacts than simply causing a lower average growth in GDP in affected countries. Malaria infected regions not only have slower growing economies, but they are also forced to spend a significant amount of their GDP on combating the disease. These costs include personal and private medical costs as well as loss 3 of income due to illness and death. This financial burden is most severe for people in the lowest income brackets. With most of their disposable income going towards prevention and treatment of malaria there is little money for schooling, migration, or savings resulting in serious social costs. The lack of proper education is a major obstacle in combating not only malaria but also sexually transmitted diseases like HIV. Limited female education and low availability of birth control, combined with high infant mortality rates, have led to high fertility rates. In order to assure a certain number of surviving heirs, couples are forced to have large numbers of children. The net result is large population growth in poor malaria stricken countries, causing a magnification of the problem.8-11 Despite the obstacles facing those areas impacted by malaria, modern medicine has afforded ways to combat this disease. Currently, there are three major classes of anti-malarial drugs—the quinoline family, the artemisinin family, and all other antimalarials12 (Figure 3). The need for so many classes of antimalarial drugs originates from the ability of the malaria parasite to develop drug resistance. The two most widely used antimalarial drugs are chloroquine and the antifolate sulphadoxine/pyrimethamine. Chloroquine is a member of the quinoline family of antimalaria drugs. The quinoline family of compounds draws their origin from quinine, a centuries old malaria treatment isolated from the bark of Cinchona.13 While quinine has excellent antimalarial activity, it requires multiple doses per day over 7 days. This long treatment schedule has led to problems associated with toxicity. Structural elucidation of quinine and medicinal chemistry studies led to derivatives including chloroquine and amodiaquine, which only have to be administered over 3 days, reducing the chances of a toxic build-up in the body.13 This short treatment period, combined with the malaria parasite’s low propensity to develop resistance to it, have made chloroquine the drug of choice for many years.14 4 Figure 312: Overview of Antimalarials Drugs While there are four different species of the malaria parasite, (Plasmodium vivax, Plasmodium ovale, Plasmodium malariae, and Plasomodium falciparum) Plasmodium falciparum is the strain that is responsible for severe malaria that results in death.15 Plasmodium falciparum’s life cycle within the human body consists of several distinct phases. Initially, the parasite takes up residence in hepatocytes, where it quickly develops and causes the release of merozoites. The newly released merozoites then invade erythrocytes. Once inside the erythrocytes the malaria parasite goes through a series of evolutions resulting in the eventual release of more merozoites, which then infect more cells (Figure 4).15 Malaria symptoms manifest during the phase of the Plasmodium’s life cycle in which it inhabits the erythrocyte. The parasites’ survival at this stage is dependent on modifications to the cells metabolic processes. These modifications, while ensuring the parasites’ survival, also allow for drugs to differentiate between infected cells and healthy cells. This makes them 5 susceptible to chemotherapeutic attack. While Plasmodium is present in the host erythrocyte, it degrades up to 80% of the hemoglobin present. 16 As a result, large amounts of Fe(II) heme are released and are subsequently oxidized to Fe(III) hematin. The hematin then aggregates to form a pigment called hemozoin.16 Chloroquine’s activity is believed to lie in its ability to prevent hemozoin formation by binding with heme through stacking of their planar aromatic structures.17 These chloroquine-heme complexes are toxic to the parasite, though the exact source of their toxicity is under debate (Figure 5).18-19 Figure 415: Plasmodium falciparum Cycle Within Humans Heptaocytes: a cell of the main tissue of the liver Erythrocytes: red blood cells Merozoites: a daughter cell of a protozoan parasite 6 Figure 5: Model For Formation of the Non-Covalent Heme-Chloroquine Complex While the ability of the malaria parasite to develop resistance to chloroquine is quite low, widespread usage over a long time period has resulted in increased resistance in many areas of the world. The recent globalization of chloroquine resistant malaria parasites has brought to the forefront the need for alternative treatments (Figure 6). Other drugs from the quinoline family can be substituted for chloroquine in many cases, but due to their similar structure and biological activity, resistance to these drugs tends to develop rapidly. 14 Since their isolation in the early 1970’s, the artemisinin family of antimalarials have seen unprecedented growth in their frequency of use.2,20,21 This rapid switch from the commonly used quinolines to the artemisnins is due to the aretemisinins’ remarkable ability to combat quinoline resistant strains of the malaria parasite. To date, there have been no examples of any strain of the malaria parasite showing resistance to the artemisinin family of drugs.2,22 7 Figure 612. Global Status of Resistance to Chloroquine and Sulphadoxine/pyrimethamine, the Two Most Widely Used Antimalarials Drugs. Data are From the World Heatlh Organization Their high level of activity against all strains of the malaria parasite is attributed to their mode of action. Like the quinoline family of drugs, the artemisinin family of drugs are believed to derive their activity through interaction with Fe(II) heme, but by a drastically different pathway,15 related to the unique endoperoxide bridge of the artemisinins (Figure 7). H H3C O O O HO CH3 H H3C H CH3 CH3 O O O HO 9a R = H, Dihydroartemisinin 9b R = Me, Artemether H CH3 O OR artemisinin A 9c R = Et, Arteether 10 R = CO(CH2)2COONa, Artesunate Figure 7: The Structure of Artemisinin and its Derivatives 8 It is generally accepted that the endoperoxide bridge is the source of artemisinin’s activity. Derivatives lacking the peroxide bridge (deoxyartemisinin) have shown no activity against the malaria parasite.23 There is believed to be an interaction between the peroxidic bond in artemisinin and the Fe(II) heme generated from the parasites degradation of hemoglobin within the erythrocyte. The result is cleavage of the peroxide bridge yielding carbon-centered free radicals15 (Figure 8). Figure 815. General Reactivity of Artemisinin After Reductive Activation of the Endoperoxide Function 9 The free radicals rapidly react, causing destruction of anything in the immediate proximity. The artemisinins can also target the malaria parasite in stages of its life cycle outside of the erythrocyte through other mechanisms.15 In addition, the artemisinins pose little threat to uninfected cells. Micromolar concentrations are necessary to induce toxicity to mammalian cells, while only nanomolar concentrations are required to kill the malaria parasite. These low dosage requirements have resulted in very few instances of negative side-effects from treatment.24 The only shortcoming of the artemisinin family of drugs is their relatively short half-lives in the body, on the order of 2-3 hours.25 For this reason, they are usually administered in combination with other antimalarial drugs that have longer half-lifes. The World Health Organization (WHO) now suggests artemisinin combinational therapy as the first-choice to treat malaria.2 In summary, malaria is a global epidemic resulting in nearly 1 million deaths every year.2 Efforts to combat this devastating disease such as government control programs and distribution of bednets have done little to stop its spread. With advances in modern medicine, treatment of malaria through drug therapy is now a reality. The malaria parasite shows resistance to many of the original quinoline family of antimalarial drugs, but it has yet to show resistance to artemisinin and its derivaties. The outstanding biological properties of artemisinin have made it an important target for both isolation and synthetic chemists. 10 CHAPTER 2 ARTEMISININ: THE LAST LINE OF DEFENSE AGAINST MALARIA Artemisinin 1, a unique sesquiterpene lactone endoperoxide, is an efficient and universal antimalarial drug. It is also the precursor to a larger family of more potent antimalarials that are derivatized at C(10), which includes artemether 9, artesunate 10, artemisone28 11, and many others26-28 (Figure 9). This class of drugs has received considerable attention due to their effectiveness against all strains of the malaria parasite. 2,22,27 Amazingly, despite the fact that artemisinin-based combinational and monotherapies have been in use for over a decade, there have been no reported cases of the malaria parasite showing resistance in humans.22 As a result, the demand for artemisinin has increased significantly. H Me H H3C H O O CH3 O O O HO H H H O CH3 O O O ()-arteannuin B Me artemisinin 1 (+)-dihydro-epi-deoxyarteannuin B H H H3C O O O HO CH3 H H3C H CH3 OCH3 9 O O O HO CH3 H3C O O O HO H CH3 OCOCH2COOH 10 H CH3 N O S 11 Figure 9: Artemisinin and Related Compounds 11 CH3 O Artemisinin is isolated from the leaves of Artemisia annua, a leafy plant native to China and Vietnam commonly referred to as sweet wormwood (Figure 10). The Chinese have used cold teas made from the leaves of A. annua to treat malaria and other ailments since as early as the 2nd century B.C.29 In spite of its widespread use, it was not until 1972 during a comprehensive investigation into Chinese herbal medicine ordered by Chairman Mao TseTsung that artemisinin was isolated and identified as the active ingredient. 23 It took another seven years before the structure was finally elucidated.30 Today, the three main sources of artemisinin are direct isolation from the leaves of Artemisia annua, bioengineering, and chemical synthesis. Figure 10: Artemisinin is a Natural Product Extracted from Artemisia annua or Sweet Wormwood (Qinghao). cmbi.bjmu.edu.cn/.../200382031.jpg Direct isolation from the plant is the main source of artemisinin. Artemisinin content in the plant ranges from 0.01 to 1.4 wt % based on dry leaf mass, depending on which species of A. annua is being examined.31 An estimated 120 million courses of artemisinin-based combinational therapies are needed every year to combat the malaria epidemic. 2 An average 12 course of treatment comprises 20 tablets, each contain 20 mg of artemether 9, which is obtained from artemisinin in a 60 % chemical yield.28 This information, combined with the average amount of leaf harvested per acre in a given season, predicts that 35,000 acres of A. annua plantations are needed to meet the global need. 31 On a global scale this is a relatively small portion of land. However, these calculations assume an isolation efficiency of 100 %, which is far from the actual isolation efficiency of 60 %. The largest processing centers for the extraction of artemisinin from Artemisia annua are located in China and Vietnam. A mixture of hexanes and petroleum ether is used to extract artemisinin from the leaves of the plant. The advantages are the simplicity of the process and the low capital cost necessary for start up and maintenance. The disadvantage is that this technique is very hazardous to both the workers and the environment. These highly flammable of solvents have low vapor pressure, and the generation explosive hydrocarbon/oxygen mixtures is problematic. Emission of greenhouse gases, smog generation, and the entrapment of hexanes in the resulting biomass, which is commonly used as animal feed, also detract from this method. In an attempt to make the extraction process more efficient and environmentally friendly, other extraction techniques have been developed. These include extraction with EtOH,32 supercritical CO2,33,34 hydrofluorocarbons,31 and ionic liquids.31 These extraction techniques have not yet been fully investigated and have not made it beyond laboratory scale. Even with the advances in isolation techniques, the viability of using only cultured A. annua as the source for artemisinin remains cost prohibitive. 35 An alternative to agricultural drug production, which has long production cycles and is highly dependent on weather and climate, is biosynthesis. Biosynthesis is the process in which chemical compounds are created from simpler starting materials with-in a living organism. Biosynthesis has the advantages of being easier to scale, requiring less space, and being more efficient in terms of product produced per kg of starting material. 36 All of these factors point to the possibility of using biosynthesis to create an affordable source of artemisinin. Unfortunately, the direct bioconversion of simple starting materials to artemisinin has yet to be achieved.37 Most efforts aim at the bioconversion of simple chemicals into artemisinic acid, another constituent of A. annua, which can then be transformed chemically to artemisinin. 38 The biosynthesis of artemisinic acid can be achieved by the use of engineered yeast36,39 or through enzyme manipulation of other heterologous hosts. 40,41 13 The most efficient yeast developed for the production of artemisinic acid is Saccharomyces cerevisiae.36,39 The yeast was engineered by Keasling and co-workers to perform three key transformations. The first was the conversion of simple sugars into farnesyl pyrophosphate (FPP) using a modified biosynthetic pathway. This new pathway produced higher yields of the desired FPP as compared to the naturally occurring, unmodified pathway. The second step was the conversion of farnesyl pyrophosphate into amorphadiene. To achieve this transformation the amorphadiene synthase gene (ADS) from A. annua was isolated and then expressed into the high FPP producer. The third step involved cloning of a new cytochrome P450 which could carry out a three-step oxidation of amorphadiene into artemisinic acid (Figure 11).39 The transgenic yeast produced artemisinic acid at an efficiency of 100 mg/L. To help gauge that number, one needs to look at the natural source of artemisinic acid. Artemisinic acid can be isolated in 1.9 % relative to the biomass of A. annua compared with 4.5 % from the yeast. The yeast is therefore capable of a productivity that is over two times greater than the plant. In addition, the yeast can produce artemisinic acid in 45 days compared to several months for A. annua.39 The efficiency of the process can be greatly improved by moving from laboratory to industrial scale. The use of bioreactors with the same yeast resulted in a 25-fold improvement from 100 mg/L to 2.5 g/L isolated yield.36 There has also been interest in the bioconversion of arteannuin B (refer to Figure 9) to artemisinin. Arteannuin B can also be isolated from Artemisia annua. It occurs at a relative abundance of three times that of artemisinin. 42 In addition, there are several synthetic routes to arteannuin B.43,44 Unfortunately, all attempts to convert arteannuin B to artemisinin through enzymatic or microbial transformations have failed.37 An enzyme involved in the bioconversion of arteannuin B to artemisinin in A. annua has been isolated and purified,45 but all attempts to express it into a heterologous host have been unsuccessful.37 The only success has been the in vitro bioconversion of arteannuin B to artemisinin by incubation with an extract from A. annua L.42 14 Figure 1139: Schematic Representation of the Engineered Artemisinic Acid Biosynthetic Pathway in S. cerevisiae 15 In addition to extraction and bioengineering efforts, access to artemisinin through total synthesis is being explored.46-53 The reported syntheses all use similar end game strategies, either employing a singlet oxygen/acid rearrangement 46,47,50,53 of ketone 12 or an ozone/acid rearrangement48,49,51 of vinyl silane 13 to install the endoperoxide functionality (Figure 12). O O O21, 78 oC MeOH, hv Path A H MeO NaO2C methylene blue HOO MeO MeO HO2C 12a 12 O O O O3/O2 78 oC Path B Me3Si HO2C 0 oC, H3C 24 h, 30% H2SO4 H2O HOO O CH2Cl2; SiO2 HCO2H CH2Cl2 35% H O O O HO CH3 H CH3 O artemisinin 1 HO2C 13 13a Figure 12: Early Approaches Toward Artemisinin, Path A was Explored by Hofheinz46 and Later Modified by Yadav,53 Path B was Explored by Avery51 Where the syntheses differ is in their construction of precursors 12 and 13. The first reported total synthesis was completed by Schmid and Hofheinz. 46 The synthesis begins with commercially available ()-isopulegol 14, which is converted into ketone 15 through a protection/hydroboration/oxidation sequence in 58 % yield over 5 steps. Ketone 15 was then alkylated with the StorkJung vinylsilane54 16 giving intermediate 17 as a 6:1 mixture of diastereomers in 62 % yield (Figure 13). For a full discussion on the StorkJung vinylsilane refer to chapter 3. 16 5 steps 1. LDA, THF, 0 oC HO O H H 85% yield 14 Me3Si O 2. 16 62% 6:1 mix of diasteromers PhCH2O 15 H H PhCH2O 17 Me3Si I 16 Figure 13: Construction of the Schmid and Hofheinz Singlet Oxygen Precursor 17 A large excess of lithium methoxy(trimethylsilyl)methylide was added to 17, giving alcohol 18 as a 8:1 mixture of diastereomers. Lactonization followed by unmasking of the ketone from the vinylsilane gave bicyclic intermediate 19. Subsequent desilylation triggered enol ether formation and ring opening to give artemisinin precursor 20. Treatmeant of 20 with molecular oxygen, methylene blue and h followed by addition of formic acid gave artemisinin 1 in 30% yield (Figure 14). 17 10 equiv. Me3Si Li H MeO THF, -78 oC 89%, 8:1 d.r. O Me3Si 1. Li, NH3 2. PCC, CH2Cl2 HO MeO H H Me3Si 3. m-CPBA, CH2Cl2 4. TFA, CH2Cl2 54% over 4 steps PhCH2O 18 H MeO Me3Si O O O H 19 H 1. O2, methylene blue methanol, hv, -78 oC Bu4NF, THF, r.t., 2 h 95% yield H H MeO HO2C 2. formic acid, CH2Cl2, 0 oC, 24 hr, 30% yield H3C O O O HO CH3 H CH3 O 20 artemisinin 1 Figure 14: Schmid and Hofheinz End Game Strategy 17 The next three total syntheses of artemisinin completed by the Avery, 48,49,51 Zhou,47 and Yadav53 labs all used similar alkylation strategies to obtain their singlet oxygen or ozone rearrangement precursors. The only exception was a formal synthesis completed by Ravindranathan and co-workers, in which an intra-molecular DielsAlder reaction was employed to give access to the singlet oxygen precursor. 50 The discovery by Roth and Action in 1989 that dihydroartemisinic acid 21 could be converted directly to artemisinin 1 through exposure to singlet oxygen instantly made dihydroartemisinic acid 21 an important synthetic target (Figure 15).55 The first group to take advantage of this new access point to artemisinin 1 was the Liu lab,52 though other groups soon followed.56 Immediately recognizing the great potential for accessing artemisinin through 21, we also began developing a short and efficient route to dihydroartemisinic acid 21. Our first proposed pathway to 21 will be discussed in chapter 3. H 1. O2, hv, CH2Cl2 78 oC, methylene blue H H HO2C 2. CH2Cl2 replaced with petroleum ether, 4 days 17% 21 H3C O O O HO CH3 H CH3 O artemisinin 1 Figure 15: First Example of Dihydroartemisinic Acid 21 Being Converted into Artemisinin 1 In addition to accessing artemisinin through artemisinic acid 21, we also hypothesized that it may be possible to convert (+)-dihydro-epi-deoxyarteannuin B 22 into artemisinin 1 through a dyotropic ozonide rearrangement (Figure 16).57-60 Generation of the ozonide 23 followed by treatment with a Lewis acid should cause the lactone to open giving intermediate 24. A 1,2-shift of the ozonide bridge followed by ring closure would generate artemisinin 1. 18 H O3 CH2Cl2; H O O then Lewis acid O O O O O O O O O 23 22 LA H O O O H3C O LA O O O O HO O 24 CH3 H CH3 O 25 artemisinin 1 Figure 16: Our Proposal for Generating the Key Endoperoxide With our end game strategies in place, we turned our attention to the synthesis of both artemisinic acid 21 and (+)-dihydro-epi-deoxyarteannuin B 22. Chemical synthesis of these two molecules would allow us to test our proposed ozonide rearrangement and would also provide general contributions to artemisinin research as a whole. 19 CHAPTER 3 SYNTHESIS OF (+)-DIHYDRO-EPI-DEOXYARTEANNUIN B AND ()-DIHYDROARTEMISINIC ACID (+)-Dihydro-epi-deoxyarteannuin B 22, which also occurs naturally in Artemisia annua,61 is a key intermediate in our long term efforts to develop an efficient synthetic route to artemisinin 1. Both artemisinin and 22 derive their origins from dihydroartemisinic acid 21, another extract from A. annua, through a nonenzymatic autooxidation process (Figure 17).62-63 Both 21 and 22 have also been converted chemically into semisynthetic 1.55,64 H H H3C O H H O O O HO H CH3 O O (+)-dihydro-epi-deoxyarteannuin B 22 CH3 ()-artemisinin 1 H HO2C H H ()-dihydroartemisinic acid 21 Figure 17: Our Synthetic Targets The common feature in both 21 and 22 is the decalin ring system. An efficient way to establish the decalin ring system is therefore paramount in developing an economical approach to artemisinin 1. Several groups have approached the decalin ring system through the DielsAlder reaction.50,65,66 The Ravindranathan50 and Meinwald65 labs both used intramolecular DielsAlder reactions to establish the decalin system (Figure 18). 20 Ravindranathan and co-workers further elaborated 27 to complete a formal synthesis of artemisinin 1. However, due to the low yield and poor selectivity in their key DielsAlder step the overall efficiency of the synthesis was quite low. 50 H toluene, 210 oC sealed tube, 72 h Eq (1) H O 25-30% 3:2 mix of epimers 26 27 OH 28 H H benzene, 170 oC Eq (2) H O sealed tube, 2 days, 95% 2:1 mix of epimers OH H 29 Figure 18: Eq (1) Ravindranathan Intramolecular Diels-Alder,50 Eq (2) Meinwald Intramolecular Diels-Alder65 Avery and co-workers chose to use an aldol cyclization of ketone 30 to generate bicyclic enone 31 in their pursuit of artemisinin derivatives.67 The aldol condensation proved to be much more difficult than the authors had envisioned. Under normal aldol conditions they saw significant epimerization of the propanol side chain. They were able to combat the epimerization by incorporation of L-proline into the reaction mixture, but yields were still quite low (Figure 19).67 O L-proline, MeOH O TIPSO H H 43% O TIPSO H H 31 30 Figure 19: Avery and Co-Workers’ Strategy to Avoid Epimerization 21 In the course of their investigation into electroreductive cyclizations,68 Schwaebe and Little investigated the electroreductive cyclization of epoxy ketone 32 to give bicycle 33.69 The reaction proceeded in high yield, but there was no control of stereochemistry (Eq 4, Figure 20). Dreiding and co-workers experienced a similar problem in their Zn-Cu cyclization of ketone 36. Dreiding was able to access the tricyclic core of artemisinic acid in a single operation. Unfortunately, none of the stereoisomers obtained were consistent with naturally occurring artemisinic acid (Eq 6, Figure 20).70 Schwaebe and Little were later able to overcome the selectivity problem by using samarium iodide (Eq 5, Figure 20).69 H CO2Me Eq (4) O 4e, 4HD (78-95%) O 32 HO HO MeO2C 33 H CO2Me Eq (5) O SmI2, THF/MeOH 0 oC (>95%) 34 HO 35 CO2Me H CH2Br CO2C2H5 Eq (6) O Zn(Cu) THF O 36 37 O Figure 20: Electrocyclic Strategies to the Decalin Ring System Other methods employed to access the arteannuin ring system include an anodic coupling reaction developed by Wu and Moeller71 (Eq 7, Figure 21) and a tandem oxyCope/ene reaction developed by Barriault and Deon72 (Eq 8, Figure 21). While the anodic coupling reaction has the advantage of providing direct access to the tricyclic core of the arteannuins in a single step, preparation of 38, the coupling precursor, is cumbersome.71 The 22 tandem oxy-Cope/ene reaction, on the other hand, proceeds with excellent diastereoselectivity and builds complexity quickly. With this as their key step, Barriault and Deon were able to complete the total synthesis of (+)-arteannuin M 42 in ten steps with an overall yield of 14.1%.72 MeO O Eq (7) H RVC anode carbon cathode 0.1 M LiClO4 O 20% MeOH/CH2Cl2 2,6-lutadiene 8 mA/ 2.4 F/mole undivided cell R 38a. R = H 38b. R = Me H MeO R 39a. R = H, 70% 39b. R = Me, 70% H H o DBU, toluene, 220 C Eq (8) O HO HO O 55-60% 40 OH 41 ODPS HO H ODPS O (+)-arteannuin M 42 Figure 21: Other Approaches Towards the Decalin Rings System In our retro synthetic analysis of dihydro-epi-deoxyarteannuin B 22, we planned on constructing the decalin ring system through a ring closing metathesis of diene 45. Diene 45 would be constructed from ketone 44 by olefination followed by oxidation of the primary alcohol to trigger lactonization. Ketone 44 was envisioned to be prepared from silyloxy menthone 43 by alkylation with a methyl vinyl ketone equivalent followed by vinyl Grignard addition. The starting material for our synthesis was isopulegol 14 (Figure 22). 23 O H H H O O O (+)-dihydro-epi-deoxyarteannuin B 22 45 O OTIPS H 43 OH OTIPS H O 44 OH isopulegol 14 Figure 22: Our Retro-Synthetic Analysis For the conversion of isopulegol 14 to ketone 43, we planned to modify a known literature procedure and expected few problems.67 Our first anticipated challenge was the alkylation of ketone 43. We needed to prepare a Michael adduct between methyl vinyl ketone (MVK) and an unstabilized cyclohexanone enolate. Unfortunately, methyl vinyl ketone is a poor electrophile for this transformation due to competing polymerization. To circumvent the competing polymerization, a number of MVK equivalents73,74 have been developed for carrying out this transformation. The StorkJung vinylsilane54 16 was identified as the MVK equivalent that best met our needs (Figure 23). The weakness of the StorkJung vinylsilane 16 is its lengthy preparation.75-80 Therefore, we needed to develop a short and efficient route to prepare 16 on large scale. Me Me3Si I 16 Figure 23: The StorkJung Vinylsilane 24 The original synthesis of 16 by Stork and Jung can be seen in Figure 24.75 The synthesis begins with propargyl alcohol 46, which is converted to the trimethylsilyl acetylene 47 before regioselective reduction of the triple bond using lithium aluminum hydride. Subsequent iodination gave the (Z)-iodo silane 48 exclusively. Organocuprate addition followed by a twostep conversion of the primary alcohol to the iodide finished the synthesis. While several slightly modified procedures have been developed,76-80 the StorkJung method remains the most commonly used procedure. There are a couple notable modifications of the original StorkJung procedure.75 The first is a method developed by Zweifel and Lewis that allowed for the preparation of the (E)iodo silane 52 by use of iso-butylaluminum hydride instead of lithium aluminum hydride as the reducing agent.80 Second, Sato and co-workers showed that (E)-iodo-alcohols 54 could be prepared through a titanium-catalyzed hydromagnesiation of propargyl alcohol 53 followed by trapping with iodine (Figure 25).76,77 H BuLi; TMSCl SiMe3 LiAlH4, NaOMe; I2 90% OH 46 Me SiMe3 SiMe3 CuI, MeLi 68% 96% OH 48 OH 47 Ph3P, CCl4 I SiMe3 Me 60% NaI butanone Me SiMe3 80% OH Cl I 49 50 16 Figure 24: The Original Synthesis of the StorkJung Vinylsilane75 16 25 SiMe3 Eq (9) HO 1) LiAlH4 NaOMe H 2) I2 HO 47 Eq (10) n-Bu SiMe3 Stork and Jung75 I 48 SiMe3 1) i-Bu2AlH Et2O n-Bu Zweifel and Lewis80 2) I2 51 SiMe3 H I 52 Eq (11) n-Bu HO 53 1) 2 equiv i BuMgCl HO (5-C5H5)2TiCl2 n-Bu H Sato and co-workers76,77 I 54 2) I2 Figure 25: Early Modifications of the StorkJung Synthesis Our lab envisioned constructing the StorkJung vinyl silane 16 by the use of a titaniumcatalyzed hydromagnesiation of 2-butynol 55 followed by trapping with trimethylsilyl chloride, based on Sato’s protocol.76 The optimized conditions can be seen in Figure 26.81 It was found that 2.3 equivalents of iBuMgCl and 5 mol% of Cp2TiCl2 in diethyl ether at 60 oC gave the cleanest reaction and the highest yield. Trapping with trimethylsilyl chloride could be done with or without the toxic hexamethylphosphoramide (HMPA) additive, however yields decrease by 10-15% without it. Subsequent conversion of the vinyl alcohol to the vinyl iodide by triphenylphosphine and N-iodosuccinimide gave us a convenient two-step protocol for the generation of the StorkJung vinylsilane 16.81 2.3 equiv i-BuMgCl, Et2O 5 mol% Cp2TiCl2, HO Me 55 4.6 equiv HMPA, Et2O 3.0 equiv TMSCl, 70-76% SiMe3 HO Me 49 1.1 equiv Ph3P 1.1 equiv NIS CH2Cl2 0 oC to rt 82-94% SiMe3 I Me 16 Figure 26: Dudley Lab Succint Method for Preparation of StorkJung Vinylsilane 16 26 With a short and efficient way to prepare the StorkJung vinylsilane 16, we turned our attention to the preparation of the second alkylation partner, ketone 43. The synthesis begins with the hydroboration of isopulegol 14. The first concern was the large price difference between crude ($55 for 1 L, Aldrich) and pure isopulegol ($32 for 1 mL, Aldrich). The diastereoselective hydroboration of refined isopulegol was known to provide a crystalline diol 56.82 This led us to develope a recrystallization procedure which allowed us to start from bulk isopulegol which is over 500 times cheaper than the pure form (Figure 27). pure OH B2H6, THF; H2O2, 94% 95:5 d.r. (ref 82) ca. 3:1 dr B2H6, THF; H2O2, 57% OH OH H H pure crystals (our work) 56 14 OH 14 Figure 27: Hydroboration of Bulk Isopulegol vs. Pure Isopulegol Monoprotection of the primary alcohol of 56 with triisopropylsilyl chloride (TIPSCl) followed by Swern oxidation83 gave us silyloxymenthone 43 (Figure 28). DMSO, (COCl)2 TIPSCl, NEt3 OH OH H H 56 DMAP, CH2Cl2 86% OH OTIPS H H NEt3, CH2Cl2 86% 57 O OTIPS H H 43 Figure 28: Conversion of Diol 56 to Ketone 43 27 Coupling of silyloxymenthone 43 with the StorkJung vinylsilane 16 proved to be more difficult than we had anticipated (Table 1). Initial attempts at alkylation of 43 (Z = OTIPS) under standard conditions yielded 59 in a modest 45% yield (Table 1, Entry 6). Similar alkyations of menthone (58, Z = H) also proved problematic (Table 1, Entries 1 and 3). 84-86 Table 1. Allylation of Menthone and Silyloxymenthone 43 R LDA O Z H H RX OLi Z H H THF B C D Enolate conformations: (preferred) CH3 CH3 H3C O MLn Z O Z H H additive(s) LnM O H3C Z A1,2 interaction Entry Z 1 H 2 RX Additive(s) 58 allyl bromide none 60 1782 OBn 15 16 none 61 5346 3 H 58 allyl iodide HMPA 60 53 4 H 58 allyl iodide HMPA, Et2Zn 60 ~80a 5 OTIPS 43 allyl iodide HMPA, Et2Zn 62 80 6 OTIPS 43 16 HMPA 59 45 7 OTIPS 43 16 HMPA, Et2Zn 59 89 a Substrate Product Estimated by 1H NMR spectroscopy. SiMe3 SiMe3 I 16 59 O (i-Pr)3SiO 28 Yield (%) We believe the problem lies in the conformation of enolate C. A1,2 strain forces the enolate C to adopt a conformation in which the adjoining alkyl substitutents must adopt a pseudoaxial orientation, blocking both the top and bottom faces (Table 1). The use of diethylzinc as an additive according to Noyori’s protocol 87 provided for substantial improvements (Table 1, cf. Entries 3 and 4; 6 and 7). Optimal conditions were determined to be 1.0 equivalents of ketone 43 plus 1.1 equivalents lithium diisopropylamide, 1.0 equivalents of hexamethyldiphosphoramide (HMPA), 1.0 equivalents of diethylzinc, and 1.5 equivalents of the StorkJung vinylsilane 16 from 78 oC to room temperature for eight hours giving ketone 59 in 89% yield. Addition of vinyl Grignard to the sterically congested ketone 59 failed to give the desired tertiary alcohol. Fortunately, the use of the organocerium reagent88 made from freshly prepared vinylmagnesium bromide and freshly activated CeCl3 cleanly added to give the desired alcohol 63 in up to 91% yield. An additional three-step procedure beginning with the addition of lithium trimethylsilylacetylide, followed by deprotection and reduction also afforded 63 in a 85% percent overall yield. The three step process avoids the need for the careful preparation of the organocerium reagent and can be performed in a shorter amount of time with less technical difficulty (Figure 29). SiMe3 SiMe3 O OTIPS H H 59 1.Li (i-Pr)3SiO 59 O conformation in which the ketone is the least hindered vinylMgBr CeCl3 THF up to 91% SiMe3 OH OTIPS H H 63 SiMe3 THF; 2. K2CO3, MeOH; 3. H2 Pd/BaSO4, pyridine, EtOH ca. 85% overall Figure 29: Vinyl Addition to Silyloxymenthone 59 Removal of the silyl ether under acidic conditions followed by oxidation of the resulting diol yielded lactone 64 in excellent yield. The vinylsilane was then selectively epoxidized over 29 the terminal olefin with m-CPBA, presumably due to the shielding effect of the lactone. Subsequent treatment of epoxide 65 with trifluoroacetic acid resulted in formation of ketone 66. Waiting until this point in the synthesis to unmask the ketone allowed us to avoid the need for lengthy protecting group strategies. Wittig methylenation 97 of ketone 66 proceeded with moderate yield; however, the most disappointing development was that all attempts at effecting a ring-closing metathesis (RCM)89 reaction on the resulting diene 45 failed (Figure 30). O SiMe3 OH OTIPS H H SiMe3 1. HCl MeOH 2. TPAP NMO >95% overall 63 SiMe3 m-CPBA; H O CF3CO2H >95% O 64 O H O 65 O H PH3P=CH2 H O ca. 30-40% RCM attempts H O 66 O O 45 Grubbs' cat H O O 22 Figure 30: Elaboration to First Metathesis Precursor 63-22 The failed ring-closing metathesis may be attributed to steric congestion around the vinyl substituent. In addition to steric congestion, the spiro-tricyclic product 22 may be significantly more strained than the diene precursor 45 prohibiting cyclization. Reductive opening of the lactone with lithium aluminum hydride did allow for the ring-closing metathesis to proceed in low yield. Subsequent tetrapropylammonium perruthenate (TPAP) oxidation successfully yielded (+)-dihydro-epi-deoxyarteannuin B 22 (Figure 31). The lower yield associated with the final lactonization step compared with the earlier lactonization step (63 to 64) lends credence to our hypothesis that the failed RCM is partially due to mild strain associated with the spiro-lactonization step. Cross metathesis by-products resulting from the 30 ruthenium catalyst reacting with the mono-substituted alkene but then failing to cyclize led us to consider the possibility of using Hoye’s relay ring closing metathesis90 (RRCM) strategy to force the catalyst onto the disubstituted olefin. Olefination of ketone 66 with phosphonium salt 68 (prepared from 5-hexen-1-ol in two steps) provided the desired RRCM substrate 69, albeit in modest yield. Despite the moderate yield in the olefination step, vast improvements were seen in the subsequent relay ring closing metathesis step (Figure 31). H 1.LiAlH4 92% H H 2.RCM 37% O TPAP NMO 62% OH OH 67 O 45 H Ph3P I H 68 relay RCM O 68, BuLi H O 17-37% (40% brsm) O 22 Grubbs' cat. 74-90% O H O 69 O 66 O Figure 31: Ring Closing Metathesis (RCM) Strategies In summary, we have successfully completed the total synthesis of (+)-dihydro-epideoxyarteannuin B 22 from bulk isopulegol 14.91 We were able to develop an efficient alkylation strategy based on Noyori’s protocol 87 that allowed us to alkylate menthone derivatives in high yield. These alkylated derivatives can serve as valuable building blocks in the synthesis of a variety of artemisinin related compounds. Two solutions were developed to overcome the initial failure of the late stage ring closing metathesis. These studies will provide 31 valuable information for our future research into the chemical synthesis of members of the artemisinin family of antimalarial compounds. Upon successful completion of (+)-dihydro-epi-deoxyarteannuin B, our next course of action was to approach artemisinin through the known oxidation of dihydroartemisinic acid 21.55 Prior approaches to the decalin ring system of dihydroartemisinic acid 21 were discussed previously in our discussion of (+)-dihydro-epi-deoxyarteannuin B 22.50,65-72 Our proposed synthesis of dihydroartemisinic acid 21 can be seen in Figure 32. We planned to begin with the same silyloxymenthone derivative 43 that we had prepared previously for our synthesis of (+)-dihydro-epi-deoxyarteannuin B 22.91 Alkylation with allyl iodide followed by olefination would give us α,β-unsaturated ester 70. We then wanted to complete a one pot methylenation and ring-closing metathesis based on Rainier’s work with the Takai reagent.92 Hydrolysis of the silyl ether followed by an acidic oxidation was intended to give a mixture of carboxylic acids 72 and 21. Carboxylic acid 21 can then be oxidized to artemisinin using singlet oxygen.55 1.LDA, THF H H 43 O 2.Et2Zn, HMPA OTIPS allyl iodide 80% O OTIPS 71 olefination O OTIPS H H H H 62 MeLi, CuCN DIBAL, THF OTIPS 70 H 1.TiCl4, PbCl2 Zn, CH2Br2 COCH3 H H+ H 72 H CO2H H H CO2H dihydroartemisinic acid 21 Figure 32: Proposed Synthesis of Dihydroartemisinic Acid 21 With the alkylation conditions for the conversion of 43 to 62 having been worked out previously91 we immediately began investigations into the olefination of ketone 62. We first 32 looked at the olefination of a model system 60, which lacked the triisopropylsilyl moiety. Ketone 60 could be made from ()-menthone 58 by alkylation with allyl iodide (Figure 33). To our surprise, the HornerWadsworthEmmons (HWE)94 olefination failed to give any of the desired product. Believing that the steric hindrance of the ketone may be to blame for the lack of reactivity, we next examined the HWE olefination of ()-menthone 58. Again the HWE olefination provided no reaction (Figure 33). A search of the literature yielded no examples of menthone or similar derivatives being olefinated by the use of stabilized Wittig or HWE reagents. LDA, Et2Zn HMPA, THF O allyl iodide 50-85% 58 60 O 60 (OEt)2P(O)CH2P(O)OEt No Reaction NaH (OEt)2P(O)CH2P(O)OEt 58 No Reaction NaH Figure 33: Olefination Attempts on Menthone and Menthone Derivatives The neighboring alkyl groups of these ketones (58 and 60) screen against nucleophilic attack. Thus, we found ourselves in need of an efficient way to olefinate hindered ketones. This led us to develop an alternative olefination strategy sequence, which began with acetylide addition followed by MeyerSchuster rearrangement—a formal 1,3 hydroxy shift of a propargyl alcohol to the corresponding enone (Figure 34).116-117 Acetylide additions are relatively insensitive to steric congestion, and it was believed that acetylide addition to ketone 60 should proceed smoothly (Figure 34). Rearrangement of 33 the resulting propargyl alcohol to the α,β-unsaturated enone via the MeyerSchuster rearrangement would complete our desired olefination. Unfortunately, the traditional MeyerSchuster reaction is limited in scope and often requires harsh reaction conditions. 117-130 We therefore need to develop mild and general reaction conditions to effect this transformation. R O O Meyer-Schuster OH R n-BuLi R 60 73 OH Eq (15) R3 R1 74 strong acid E R3 "Meyer-Schuster" R1 R2 O R2 F Figure 34: Proposed Olefination Strategy In summary, we designed a concise route to dihydroartemisinic acid 21. In order to test the feasibility of this route, we first have to develop a method for the olefination of hindered ketones. We proposed a two-step olefination consisting of acetylide addition followed by MeyerSchuster rearrangement. The scope of the original MeyerSchuster rearrangement is limited due to harsh reaction conditions. Our efforts into developing mild reaction conditions for this powerful transformation will be discussed fully in Part II. Addendum: We have successfully implemented the two-step olefination strategy outlined above. Ethoxyacetylene cleanly added to ketone 60 with the use of n-BuLi in THF to give propargyl alcohol 73. Treatment of propargyl alcohol 73 with 5 mol % AuCl3 and 5.0 equivalents of ethanol in methylenechloride at room temperature gave what appears to be the desired α,β34 unsaturated ester 74 (Figure 35). These are preliminary results and are based on analysis of only the 1H NMR (spectra immediately follow). However, these initial findings support the feasibility of the approach. R n-BuLi O OH -78 oCr.t. 12 h, 80% R O CH2Cl2, rt, 5 min 98% R 74 73 60 5 mol% AuCl3 5.0 equiv. EtOH R = OEt Figure 35: Implementation of Our Olefination Strategy Towards Dihydroartemisinic Acid 21 35 36 37 CHAPTER 4 EXPERIMENTAL: (+)-DIHYDRO-EPI-DEOXYARTEANNUIN B GENERAL EXPERIMENTAL PROCEDURES: 1 H NMR and 13 C NMR spectra were recorded on a 300 MHz spectrometer using CDCl3 as the deuterated solvent. The chemical shifts () are reported in parts per million (ppm) relative to the residual CHCl3 peak (7.26 ppm for 1H NMR and 77.0 ppm for 13 C NMR for all compounds except for 22, which was referenced to 7.27 ppm for 1H NMR and 77.2 ppm for 13 C NMR). The coupling constants (J) are reported in Hertz (Hz). IR spectra were recorded on an FTIR spectrometer on NaCl discs. Mass spectra were recorded using chemical ionization (CI), electron ionization (EI), fast atom bombardment (FAB), or electrospray ionization (ESI). Yields refer to isolated material judged to be ≥95 % pure by 1H NMR spectroscopy following silica gel chromatography. All chemical were used as received unless otherwise stated. Tetrahydrofuran (THF) and diethyl ether (Et2O) were purified by passing through a column of activated alumina. The n-BuLi solutions were titrated against a known amount menthol dissolved in tetrahydrofuran using 1,10-phenanthroline as the indicator. The purifications were performed by flash chromatography using silica gel F-254 (230-499 mesh particle size). HMPA was distilled under reduced pressure over CaH2, and stored under an inert atmosphere. Diisopropylamine was distilled over CaH2, under an inert atmosphere, and used fresh. 38 SiMe3 HO Me 49 (E)-3-Trimethylsilanylbut-2-en-1-ol [49] To an oven-dried, three-necked, round-bottom flask equipped with a pressure-equalizing dropping funnel, reflux condenser, and septum was added isobutylmagnesium chloride in diethyl ether (46.0 mL, 92.0 mmol) under argon. To the Grignard solution was added bis(cyclopentadienyl)titanium dichloride (0.50 g, 2.0 mmol) at 0 oC. The reaction was held at 0 oC for 30 min then 2-butyn-1-ol (3.0 mL, 40.0 mmol) was added dropwise as a solution in diethyl ether (100 mL). The reaction was then heated to reflux and held for 16 hr. The reaction was then allowed to cool to room temperature and hexamethylphosphoramide (19.0 mL, 109 mmol) was added dropwise as a solution of THF (125 mL) followed by chlorotrimethylsilane (14.0 mL, 109 mmol). The reaction was heated to reflux and held for 16 hr, then allowed to cool to room temperature. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted three times with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4 and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 20/1-5/1) to give 49 as a pale yellow oil (4.38 g, 76%): H NMR (300 MHz, CDCl3) 0.07 (s, 9H), 1.71 (dt, J = 1.7, 0.8 Hz, 3H), 4.27 (dd, J = 6.0, 0.8 1 Hz, 2H), 5.88 (tq, J = 5.8, 1.7 Hz, 1H). C NMR (75 MHz, CDCl3) -2.4, 14.6, 59.5, 137.5, 13 139.2. IR (neat) 3324, 2954, 1621, 1440, 1359, 1247 cm-1; MS m/z 143.1 [65, M – H]+. CH: Calcd for C7H16OSi: C, 58.27; H, 11.18. Found: C, 58.04; H, 10.88. 39 40 41 SiMe3 I Me 16 ((E)-3-Iodo-1-methylpropenyl)trimethylsilane [16] (E)-3-Trimethylsilanylbut-2-en-1-ol (49, 0.20 g, 1.4 mmol, 1.0 equiv) and triphenylphosphine (0.40 g, 1.5 mmol, 1.1 equiv) were weighed into an oven-dried, two-necked, round-bottomed flask equipped with an argon inlet and septum. Methylenechloride (3.5 mL) was added via syringe followed by the addition of N-iodosuccinimide (0.34 g, 1.5 mmol, 1.1 equiv, freshly recrystallized from dioxane/carbontetrachloride). The flask was wrapped in aluminum foil and the reaction mixture was stirred for 1 h in an ice bath and then 3 h at room temperature. When the reaction was complete (as determined by TLC), 5 mL of hexanes were added and the mixture was immediately filtered through 4 g of silica gel with the aid of 200 mL of hexanes. The filtrate was concentrated in vacuo give a thick yellow oil. (Note: Material at this stage was judged to be >95% pure by 1H NMR and was suitable for use.) Subsequent filtration through 2 g of neutral alumina with 100-150 mL of hexanes and concentration in vacuo afforded 0.29 g (82%) of 1 as a pale yellow liquid: H NMR (300 MHz, CDCl3) 0.06 (s, 9H), 1.71 (d, J = 1.8 Hz, 3H), 3.93 (d, J = 8.4 Hz, 2H), 1 6.02 (tq, J = 8.4, 1.8 Hz, 1H); C NMR (75 MHz, CDCl3) -2.5, 1.2, 13.7, 134.2, 143.2; IR 13 (neat) 2954, 1604, 1437, 1247, 1143, 835 cm-1. MS m/z 254.2 [12, M]+. 42 43 44 1. BH3 THF 2. H2O2, NaOH OH Isopulegol 14 3. NEt3, TIPSCl DMAP, CH2Cl2 4. DMSO, (COCl)2, NEt3, CH2Cl2 O OTIPS 43 (2S,5R)-5-Methyl-2-((R)-1-methyl-2-triisopropylsilanyloxy-ethyl)-cyclohexanone [43] For detailed procedures and characterization data in support of the conversion of a crude mixture of isopulegol diastereomers to analytically pure 43 refer to Avery, M. A.; Fan, P.; Karle, J. M.; Bonk, J. D.; Miller, R.; Goins, D. K. J. Med. Chem. 1996, 39, 18851897. H NMR: 3.68 (dd, J = 5.1, 9.6 Hz, 1H), 3.61 (dd, J = 5.6, 9.6 Hz, 1H), 2.33 (m, 1H), 2.30 (m, 1 1H), 2.12 (ddd, J = 3.0, 6.0, 9.0 Hz, 1H), 1.95-2.04 (m, 2H), 1.75-1.92 (m, 2H), 1.29-1.51 (m, 2H), 1.04 (m, 21H), 0.99 (d, J = 6.4 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H). 1 H NMR spectra for intermediates are given. 45 46 47 48 SiMe3 O OTIPS 59 (2S, 3R, 6S)-3-Methyl-6-(1-methyl-2-triisopropylsilanyloxy-ethyl)-2-((E)-3-trimethylsilanylbut-2-enyl)-cyclohexanone [59] To a THF solution (6 mL) of n-BuLi (1.44 mL, 3.38 mmol, 2.35 M) was added diisopropylamine (0.52 mL, 3.68 mmol) at –78 °C under an argon atmosphere. The reaction mixture was held at –78 °C for 15 min. then allowed to warm to room temperature and keep there for 30 min before being recooled to –78 °C. Next, 43 (1.00 g, 3.07 mmol) was added dropwise as a solution in THF (2.0 mL). The reaction was held at –78 °C for 40 min. before being allowed to warm to 0 °C and held for 30 min., after which the reaction was recooled to –78 °C. Hexamethylphosphoramide (0.53 mL, 3.07 mmol) was added, the reaction mixture was stirred for 5 min, and then diethyl zinc (0.31 mL, 3.07 mmol) was added. A solution of 16 (1.19 g, 4.6 mmol) in 2 mL of THF was added dropwise and the reaction mixture was allowed to warm to room temperature and stirred overnight. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 50/1) to give 59 as a clear colorless oil (1.24 g, 89%). H NMR (300 MHz, CDCl3) 0.00 (s, 9H), 0.95 (d, J = 6.7 Hz, 3H), 1.03-1.06 (m, 21H), 1.33- 1 1.49 (m, 2H), 1.55 (s, 3H), 1.64-1.68 (m, 3H), 1.82-1.99 (m, 3H), 2.05-2.26 (m, 3H), 2.34-2.48 (m, 2H), 3.65 (d, J = 4.8 Hz, 2H), 5.64 (tq, J = 6.5, 1.7 Hz, 1 H); 13 C NMR (75 MHz, CDCl3) - 2.1, 12.0, 14.4, 15.7, 18.0, 20.7, 25.4, 31.1, 35.1, 41.3, 53.3, 58.4, 65.7, 136.0, 137.9, 213.0; IR (neat) 2953, 2866, 1712, 1617, 1463, 1246, 1098, 836 cm-1; HRMS (ESI) Calcd for C26H52O2Si2 (M + Na+) 475.3404. Found 475.3388. 49 50 51 O 60 (2S, 3R, 6R)-2-Allyl-6-isopropyl-3-methyl-cyclohexanone [60] To a THF solution (4 mL) of n-BuLi (2.31 mL, 4.27 mmol, 1.85 M) was added diisopropylamine (0.65 mL, 4.66 mmol) at –78 °C under an argon atmosphere. The reaction mixture was held at –78 °C for 15 min. then allowed to warm to room temperature and keep there for 30 min before being recooled to –78 °C. Next, ()-menthone (0.67 mL, 3.88 mmol) was added dropwise as a solution in THF (2.0 mL). The reaction was held at –78 °C for 40 min. before being allowed to warm to 0 °C and held for 30 min., after which the reaction was recooled to –78 °C. Hexamethylphosphoramide (0.68 mL, 3.88 mmol) was added, the reaction mixture was stirred for 5 min, and then diethyl zinc (5.62 mL, 3.88 mmol, 0.69 M) was added. A solution of allyl iodide (1.07 mL, 11.67 mmol) in 2 mL of THF was added dropwise and the reaction mixture was allowed to warm to room temperature and stirred overnight. Saturated aqueous NH 4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 50/1) to give 60 as a clear colorless oil (0.64 g, 85%). H NMR (300 MHz, CDCl3) 0.87 (dd, J = 13.3, 6.5 Hz, 6H), 1.05 (d, J = 6.2 Hz, 2H), 1.21- 1 1.67 (m, 4H), 1.87 (dq, J = 8.7, 2.7 Hz, 1H), 1.99-2.16 (m, 4H), 2.27-2.38 (m, 2H), 4.91-5.05 (m, 2H), 5.84 (ddt, J = 17.1, 10.2, 6.8 Hz, 1H). For full characterization refer to: Nakashima, K.; Imoto, M.; Sono, M.; Tori, M.; Nagashima, F.; Asakawa, Y. Molecules 2002, 7(7), 517527. 52 53 O OTIPS 62 (2S,3R,6R)-2-Allyl-3-methyl-6-(R-1-methyl-2-triisopropylsilyoxyethyl)-cyclohexanone [62] To a THF solution (6 mL) of n-BuLi (1.54 mL, 3.23 mmol, 2.1 M) was added diisopropylamine (0.49 mL, 3.52 mmol) at –78 °C under an argon atmosphere. The reaction mixture was held at –78 °C for 15 min. then allowed to warm to room temperature and keep there for 30 min before being recooled to –78 °C. Next, 43 (0.96 g, 2.94 mmol) was added dropwise as a solution in THF (2.0 mL). The reaction was held at –78 °C for 40 min. before being allowed to warm to 0 °C and held for 30 min., after which the reaction was recooled to –78 °C. Hexamethylphosphoramide (0.61 mL, 3.52 mmol) was added, the reaction mixture was stirred for 5 min, and then diethyl zinc (0.36 mL, 3.52 mmol) was added. A solution of allyl iodide (1.0 mL, 10.95 mmol) in 2 mL of THF was added dropwise and the reaction mixture was allowed to warm to room temperature and stirred overnight. Saturated aqueous NH 4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 50/1) to give 62 as a clear colorless oil (0.86 g, 80%). H NMR (300 MHz, CDCl3) 0.91-1.14 (m, 21H), 1.20-1.68 (m, 7H), 1.80-2.02 (m, 4H), 2.20- 1 2.49 (m, 5H), 3.66 (dd, J = 4.9, 1.8 Hz, 2H), 4.89-5.06 (m, 2H), 5.84 (ddt, J = 17.1, 10.1, 6.8 Hz, 1H) 13 C NMR (75 MHz, CDCl3) 5.3, 11.5, 15.6, 18.0, 20.5, 29.7, 30.5, 35.0, 40.4, 53.1, 57.7, 62.5, 65.6, 115.3, 137.3, 212.9. 54 55 56 SiMe3 OH OTIPS 63 (1S,2S,3R,6S)-3-Methyl-6-((R)-1-methyl-2-triisopropylsilanyloxy-ethyl)-2-((E)-3trimethylsilanyl-but-2-enyl-1-vinyl-cyclohexanol [63] Procedure 1: A flask containing CeCl3 (0.066 g, 0.27 mmol) was heated to 140 °C under vacuum for 3 hr then cooled to 0 °C and placed under an argon atmosphere. THF (0.9 mL) was added and the mixture was allowed to stir for 2 hr. The resulting slurry was then cooled to –78 °C and vinylmagnesium bromide (2.5 mL, 0.94 mmol, 0.37 M) in THF was added. After 30 min, a solution of 59 (0.06 g, 0.13 mmol) in 0.5 mL of THF was added dropwise by syringe. The reaction mixture was held at –78 °C for 1 h and then allowed to warm to –15 °C and held for 1 h. Cold saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with diethyl ether. The organic layer was dried over Na2SO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 50/1) to give 63 as clear colorless oil (0.56 g, 87%). Procedure 2: To a THF solution (6 mL) of trimethylsilylacetylene (0.20 mL, 1.4 mmol) was added n-BuLi (0.51 mL, 1.1 mmol, 2.1 M) dropwise at –78 °C under an argon atmosphere. The excess dry ice was removed and the reaction was allowed to slowly warm to 0 °C over 1.5 hr, then recooled to –78 °C. Next, 59 (0.324 g, 0.72 mmol) was added dropwise as a solution in THF (2.0 mL) and the reaction was allowed to slowly warm to room temperature. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with diethyl ether. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO 4, filtered, and concentrated. The crude mixture was then added to a methanol solution saturated with potassium carbonate (7.0 mL) and left overnight. The reaction mixture was diluted with water and extracted with diethyl ether. The organic layer was washed with saturated aqueous sodium bicarbonate and brine. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate 57 = 20/1) to give a clear colorless oil (0.316 g), tentatively assigned as the propargyl alcohol SiMe3 corresponding to acetylide addition of ketone 59; OH OTIPS 93%. A 15-mL round- bottomed flask was then charged with 5% Pd on BaSO 4 (31 mg, 0.014 mmol) under an atmosphere of hydrogen. The palladium turned a dark black color. Methanol (4.0 mL) and pyridine (0.07 mL, 0.87 mmol) were then added and the solution was allowed to mix for 15 min. The propargyl alcohol (0.295 g, 0.62 mmol) obtained earlier was then added as a solution in methanol (2.0 mL). After 18 hr the reaction mixture was filtered through a plug of Celite (hexanes/ethyl acetate = 20/1) and was then purified using silica gel column chromatography (hexanes/ethyl acetate = 100/1) to give 63 as clear colorless oil (0.274 g, 93%). H NMR (300 MHz, CDCl3) 0.00 (s, 9H), 0.77 (d, J = 7.2 Hz, 3H), 0.85 (d, J = 6.4 Hz, 3H), 1 1.03-1.13 (m, 21H), 1.32 (dd, J = 13.0, 3.3 Hz, 1H), 1.44 (dq, J = 12.5, 3.3 Hz, 1H), 1.55-1.79 (m, 8H), 1.96-2.05 (m, 1H), 2.16-2.23 (m, 1H), 2.32-2.40 (m, 1H), 3.35 (dd, J = 10.1, 4.0 Hz, 1H), 3.46 (dd, J = 10.3, 10.3 Hz, 1H), 4.37 (d, J = 1.3 Hz, 1H), 5.18 (dd, J = 10.5, 2.4 Hz, 1H), 5.32 (dd, J = 17.2, 10.5 Hz, 1H), 5.62 (dd, J = 17.2, 10.5 Hz, 1H), 5.73 (tq, J = 6.4, 1.6 Hz, 1H); C NMR (75 MHz, CDCl3) -2.1, 11.9, 14.5, 18.0, 18.6, 20.6, 20.8, 28.6, 33.3, 34.5, 36.3, 13 51.5, 52.5, 65.6, 77.6, 114.1, 132.8, 142.3, 145.7; IR (neat) 3390, 2950, 2868, 1614, 1462, 1246, 1056, 835 cm-1; HRMS (FAB) Calcd for C28H56O2Si2 (M + Na+) 503.3717. Found 503.3711. [α]546 = –7.92° (c 0.001, CHCl3). 58 59 60 SiMe3 O 64 O (3R, 3aS, 6R, 7S, 7aS)-3,6-Dimethyl-7-((E)-3-trimethylsilanyl-but-2-enyl)-7a-vinyl- hexahydro-benzofuran-2-one [64] To a methanol solution (8.5 mL) of 63 (0.113 g, 0.234 mmol) was added conc. HCl (88 μL, 2.87 mmol) at 0 °C under an argon atmosphere. After 2 hr the reaction was quenched with aqueous sodium bicarbonate and extracted with diethyl ether. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 10/1-5/1) to give a diol intermediate as a white crystalline solid (0.076 g, 0.234 mmol). 1H NMR (300 MHz, CDCl3) 0.03 (s, 9H), 0.78 (d, J = 7.3 Hz, 3H), 0.96 (d, J = 6.4 Hz, 3H), 1.04-1.14 (m, 1H), 1.18-1.30 (m, 3H), 1.48-1.60 (m, 3H), 1.64 (t, J = 1.5 Hz, 3H), 1.82-1.88 (m, 2H), 2.15-2.24 (m, 2H), 2.33-2.42 (m, 1H), 3.26 (dd, J = 11.0, 3.5 Hz, 1H), 3.39 (dd, J = 10.6, 10.6 Hz, 1H), 5.25 (ddd, J = 18.8, 14.1, 1.5 Hz, 2H), 5.66 (dd, J = 17.3, 10.8 Hz, 1H), 5.82-5.86 (m, 1H); C NMR (75 MHz, CDCl3) -2.2, 14.9, 18.6, 13 20.3, 20.5, 27.4, 31.5, 34.4, 36.0, 50.9, 52.2, 64.0, 79.3, 114.4, 137.4, 138.4, 144.3; IR (neat) 3306, 2953, 1614, 1456, 1246, 835 cm-1. HRMS (EI) Calcd for C19H36O2Si (M+) 324.2485. Found 324.2484. [α]546 = –9.80° (0.0012, CHCl3); mp 6567 °C. To a mixture of the diol (0.076 g, 0.234 mmol), 4 Å molecular sieves (0.10 g), and N-methylmorpholine oxide (0.082 g, 0.7 mmol) in CH2Cl2 (2.0 mL) was added tetra-n-propylammonium perruthenate (0.008 g, 0.023 mmol) under an argon atmosphere. After 2.5 hr the reaction mixture was flushed through a plug of SiO2 (hexanes/ethyl acetate = 10/1) to give lactone 64 as a white crystalline solid (0.075 g, 100%). 1H NMR (300 MHz, CDCl3) 0.01 (s, 9H), 0.92 (d, J = 6.5 Hz, 3H), 1.08 (d, J = 7.2 Hz, 3H), 1.20-1.26 (m, 2H), 1.39-1.43 (m, 1H), 1.55-1.74 (m, 6H), 2.08-2.18 (m, 2H), 2.40-2.48 (m, 1H), 2.94 (apparent quint., J = 6.9 Hz, 1H), 5.22-5.32 (m, 2H), 5.63-5.72 (m, 2H); C NMR (75 MHz, CDCl3) -2.2, 9.1, 14.5, 20.7, 24.1, 28.1, 32.9, 33.7, 39.1, 43.4, 49.3, 87.5, 13 115.4, 134.7, 140.3, 140.4, 179.8; IR (neat) 2952, 1778, 1615, 1455, 1246, 1178, 835 cm-1; HRMS (EI) Calcd for C19H32O2Si (M+) 320.2172. Found 320.2168. [α]546 = –14.58° (c 0.0024, CHCl3); mp 7476°C. 61 62 63 64 65 O O 66 O (3R, 3aS, 6R, 7S, 7aS)-3,6-Dimethyl-7-(3-oxo-butyl)-7a-vinyl-hexahydro-benzofuran-2-one [66] To a CH2Cl2 solution (5.0 mL) of 64 (0.075 g, 0.234 mmol) was added m-CPBA (0.064 g, 0.281 mmol) at 0 °C under an argon atmosphere. After 2 hr the reaction was quenched with aqueous sodium bicarbonate and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered, and concentrated. The resulting residue was dissolved in CH2Cl2 (3.0 mL) and trifluoroacetic acid (60 μL, 0.8 mmol) was added. After 1 h the reaction was quenched with aqueous sodium bicarbonate and extracted with CH2Cl2. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 8/1) to give 66 as a white crystalline solid (0.062 g, 100%). H NMR (300 MHz, CDCl3) 0.94 (d, J = 6.5 Hz, 3H), 1.08 (d, J = 7.2 Hz, 3H), 1.19-1.25 (m, 1 2H), 1.39-1.51 (m, 1H), 1.59-1.94 (m, 6H), 2.11 (s, 3H), 2.30-2.41 (m, 1H), 2.63-2.74 (m, 1H), 2.90 (apparent quint., J = 6.9 Hz, 1H), 5.29 (m, 2H), 5.66 (dd, J = 17.3, 10.7 Hz); 13 C NMR (75 MHz, CDCl3) 9.0, 20.1, 21.1, 24.0, 29.9, 30.5, 32.7, 38.7, 41.1, 43.5, 46.9, 87.6, 115.5, 140.1, 179.7, 209.1; IR (neat) 2929, 2359, 1771, 1714, 1448, 1177, 947 cm-1; HRMS (CI) Calcd for C16H24O3 (M + H+) 265.1804. Found 265.1791. [α]546 = –11.90° (c 0.0016, CHCl3); mp 6466 °C. 66 67 68 O 45 O (3R, 3aS, 6R, 7S, 7aS)-3,6-Dimethyl-7-(3-methyl-but-3-enyl)-7a-vinyl-hexahydro- benzofuran-2-one [45] To a THF solution (1.0 mL) of methyltriphenylphosphonium bromide (0.073 g, 0.2 mmol) was added n-BuLi (0.13 mL, 0.18 mmol, 1.4 M) at –40 °C under an argon atmosphere. The solution turned a bright yellow. The mixture was allowed to warm to 0 °C over 1 h then 66 (0.027 g, 0.10 mmol) was added as a solution in THF (1.0 mL). The mixture was then allowed to warm to room temperature and left overnight. The reaction was quenched with water and extracted with diethyl ether. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 20/1) to give 45 (0.008 g, 31%) as a clear colorless oil. H NMR (300 MHz, CDCl3) 0.98 (d, J = 6.5 Hz, 3H), 1.08 (d, J = 7.2 Hz, 3H), 1.01-1.13 (m, 1 3H), 1.16-1.31 (m, 1H), 1.40-1.50 (m, 2H), 1.67-1.80 (m, 6H), 1.87-1.97 (m, 1H), 2.05-2.16 (m, 2H), 2.93 (apparent quint., J = 6.9 Hz, 1H), 4.63 (d, J = 7.1 Hz, 1H), 5.29 (ddd, J = 18.8, 14.1, 1.5 Hz, 2H), 5.66 (dd, J = 17.3, 10.8 Hz, 1H); 13 C NMR (75 MHz, CDCl3) 9.1, 20.4, 22.4, 24.1, 26.5, 32.4, 32.9, 37.6, 39.2, 43.4, 48.0, 87.7, 109.5, 115.3, 140.2, 146.3, 179.8; IR (neat) 3072, 2933, 1778, 1646, 1179, 945 cm-1; HRMS (CI) Calcd for C16H26O2 (M + H+) 263.2011. Found 263.2005. 69 70 71 I PPh3 68 Hex-5-enyl-triphenyl-phosphonium iodide [68] To a nitromethane solution (35 mL) of 6-iodo-1-hexene (3.1 g, 14.75 mmol) was added triphenylphosphine (3.9 g, 14.75 mmol). The mixture was heated to 50 °C for 18 hr then cooled to room temperature. The mixture was then diluted with diethyl ether (50 mL) and a white solid crashed out. The precipitate was gravity filtered and rinsed with diethyl ether (30 mL) to give 68 (4.7 g, 67 %) as a white crystalline solid. H NMR (300 MHz, CDCl3) 1.52-1.80 (m, 4H), 2.02 (apparent q, J = 6.9 Hz), 3.52-3.62 (m, 1 2H), 4.81-4.91 (m, 2H), 5.62 (ddt, J = 16.9, 10.1, 6.7 Hz, 1H), 7.63-7.79 (m, 15H); 13 C NMR (75 MHz, CDCl3) 21.5, 21.6, 22.4, 23.1, 28.9, 29.1, 32.7, 115.2, 117.3, 118.4, 130.3, 130.5, 133.4, 133.5, 135.0, 137.4; mp 166168 °C. 72 73 74 O 69 O (3R, 3aS, 6R, 7S, 7aS)-3,6-dimethyl-7-((Z)-3-methyl-nona-3,8-dienyl)-7a-vinyl-hexahydrobenzofuran-2-one [69] To a diethyl ether solution (2.0 mL) of hex-5-enyl-triphenyl-phosphonium iodide 68 (0.137 g, 0.29 mmol) was added n-BuLi (0.124 mL, 0.248 mmol, 2.0 M) at –78 °C under an argon atmosphere. The mixture was then allowed to warm to room temperature and then recooled to –78 °C. Next, 66 (7.6 mg, 0.029 mmol) was added as a solution in diethyl ether (1.0 mL) and allowed to warm to room temperature. After 4 hr the reaction was quenched with water and extracted with diethyl ether. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 20/1) to give 69 (3.5 mg, 37%) as a white crystalline solid. H NMR (300 MHz, CDCl3, Diagnostic Peaks) 0.98 (d, J = 6.5 Hz, 3H), 1.08 (d, J = 7.0 Hz, 1 3H), 2.93 (apparent quint., J = 6.9 Hz, 1H), 4.92-5.09 (m, 3H), 5.24-5.36 (m, 2H), 5.60-5.69 (m, 1H), 5.71-5.88 (m, 1H); HRMS (ESI) Calcd for C22H34O2 (M + Na+) 353.2457. Found 353.2460. 75 76 O O 22 (+)-Dihydro-epi-deoxyarteannuin B [22] To a CH2Cl2 (1.5 mL) solution of 69 (8.0 mg, 0.024 mmol) under an argon atmosphere in a 1dram vial was added Grubbs 2nd generation catalyst (2.0 mg, 0.0024 mmol). The sealed vial was heated to 40 °C for 4 hr then cooled to room temperature and flushed through a plug of SiO2. The resulting eluent was concentrated and purified using silica gel column chromatography (hexanes/ethyl acetate = 10/1) to give 22 (4.4 mg, 77%) as a white crystalline solid. H NMR (300 MHz, CDCl3) 0.95 (d, J = 6.5 Hz, 3H), 0.97-1.09 (m, 1H), 1.16 (d, J = 7.1 Hz, 1 3H), 1.17-1.25 (m, 2H), 1.37-1.48 (m, 1H), 1.62-1.76 (m, 6H), 1.85-1.93 (m, 1H), 2.02-2.13 (m, 3H), 3.15 (apparent quint., J = 6.9 Hz, 1H), 5.65 (q, J = 1.5 Hz, 1H); 13C NMR (75 MHz, CDCl3) 9.5, 19.9, 23.7, 24.0, 29.9, 31.1, 32.7, 39.9, 43.0, 46.9, 83.4, 122.0, 142.5, 179.5; IR (neat) 2929, 1764, 1449, 1162, 909 cm-1; HRMS (ESI) Calcd for C15H22O2 (M + Na+) 257.1518. Found 257.1518. [α]546 = 14.2° (c 0.00056, CHCl3); mp 6870 °C. Copies of NMR spectra that support our structural and stereochemical assignments are provided on pages 7893. 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 PART II: THE MEYER-SCHUSTER REACTION AND BEYOND CHAPTER 5: EVOLUTION OF THE MEYER-SCHUSTER REACTION In the course of our investigation into the total synthesis of dihydroartemisinic acid 21, we encountered the need to convert ketone 60 into α,β-unsaturated enone 74 (Figure 36). Initial attempts at olefination of 60 by the HornerWadsworthEmmons reaction proved unsuccessful, with starting material being recovered in all cases. The lack of reactivity was attributed to the steric hinderance around the ketone. Therefore, an alternative method to access α,β-unsaturated carbonyl 74 was needed. O olefination O H H R HO2C 60 dihydroartemisinic acid 21 74 Figure 36: Strategy Towards Dihydroartemisinic Acid 21 α,β-Unsaturated carbonyl compounds are common building blocks in organic synthesis.95 Their construction is most commonly achieved by the homologation of aldehydes and ketones through aldol condensations,96 Wittig olefinations,97 HornerWadsworthEmmons (HWE) olefinations,97 or other olefination methods.98 The aldol condensation is the most atom economical99 of these approaches, producing only water as a by-product. While the aldol 94 condensation avoids the need for the use of toxic stoichiometric phosphines, phosphine oxides, and phosphonates associated with the Wittig and HWE reactions, it often provides inferior yields. In addition, all of the aforementioned olefination methods are sensitive to steric congestion around the carbonyl. As a consequence, they often fail to olefinate hindered carbonyl compounds, as illustrated in our failed olefination attempts of 60. Therefore, alternative and atom-economical means of converting carbonyls into their homologated α,β-unsaturated carbonyl analogs was desirable. One potential method would be an addition/rearrangement sequence between a carbonyl and an acetylenic -bond (Figure 37). (a) stepwise annulation / electrocylic ring-opening O R1 R2 [2 +2] annulation Y R3 R3 Y R2 ring-opening 1 R G R2 O 4 electrocyclic O R1 Y R3 H (b) nucleophilic 1,2-addition / rearrangement acetylide addition O R1 R3 R2 Y R2 R1 G R3 OH R I R3 R2 3 O elimination R2 R1 3 rearrangement R R1 H E Rupe rearrangement105 R2 O MeyerSchuster R1 F redox isomerization Ir106 Pd107,108 Rh109,110 Ru111,112 R2=H O R1 R3 K J Figure 37: Atom-Economical Carbonyl Olefination Strategies The nature of the alkyne utilized dictates which reaction pathway will be followed. Highly electron-rich alkynes (i.e., ynolates,100 ynamine,101,102 or ynamides,103,104 Figure 37a) undergo a formal [2+2] cycloaddition with the carbonyl to produce an oxatene intermediate, which then 95 undergoes electrocyclic ring opening to provide the homologated enoate/enamide H. The use of terminal alkynes opens the door to an alternative pathway. The acetylide can add to ketone G via a 1,2-nucleophilic addition to give propargyl alcohol E. The resulting propargyl alcohol E is capable of undergoing a variety of reactions,105-112 including the desired MeyerSchuster rearrangement to give α,β-unsaturated carbonyl compound F (Figure 37b). To a certain extent, control over which pathway the propargyl alcohol follows can be achieved by careful selection of the reaction conditions. However, development of precise reaction conditions allowing for only the desired MeyerSchuster reaction to take place is paramount to the success of the proposed two-stage olefination method. A full understanding of the MeyerSchuster reaction is a prerequisite to the development of new reaction conditions. The MeyerSchuster reaction is a powerful rearrangement that converts propargyl alcohols into α,β-enones. The process formally involves a 1,3-shift of the hydroxyl moiety followed by tautomerization of the presumed allenol intermediate L (Figure 38).113-115 R2 OH R1 R2 • 1 R R2 3 R E R3 OH L O R1 R3 H F Figure 38: Meyer and Schuster’s Rearrangement The reaction was first reported by Meyer and Schuster in 1922, and it was conducted in acidic media at elevated temperatures.116 The requirements of strong acid and high temperatures severely limited the scope of the MeyerSchuster reaction.117 Substrates were confined to propargyl alcohols that lacked -hydrogens. The presence of -protons allowed for the Rupe rearrangement105 (Figure 39), which generally took precedence over the MeyerSchuster reaction.117 96 H H R2 R1 O H H R3 R2 R2 + H2O R1 R1 M R2 N R3 O R1 R3 R3 I O Figure 39: Competing Rupe Rearrangement Initial attempts to increase the efficiency and the scope of the MeyerSchuster reaction focused on activation of the hydroxyl moiety by transition metal catalysts. Vanadium was the first transition metal to show promise. Chabardes and co-workers demonstrated that trialkyl orthovanadates could be used to prepare aliphatic α,β-unsaturated aldehydes from propargyl alcohols with a terminal alkyne.118 While avoiding the need for strong acid, temperatures in excess of 140 oC were still necessary to promote rearrangement. At these high temperatures catalyst degradation was problematic. Catalyst decomposition was partially addressed by Pauling and co-workers, who developed more stable tris[triarylsilyl] vanadates.119 The tris[triarylsilyl] vanadate catalysts were also limited to the preparation of α,β-unsaturated aldehydes.120 Catalyst stability was advanced further by the Vol’pin lab with the development of a polymeric silyl vanadate catalyst. 124 The polymeric catalyst showed much greater stability than its monomeric counterpart. The new catalyst allowed for lower catalyst loadings, but it did nothing to address the high temperature requirements. Despite its shortcomings, this novel vanadate chemistry was used by HoffmannLa Roche in a variety of terpenoid syntheses (Figure 40).121-123 OH CHO 4 mol % tri[triphenylsilyl] vanadate 3 mol % triphenylsilanol O 75 cat. benzoic acid xylene, 140 oC, 3 h 79%, mix of E/Z O 76 Figure 40: Hoffmann-La Roche Inc. Application of Vanadate Catalyst 121 97 The orthovanadate-catalyzed rearrangement is thought to proceed by formation of an intermediate vanadate ester Q, which can then undergo a [3,3]-sigmatropic rearrangement to give allene ester R. Allene R then undergoes trans-esterification or hydrolysis to give allenol L, which tautormerizes to the α,β-enone F (Figure 41). R1 H R2 O R3 OR OR V O E OR P transEsterification ROH O Q R3 R3 R2 [3,3]-sigmatropic OR V OR rearrangement O R2 O tautomerization • R1 • R1 R2 R R3 transEsterification R1 OH R3 L O O V OR OR R1 R2 F 126 Figure 41: Proposed Orthovanadate Mechanism Attempting to expand the scope of this potentially useful reaction, Chabardes and coworkers performed a comprehensive study on the use of a variety of oxo-derivatives of vanadium, molybdenum,125 rhenium, and tungsten to catalyze the rearrangement.126 Ultimately, low transformation rates, even under high dilution, and harsh reaction conditions limited the utility of the reaction.126 The early oxovanadium work saw little use outside of industrial application, until it was resurrected by Chung and Trost in 2006. They found that in the presence of suitable electrophiles (initial work focused on imines), the vanadium allene intermediate R could be trapped to yield aldol-type products T, instead of undergoing hydrolysis to the MeyerSchuster products (Figure 42).127 98 E R3 (+) X • R1 R2 R O O N O X V O N OR OR R3 R4 R4 OR V OR R2 R3 R3 X N H R4 R2 R1 S R1 T Aldol-type product H+ O O R2 R1 MeyerSchuster rearrangement product F Figure 42: Trapping of Oxo-Vanadate Allene R Other early catalyst systems that were explored to activate the propargyl alcohol included a Ti/Cu system128 (which activated both the propargyl alcohol and the alkyne) and a vanadium-pillared montmorillonite system.129 Both catalyst systems improved the yield and lowered the reaction time of the rearrangement, but neither addressed the high temperature requirements. It was not until the development of a tetrabutylammonium perrhenate(VII)/ptoluenesulfonic acid catalyst system, in the early 1990’s by the Narasaka Lab, that the rearrangement could be carried out at room temperature without sacrificing yield. 130 An attractive feature of this catalyst system is its ability to generate α,β-unsaturated acylsilanes (Figure 43) in addition to α,β-enones. O OH 10 mol% Bu4NReO4 77 Si 10 mol% p-TsOH H2O CH2Cl2, r.t. 2 hrs, 75% Si 78 Figure 43: Narasaka Lab’s Perrhenate(VII)/p-Toluenesulfonic Acid System130 Recently, there has been a reemergence in the use of high oxidation-state metal-oxo complexes to catalyze the MeyerSchuster rearrangement. In their investigation into the 99 rhenium(V)-catalyzed nucleophilic substitution of propargyl alcohols, the Toste Lab noticed the formation of MeyerSchuster by-products.131 Using this as a starting point, Vidari and coworkers developed an efficient rhenium(V)-oxo catalyst to effect the MeyerSchuster rearrangement of both alkyl and aryl substituted alkynols132 (Figure 44). [ReOCl3(OPPh3)(SMe2)] (5 mol%) DME, 80 oC, 36 h, yield 91% d.r. 6:1 OH (6S)-79 O (S)--ionone 95% ee 80 Figure 44: Vidari’s Lab Extenstion of Toste’s Methodology132 The rhenium-catalyzed reactions proceeded with high (E)-selectivity, which can be attributed to the isomerization of the (Z)-isomer to the more stable (E)-isomer under the reaction conditions.133 Terminal alkynes leading to the corresponding α,β-unsaturated enals were also supported by this catalyst system. Due to the high oxo-philicity of the rhenium catalyst, these reactions were performed under strictly anhydrous conditions. The ability of the rhenium catalyst to be recycled multiple times, without loss of catalytic efficiency, added to the appeal of the method.132 Primary propargyl alcohols have proven to be very difficult substrates for the MeyerSchuster reaction.117 To address this shortcoming in MeyerSchuster methodology, Akai and co-workers developed a multi-catalyst system consisting of MoO2(acac)2, AuCl(PPh3) and AgOTf, which allowed for the MeyerSchuster rearrangement to proceed smoothly under mild conditions.134 Selected examples can be seen in Table 2. 100 Table 2: Mo-Au Combo Catalysis for Rearrangement of E into α,β-unsaturated Ketones F R1 R2 MoO2(acac)2 AuCl(PPh3)-Ag(OTf) (1 mol% each) OH E1-6 R3 R1 O R2 toluene, rt, 2 h R3 F1-6 Substrate E Entry Product F R1 R2 R3 Time (h) Isolated Yield (%) 1 E1 H H (CH2)2Ph 0.5 F1 93 2 E2 H H n-C7H15 0.5 F2 84 3 E3 H H Ph 1 F3 61 4 E4 H Me n-C6H13 0.25 F4 97 (E/Z 93:7) 5 E5 Me Me Ph 2.5 F5 90 6 E6 Me Ph(CH2)2 n-C4H9 2 F6 94 (E/Z 67:33) Primary (entries 1-3), secondary (entry 4), and tertiary (entries 5-6) propargyl alcohols all rearrange smoothly and in high yield. The low reaction times, typically less than an hour, and the ability of the rearrangement to proceed at room temperature can be attributed to the double activation of the propargyl alcohol. The Au and Ag catalysts activate the alkyne system, while the Mo catalyst activates the alcohol. 125,126,135-137 The catalytic activity of all the transition metal catalysts discussed thus far originates from formation of a propargyl metal ester, which then undergoes rearrangement and transesterification to give the formal MeyerSchuster rearrangement product (refer back to Figure 41). The transition metal oxo-catalysts are acting as ―hard‖ Lewis acids, preferring to coordinate to the more Lewis basic lone pair of the oxygen than the ―soft‖ -system of the alkyne (Figure 45). 101 R2 non-bonded electons on oxygen: 1 hard Lewis base R R3 OH -bonded electron of alkyne: soft Lewis base Figure 45: Lewis Basic Sites on a Propargyl Alcohol The drawback to these ―hard‖ Lewis acid catalysts is they generally require harsh reaction conditions to carry out the catalytic cycle. An alternative approach would be first to functionalize the propargyl alcohol as an ether or ester. Ideally, this pre-functionalization would allow for the rearrangement to proceed under mild reaction conditions, through activation of the alkyne -system by a ―soft‖ Lewis acid (Figure 46). There are several examples of propargyl ethers being successfully converted into α,βunsaturated carbonyl compounds.138,139 These processes typically involve hydration of the alkyne, followed by β-elimination of the ether alkoxide to generate the α,β-unsaturated carbonyl. Although the end result is the same, the reactions are not strictly MeyerSchuster rearrangements, and they involve distinct synthetic challenges. [M] R1 R2 O R3 [M] R3 O [3,3]-sigmatropic rearrangement U • R1 R2 O O V Figure 46: ―Soft‖ Lewis Acid Activation In contrast, propargyl acetates can undergo a 1,3-migration140-146 of the acetate, yielding formal MeyerSchuster products after hydrolysis (Figure 47, Eq. 16), or a 1,2migration147-150 leading to metal carbenes, which have their own unique chemistry. 102 The Yamada lab has shown that covalent activation of the propargyl alcohol as a carbonate and the rearrangement/hydrolysis sequence can be merged into a single operation using high-pressure carbon dioxide, base, and a silver catalyst (Figure 47, Eq 17).151 However, functionalization of the propargyl alcohol in a separate step is much more common. R2 R1 R3 R3 2 1R R propargyl acetate preparation OH "catalyst" rearrangement/ hydrolysis O O U E R2 O R1 R3 H F Eq (16) Standard Two-Step Conversion with Isolation of U R1 2 R OH E R3 cat. Ag+ CO2, DBU RT R3 R2 R1 R1 O O W R2 rearrangement O- Eq (17) Yamada Lab's Single Operation Conversion O C O O R3 H F 151 Figure 47: Two-Step vs. Single-Step Preparation of -Enones via Functionalization of the Propargyl Alcohol Cationic gold catalysts have received attention for their exceptional ability to activate carbon-carbon triple bonds.135-137 Zhang and co-workers exploited the affinity of gold salts towards acetylenic -bonds to efficiently convert propargylic esters to α,β-unsaturated carbonyl derivatives at room temperature.152 In the initial examination of the reaction, Zhang et al. focused on optimization of the rearrangement of propargyl esters derived from aldehydes, because they were less likely to undergo elimination to form enynes than propargyl esters derived from ketones. A variety of Au(I) and Au(III) catalysts were screened. Au(PPh 3)NTf2 provided the best results, and all further studies were conducted with this catalyst. Optimization allowed for catalyst loading to be lowered to 2 mol %, without sacrificing yield or selectivity; in almost all cases complete (E)-selectivity was achieved (Figure 48).152 103 OAc O Me 3 81 Me Au(PPh3)NTf2 (2 mol%) 2-butanone, r.t., 92% yield E only Me 3 82 Me Figure 48: Preparation of -Monosubstituted (E)-,-Unsaturated Ketones Simple extension of these optimized reaction conditions to substrates derived from ketones was not practical, with elimination to form enynes and other side reactions being substantial. Diligent optimization was required before the method was extended to the preparation of -disubstituted -unsaturated ketones. Catalyst loading had to be increased to 5 mol % and solvent conditions had to be carefully controlled (Figure 49).153 The limitation in Zhang’s methodology is that it does not work for terminal alkynes, which readily undergo Markovinkov hydration under the reaction conditions. Au(PPh3)NTf2 (5 mol%, 0.05 M in acetone) OAc O n-Bu 83 acetonitrile/H2O (80/1) 0 oC, 30 min; then r.t. 16 h 97% yield n-Bu 84 Figure 49: Preparation of -Disubstituted -Unsaturated Ketones The reaction is believed to proceed through a Au-catalyzed [3,3]-rearrangement140-146 of the propargylic ester U, followed by Au activation of the intermediate allene V giving rise to intermediate Z. Intermediate Z can then under-go hydrolysis/protiodeauration to give MeyerSchuster products F. With a suitable electrophile present, intermediate Z could be trapped to yield a wide range of products including alkenyl enol esters/carbonates, 154 αylidene-β-diketones,155 cyclopentenones,156 indoline-fused cyclobutanes,157 α-ylidene-β-keto and –malonate esters,158 indenes,159 aromatic ketones,160 and α-haloenones161,162 (Figure 50). 104 R4 R4 R1 O O Au catalyst R1 O R4 O R R2 Y R2 R2 U R3 X O 1 R3 [Au] O R4 O O R3 • R1 [Au]- R2 R3 [Au] V O 2 R O X+ = H+ hydrolysis/protiodeauration R3 F Meyer-Schuster Product O R1 4 R - [Au] X+ R1 OH R4 O R3 X+ = E+ various functional products R2 Z Figure 50: Proposed Mechanism for Au(PPh3)NTf2-Catalyzed Rearrangement152 At the same time as Zhang’s work, another gold-based catalyst system was being developed in the lab of Nolan. During their investigation of a Au(I)/Ag(I) catalyst system designed to achieve a tandem [3,3]-sigmatropic rearrangement/intramolecular hydroarylation of phenylpropargyl acetates yielding indenes,159 Nolan and co-workers noticed MeyerSchuster by-products when water was present in the reaction. Wishing to capitalize on this observation, the Nolan and Maseras labs modified the original Au(I)/Ag(I) catalyst system to convert a variety of propargyl acetates to their α,β-unsaturated carbonyl counterparts.163 The optimal conditions were determined to be 2 mol % of both (ItBu)AuCl and AgSbF6, in a 10:1 THF/H2O solvent system at 60 oC for 8 hr (Table 3). Nolan et al. found that these long reaction times could be avoided by the use of a microwave. Microwave irradiation at 80 oC allowed for the reaction to proceed in 12 minutes, without any negative effect on yield or selectivity (entry 2). A variety of substituents at the propargylic position were tolerated, including electron-rich (entry 3) and electron-deficient (entry 1) aryl groups, as well as simple alkyl groups (entry 7). Alkyl and aryl substituents on the alkyne were also tolerated. Terminal alkynes (entry 4), which have proved to be poor substrates for other methods, could also be used. However, the use of sterically bulky substituents, such as trimethylsilyl (TMS) and tert-butyl (entries 5 and 6), resulted in no reaction. In most cases, excellent (E)-selectivity was observed (e.g. entry 7).163 105 Table 3: Selected Examples of [(ItBu)AuCl]/AgSbF6 Catalyzed Rearrangement[a] OAc THF/H2O, 60 oC, 8 h 2 R R3 U7-14 Entry R1 [ltBu)AuCl]/AgSbF6 (2 mol%) R1 O N R3 R2 ltBu F7-14 Propargyl Acetate U Enone OAc N F Yield [%] F7 91 F8 90 F9 89 F10 90 F11 nr F12 nr O Bu 1 Bu F U7 F OAc O Bu 2 [b] Bu F U8 F OAc O Bu 3 Bu MeO U9 MeO OAc O H 4 H U10 OAc O Si 5 Si U11 OAc O U12 6 OAc O 82 U13 7 F13 (E:Z, 12:1) OAc O 8 Bu U14 Bu F14 94 [a] Reaction conditions: alkyne U (1 mmol), [(ItBu)AuCl]/AgSbF6 (2 mol%), THF (10 mL), H2O (1 mL). [b] performed in a microwave at 80 oC, reaction time 12 min. While Nolan and co-workers’ method proved to be fairly general, perhaps the most interesting aspect of their study was their proposed mechanism. In an attempt to understand 106 better the reaction mechanism, Nolan and Maseras investigated the reaction both experimentally and computationally.163 Based on their findings, they suggested a SN2’ type mechanism, instead of the more commonly accepted 1,3 shift of the propargyl acetate. 140-146 The gold-catalyst was believed to activate water, instead of the -system of the alkyne, to generate [(NHC)AuOH]. The AuOH complex acted as the the active catalyst (Figure 51).163 N O N N N O O H Au O 86 85 Au • O OH O 87 Figure 51: SN2’ Type Addition of Au Catalyst N N Au OH H2O 88 86 163 Nolan and co-workers later extended this method to the preparation of α,β-unsaturated carbonyls from propargyl alcohols, eliminating the functionalization step, using a similar catalyst system.164 High conversions were achieved for a variety of substrates; however, rearrangement of primary alcohols and terminal alkynes proved unsuccessful.164 The Chung lab also found success in converting propargyl alcohols into α,β-unsaturated ketones using a Au(I) catalyst system. Chung et al. screened a variety of Lewis acids including FeCl3, InCl3, GaCl3, PtCl2, Ag(OTf) and AuCl3 before selecting [Au(PPh3)](OTf). The only catalyst that gave complete conversion was [Au(PPh3)](OTf), generated from AuCl(PPh3) and Ag(OTf).165 Yields were generally moderate, and side reactions such as the Rupe rearrangement105 and enyne formation were problematic. Chung suggested a new mechanism for his Au(I) catalyst system featuring a cumulene intermediate, although it was purely speculative (Figure 52). 107 Au(1) R3 • • AA R1 R2 Figure 52: Chung’s Proposed Cumulene Intermediate165 Although cationic gold is the most commonly used ―soft‖ Lewis acid catalyst to achieve the MeyerSchuster rearrangement, many other ―soft‖ Lewis acids are also capable of catalyzing the rearrangement of propargyl acetates. During their investigation into the regioselective hydration of internal alkynes using a Hg(OTf)2 catalyst, Nishizawa and coworkers attempted to influence the reaction site by using neighboring group participation.166 They hypothesized that by placing an acetate group in the propargylic position, directed hydration could be achieved through a coordination between the mercury catalyst and the acetate functionality. To test this hypothesis, they examined the reaction of propargyl acetate 89, in water with a catalytic amount of Hg(OTf)2. Instead of obtaining the expected hydration products 91 and 92, the major product was vinyl ketone 90. Trace amounts of the dialkylated mercuric product 93 were also present (Figure 53).166 OAc 89 O cat. Hg(OTf)2 H2O 90 OAc 91 O O O Hg 93 OAc 92 O Figure 53: Attempted Regioselective Hydration166 108 Based on the initial findings, the Nishizawa lab chose to optimize the reaction for the formation of α,β-enones. A screening process revealed the optimal conditions to be 5 mol % Hg(OTf)2 with 1.5 equivalents of water in acetonitrile, at room temperature for 4 hours. Despite optimization, yields were generally moderate and side reactions could not be avoided 166 (Figure 54). OAc C7H15 O Hg(OTf)2 5 mol % 1.5 equiv H2O, CH3CN, 0.1 h C7H15 94 95 (67%) C7H15 96 (13%) Figure 54: Optimized Hg(OTf)2 Rearrangement166 In 2007, Nishizawa and co-workers further extended the method to include the formation of α,β-unsaturated esters from secondary-ethoxyalkynyl acetates. 167 The switch from aliphatic alkynes to the more electron rich ethoxy alkyne allowed for the reaction to proceed with only 1 mol % catalyst loading, compared with the 5 mol % required for aliphatic alkynes. Yields were generally high, and excellent (E)-selectivity was achieved for substrates containing alkyl substituents in the propargyl postion (Eq 18, Figure 55). However, a mixture of (E/Z)isomers were obtained when aryl substituents were introduced at the propargyl position (Eq 19, Figure 55).167 While oxygen activated alkynes, which result in α,β-unsaturated esters after rearrangement, are the most common, other heteroatoms 168 have been utilized. α,βunsaturated thioesters have been synthesized by the lab of Kataoka and Yoshimatsu. 169 They were able to convert γ-sulfur substituted propargyl alcohols to the MeyerSchuster products using polyphosphoric acid trimethylsilyl ester, albeit in low yield with significant elimination to form enynes. 109 O OAc Hg(OTf)2 1 mol % Ph 1 equiv. H2O, CH2Cl2, 2 h OAc 1 equiv. H2O, CH2Cl2, 40 min OEt 99 Eq (18) O Hg(OTf)2 1 mol % Ph OEt 98 (89%) E/Z 100:0 OEt 97 Ph Ph Eq (19) OEt 100 (77%) E/Z 50:50 Figure 55: Hg(OTf)2-Catalyzed Hydration of Ethoxypropargyl Acetates167 Very recently, the first example of a MeyerSchuster rearrangement to form an α,βunsaturated amide was realized by the Akai lab, using their aforementioned Mo/Au/Ag catalyst system (Figure 56).134 To the best of our knowledge, these two reports are the only examples of non-oxygen heteroatom activation of alkynes towards the MeyerSchuster rearrangement. OH n-C6H13 101 N Bn Ts MoO2(acac)2 AuCl(PPh3)-AgTOf (1 mol% each) toluene, rt, 2 h O n-C6H13 102 (80%) E only N Bn Ts Figure 56: α,β-Unsaturated Amide Formation134 In addition to strong acid, oxo-philic transition metals, and Lewis acid catalysis, the MeyerSchuster reaction can also be catalyzed by ruthenium complexes.170-174 The ruthenium catalysts are limited to the conversion of propargyl alcohols with a terminal acetylene. The requirement for terminal alkynes can be explained by the proposed mechanism. The reaction is a two-step process. In the first step, a carboxylic acid is added to the alkyne, presumably through a Ru-allenylidene intermediate DD, which can only be formed from a terminal alkyne. 110 The resulting enol esters FF can be isolated or cleaved with strong acid to give MeyerSchuster products GG (Figure 57). H R1 O GG R2 R1 H+ H2O CC R2 OH BB Ph O O [LnRu] R2 HO R1 R1 FF R2 OH CC [LnRu] H+ LnRu Ph O O H LnRu C C R1 R2 HO DD H R2 HO R1 O EE HO Ph 103 Figure 57: Proposed Mechanism for the Ru-Catalyzed Isomerization of Propargyl Alcohols into α,β-Unsaturated Aldehydes170 With the variety of methods now available for the mild execution of the MeyerSchuster rearrangement, it has begun to be applied to the total synthesis of complex molecules.175-178 A representative example can be found in the Weinreb lab’s efforts towards the total synthesis of the marine alkaloid chartelline A 104.178 They needed to olefinate a -lactam chemoselectively 111 in the presence of a β-lactam. They found that they could chemoselectively add lithio tertbutylacetylide to the -lactam, which set the stage for the rearrangement. Standard MeyerSchuster conditions (i.e. acid) gave the α,β-unsaturated ketone 106; however, the acidic conditions resulted in removal of the Boc protecting group. Gratifyingly, use of the milder tetrabutyl-ammonium perrhenate/p-toluenesulfonic acid catalyst system developed by the Narasaka130 lab allowed for the rearrangement to take place in quantitative yield without loss of the Boc group (Figure 58).178 Undoubtedly, the application of the MeyerSchuster reaction to other total synthesis will be soon to follow. O Cl Br Br N N Br Br N H N chartelline A 104 O O N PMP 91% N H 106 TFA, 0 oC CH2Cl2 O O N PMP N Boc 105 OH N PMP n-Bu4NReO4 p-TsOH CH2Cl2, rt 100% N Boc O 107 Figure 58: Application of the MeyerSchuster Reaction in Total Synthesis 178 In conclusion, the MeyerSchuster reaction has evolved, from a simple acid-catalyzed rearrangement of tertiary propargyl alcohols to α,β-enals, into an elegant reaction whose reactivity can be precisely controlled by activation of the alcohol and alkynyl moieties independently or in tandem. Once limited only to the formation of α,β-unsaturated ketones and aldehydes, recent advances have made access to α,β-unsaturated esters,167,179 amides,134 thioesters,169 and acylsilanes130 a reality. A vast array of metals have been used to achieve 112 this transformation, including Au, Ag, Cu, Ti, Re, V, Mo, W, and Ru. The use of the MeyerSchuster reaction has not been confined to method development; it has also found use in a variety of total synthesis.175-178 The variety of methods now available to execute the MeyerSchuster rearrangement will undoubtedly lead to an increase in the use of this powerful reaction. In the next chapter, our contributions to MeyerSchuster methodology will be discussed. Key topics will include development of a Au(III)-catalyzed rearrangement of propargyl ethynyl ethers into α,β-unsaturated esters and its use in the olefination of hindered ketones,179 efforts to control the (E/Z)-selectivity of the MeyerSchuster rearrangement,180 and the search for more affordable catalysts.181 It is important to note that our work pre-dates (and may have played a role in stimulating) much of the chemistry discussed above in Chapter 5. 113 CHAPTER 6 LEWIS ACID-CATALYZED MEYER-SCHUSTER REACTIONS: METHODOLOGY FOR THE OLEFINATION OF ALDEHYDES AND KETONES Our interest in the MeyerSchuster reaction stemmed from the desire to develop an efficient, atom-economical means to olefinate hindered ketones. As discussed in the previous chapter, classic olefination techniques such as the Wittig olefination, the HornerWadsworthEmmons (HWE) olefination, and the aldol condensation are sensitive to steric congestion around ketones. The phosphorus-based reagents used in the HornerWadsworthEmmons and HornerWittig olefinations are particularly sensitive to steric congestion and often fail to work on hindered substrates. An alternative approach would be to use a two-step olefination process, consisting of acetylide addition followed by rearrangement to the α,β-unsaturated carbonyl (Figure 59). Acetylide addition is relatively insensitive to steric congestion and is expected to proceed smoothly. Therefore, the limiting factor in the two-step olefination is the rearrangement. acetylide addition O R1 R2 G R3 Y R2 R1 R3 OH MeyerSchuster R2 O 3 rearrangement R R1 H E F Figure 59: Two-Step Olefination Strategy An obvious choice for this rearrangement is the MeyerSchuster reaction. The original MeyerSchuster reaction was conducted in acidic media at elevated temperatures. Metal oxide 114 and transition metal catalysts have also been employed to carry out the MeyerSchuster reaction, but limited scope, harsh reaction conditions, and a prevalence of side reactions have detracted from the usefulness of these methods. For a more detailed explanation of our proposed olefination strategy and a complete history of the MeyerSchuster reaction, refer to Chapter 5. Note that our initial studies and publication pre-date much of the work described in Chapter 5. In order for the two-step olefination strategy to succeed, we needed to develop mild conditions for promoting the MeyerSchuster rearrangement. Our initial approach towards the MeyerSchuster reaction derived its origins from a combination of (1) early oxo-vanadium chemistry, which showed the feasibility of using oxophilic reagents to effect the MeyerSchuster rearrangement (refer to chapter 5), and (2) the observation that Cr(VI) salts can oxidize tertiary allylic alcohols through a 1,3 rearrangement 182 (Eq 20, Figure 60). We hypothesized that these same Cr(VI) salts would be able to undergo a similar rearrangement with propargyl alcohols, to give chromate ester-allene II. Subsequent hydrolysis would yield the MeyerSchuster product F (Eq 21, Figure 60). O OH O Cr CrO3 O O H O 109 Cr Cr O 108 O O O O O OH 111 110 112 Eq (20): Cr(VI) Oxidation of Tertiary Allylic Alcohols R1 O O HO R1 R2 3 R E CrO3 Cr O R1 R2 R2 O • R3 O O II Eq (21): Our Proposed Cr(VI) Rearrangement of Propargyl Alcohols Figure 60: Cr(VI) Rearrangements 115 O Cr R3 HH R1 H+ O R2 O R3 F Our ultimate goal was to develop an efficient means to olefinate hindered ketones, so we decided to use the propargyl alcohol 113, derived from 1-hexyne addition to (-menthone 58, as our model substrate (Eq 22, Figure 61). The first chromium reagent screened was pyridinium chlorochromate (PCC) (Eq 23, Figure 61). The starting propargyl alcohol 113 was completely consumed within 24 hours, but there was no evidence of the desired product. Other catalyst systems that were capable of undergoing 1,3-rearrangements were also tested, including MnO2 and 1,1’-thiocarbonyl diimidazole (Eqs 23 and 24, Figure 61); even with elevated temperatures and extended reaction times, no reaction was observed. n-Bu n-BuLi, THF Eq (22) O 78 oC to rt 96 % 58 O "catalyst" OH n-Bu n-Bu 113 114 4.0 equiv PCC Eq (23) Complex Mixture 113 CH2Cl2, reflux Eq (24) Eq (25) 113 113 5.0 equiv MnO2 benzene, reflux S C N N N N No Reaction No Reaction DCE, reflux Figure 61: Initial Oxo-philic Approaches to the Meyer-Schuster Rearrangement In an attempt to limit the potential side reactions and to make 1H NMR interpretation more facile, propargyl alcohol 113 was replaced with 117 in further studies. Propargyl alcohol 117 was readily prepared from phenylacetylene addition to diphenyl ketone 115 (Eq 26, Figure 62). Disappointingly, treatment of 117 with PCC again resulted in a complex mixture of products (Eq 27, Figure 62). 116 O HO n-BuLi, THF Eq (26) 78 oC to rt quantitative 117 116 115 4.0 equiv PCC Eq (27) Complex Mixture 117 CH2Cl2, reflux Figure 62: Switch to Triphenyl Propargyl Alcohol 117 A concurrent report in the literature led us to consider an alternative approach. Campagne and co-workers observed MeyerSchuster by-products when attempting a gold(III)catalyzed nucleophilic substitution of propargyl alcohol 118 with ethanol (Eq 28, Figure 63).183 Based on these findings, similar conditions were applied to triphenyl propargyl alcohol 117. Propargyl alcohol 117 was treated with 10 mol % AuCl3 and 2.5 equivalents of ethanol in dichloromethane at room temperature. After 24 hrs, a 2:3 mixture of ethyl ether 121 and the desired α,β-unsaturated ketone 122 was obtained (Eq 29, Figure 63). 5.0 equiv. Ethanol 1 mol% NaAuCl4, 2H2O OH Eq (28)183 Ph O Ph C5H11 DCM, rt, 24 h Ph Ph OH 10 mol% AuCl3, DCM Ph Ph C5H11 119 60% 118 Eq (29) OEt 2.5 equiv. Ethanol rt, 24 h 117 Ph Ph 117 120 40% OEt Ph Ph O Ph Ph 121 40% Figure 63: Initial Gold(III) Experiment C5H11 122 60% Based on this initial hit, a series of experiments was conducted to determine the optimal conditions for this transformation. A quick screening of common organic solvents with 5 mol % of gold(III) chloride was performed (Table 4). Table 4: Solvent Screening With AuCl3 Catalyst Ph Ph OH Ph AuCl3, H2O Ph Solvent Ph Ph 122 117 Entry Substrate Mol % Solvent AuCl3 a O Equiv. H2O Temperature Time % C h Conversiona 0 1 117 5 CH2Cl2 6.0 25 24 0 2 117 5 H2O N.A. 25 24 0 3 117 5 H2O N.A. Reflux 24 0 4 117 5 DMF 4.0 25 24 0 5 117 5 DMF 4.0 100 24 0 6 117 8 Et2O 4.0 25 24 < 5% 7 117 15 Ethanol 0 25 24 100b 8 117 3 THF 4.0 25 24 0 9 117 3 1,4-dioxane 4.0 25 24 0 10 117 3 1,4-dioxane 4.0 60 24 0 11 117 5 CH3CN 0 25 24 < 5% 12 117 5 CH3CN 4.0 25 24 10 % 13 117 5 CH3CN 4.0 60 24 100% % conversion based on 1H NMR. b gave exclusively the ethyl ether 121 118 Surprisingly, the only two solvents that provided any conversion to the desired α,βunsaturated ketone 122 were diethyl ether (entry 6) and acetonitrile (entry 11). The lack of reactivity in dichloromethane with 6 equivalents of water and 5 mol% of AuCl 3 (entry 1) was most likely due to the lower catalyst loading; however, the switch from ethanol to water cannot be ruled out as the problem. Not wishing to increase the catalyst loading, we chose to optimize the reaction with acetonitrile as the solvent. The addition of water as an additive increased the conversion to 10%, and complete conversion could be achieved by heating the mixture to 60 o C for 24 hours (entries 12 and 13). Although complete conversion of propargyl alcohol 117 could be achieved with 5 mol % of AuCl3 in acetonitrile at 60 oC in 24 h, the reaction was not clean. There was a significant amount of an unidentified by-product present. The number of equivalents of water present in the reaction mixture was found to influence by-product formation (Table 5). Table 5: Screening of Water Equivalents Ph Ph OH Ph 117 Ph 5 mol% AuCl3 O Ph MeCN, 60 oC 24 h Ph 122 By-Product 123 Entry Substrate Equivalents of H2O Ratio of 122 to 123a 1 117 1.0 1 : 0.66 2 117 2.0 1 : 0.62 3 117 3.0 1 : 0.29 4 117 4.0 1 : 0.40 5 117 5.0 1 : 0.46 a Ratios based on integration of 1H NMR spectra for comparative purposes. Numbers do not necessarily reflect the mole fraction of the unknown product. Three equivalents of water were found to be optimal for suppression of by-product formation. Unfortunately, the by-product could not be suppressed completely. It was thought 119 that perhaps the long reaction times (24 h) were contributing to by-product formation by allowing AuCl3 to react with the solvent. The reactions were limited to 2 hours and catalyst loading was increased to achieve complete conversion (Table 6). A 10 mol % catalyst loading resulted in complete conversion, but there was still significant by-product formation (entry 3). Decreasing the temperature resulted in incomplete conversion and appeared to have little effect on by-product formation (entry 4). A catalyst loading of 20 mol % was found to allow the reaction to go to completion with little by-product formation (entry 5). Based on these results, reaction time seemed to be less important than catalyst loading in prevention of by-product formation. Table 6: Catalyst Loading X mol % AuCl3, 3.0 equiv. H2O OH Ph Ph Ph Ph Ph MeCN, 2 h Entry 1 2 3 4 5 6 a Substrate 117 117 117 117 117 117 Ph 122 117 o Mol % AuCl3 1 5 10 10 20 50 % Conversion determined by 1H NMR Temperature C 60 60 60 25 60 60 b O By-Product 123 % Conversiona < 10 50 100 50 100 100 Amount of 123b N.D. High High High Low Low Amount of 123 present determined qualitatively by 1H NMR We next examined the scope of the reaction using 20 mol % AuCl3, 3.0 equivalents of water in CH3CN at 60 oC as our standard conditions. The triphenyl tertiary propargyl alcohol 117 rearranged cleanly to afford α,β-unsaturated ketone 122 in 90 % yield (Eq 30, Figure 64). Switching to the secondary propargyl alcohol 124 proved to be detrimental. The expected α,βunsaturated ketone 125 was obtained in only a 16 % yield. The other 84 % comprised of a complex mixture of unidentified by-products (Eq 31, Figure 64). The tertiary propargyl alcohol 120 126 also proved to be a poor substrate, giving only the elimination product 127 in a 79 % yield (Eq 32, Figure 64). Even at room temperature, only the elimination product 127 was observed. Eq (30) 20 mol % AuCl3 3.0 equiv. H2O OH Ph Ph Ph 117 MeCN, 60 oC 18 hr 20 mol % AuCl3 3.0 equiv. H2O OH Eq (31) Bu a 124 OH Ph Eq (32) c 126 MeCN, 60 oC 18 hr 20 mol % AuCl3 3.0 equiv. H2O MeCN, 60 18 hr Ph Ph O Ph 122 90% O Bu 125b 16% Ph oC 127 79% a 124 was prepared from 1-hexyne addition to benzaldehyde in an 84% yield only the (E)-isomer was observed based on 1H NMR c 126 was prepared from phenyl acetylene addition to 3-pentanone in an 98% yield b Figure 64: Initial Substrate Screening with MeCN These initial results suggested that the conditions were unsuitable for substrates other than tertiary propargyl alcohols, which are incapable of undergoing elimination. In addition, the unidentified by-product was still present in all cases. Previously, we avoided the use of CH2Cl2 as a solvent, due to the lack of reactivity exhibited when 5 mol % gold catalyst loading was employed. Now using 20 mol % of AuCl3 in our reactions, CH2Cl2 was re-examined (Figure 65). Gratifyingly, the use of 20 mol % of AuCl3, in CH2Cl2, allowed for the conversion of propargyl alcohol 117 into 122 in an 87 % yield. The use of methylenechloride also increased the conversion of 124 to 125 from 16 % (in CH3CN) to 44 %. Under the new conditions, formation of 128 from 126 was also possible, although elimination to 127 was still the major 121 product. Other advantages of the CH2Cl2 system were the reaction could be carried out at room temperature, and there was no sign of any by-product formation. 20 mol % AuCl3 3.0 equiv. EtOH OH Ph Ph Eq (33) Ph 117 Ph Ph CH2Cl2, rt 18 hr Ph 122 87% 20 mol % AuCl3 3.0 equiv. EtOH OH Eq (34) O O Bu Bu 124 OH Ph Eq (35) 126 a CH2Cl2, rt, 18 hr 125a 44% 20 mol % AuCl3 3.0 equiv. EtOH Ph O Ph CH2Cl2, rt 18 hr 127 57% 128 31% only the (E)-isomer was observed based on 1H NMR Figure 65: Initial Substrate Screening with CH2Cl2 Although the switch from acetonitrile to methylenechloride eliminated by-product formation and led to higher yields, the overall efficiency of the reactions was still quite low. The gold(III) salt is presumed to catalyze the reaction through an initial activation of the -system of the alkyne.135-137 Increasing the electron density of the -system may aid the transformation by increasing gold’s affinity towards the -system. To test this theory, the ethynyl ether versions of 117 and 124 were prepared from lithium ethoxyacetylide addition to the necessary ketone. These analogs were then subjected to the reaction conditions (Figure 66). The tertiary propargyl alcohol 129 rearranged cleanly to give the α,β-unsaturated ester 130 in a 99 % yield in less than 5 minutes (Eq 36, Figure 66). Rearrangement of secondary ethoxy propargyl alcohol 132 also proceeded smoothly; however, the increased yield and lower reaction time came at the sacrifice of selectivity (Eq 37, Figure 66). 122 O Eq (36) OEt Ph n-BuLi, THF 78 oC to rt Ph 115 H 131 a Ph OEt 129 99% OH O Eq (37) 20 mol % AuCl3 5.0 equiv. EtOH Ph OH OEt 132 92% O OEt Ph 130 99% 20 mol % AuCl3 5.0 equiv. EtOH OEt n-BuLi, THF 78 oC to rt CH2Cl2, rt < 5 min Ph CH2Cl2, rt < 5 min O OEt 133a 86% Obtained as a 1:1 mixture of the (E) and (Z)-isomers Figure 66: Use of Electron-Rich Ethoxyacetylene The use of oxygen-activated alkynes expanded the scope of the reaction to a wider range of substitution patterns (Table 7). Preparation of both di- and tri-substituted olefins is supported (entries 3 and 4-7), and yields are generally excellent. Entries 1-3 highlight the evolution of the conditions. An attractive feature of these AuCl 3-catalyzed rearrangements is that they can be performed open to air with wet solvents, without sacrificing yield. The use of oxygen-activated alkynes allowed for lowering of the catalyst loading. Table 8 recounts our efforts to determine the minimum catalyst loading needed to achieve the MeyerSchuster rearrangement of ethoxyalkynyl carbinol 136. Below 5 mol % (entries 4 and 5), complete conversion is not achieved. Based on qualitative thin-layer chromatography (TLC) monitoring of the experiments, the reactions appear to proceed rapidly and then stall at a certain point. Why the reactions stall is not yet understood. Thus, 5 mol % catalyst loading was the minimum that allowed for complete conversion. The 5 mol % catalyst loading could be extended to a variety of substrates including aliphatic, aromatic, hindered, and unhindered propargyl alcohols (entries 6-8). 123 Table 7: Initial Screening of Substrates Using Oxygen Activated Alkynes R2 20 mol % AuCl3 5.0 equiv. EtOH OH R2 R1 3 E R 2 R3 R1 CH2Cl2, rt 3 O F Entry 1 R R R Substrate Time Product Yield (%)b 1a Ph H Bu 124 18 h 125 16 2 Ph H Bu 124 18 h 125 44 3 Ph H OEt 132 < 5 min 133 86 4 Ph Ph OEt 129 < 5 min 130 > 95 5 4-t-Bu- OEt 134 < 5 min 135 82 OEt 136 < 5 min 137 > 95 OEt 138 < 5 min 139 86 cyclohexyl 6 adamantyl 7 t-Bu Me a CH3CN was used as the solvent and the reaction was heated to 60 oC b Isolated yield of pure product, unless otherwise indicated Table 8: Catalyst Loading and Optimizationa R2 OH R1 JJ Entry 1 2 3 4 5 6 7 8 R1 adamantyl adamantyl adamantyl adamantyl adamantyl Ph t-Bu n-Bu R2 AuCl3, rt, 5 min OEt R2 Ph Me n-Bu 5 equiv EtOH, CH2Cl2 Substrate 136 136 136 136 136 129 138 140 a OEt R1 AuCl3 20 mol % 10 mol % 5 mol % 1 mol % 0.1 mol % 5 mol % 5 mol % 5 mol % O KK Product 137 137 137 137 137 130 139 141 Conversion (%) > 95 > 95 > 95 ca. 50b < 5b > 95 > 95 > 95 Typical procedure: Gold(III)chloride added to a solution of propargyl alcohol (1 equiv), EtOH (5 equiv), and CH2Cl2 in an open flask at room temperature. See Chapter 7 for details. b After 24 h. 124 With the rearrangement under control, attention was switched to streamlining the twostep addition/rearrangement process. It was found that the two-stage olefination could proceed without purification of the intermediate propargyl alcohol. The advantages of carrying out the rearrangement on the crude alcohol were three-fold. The time necessary to complete the overall olefination of the carbonyl was significantly reduced, the high costs associated with silica gel and solvents were reduced, and the risk of losing material in the intermediate purification process was eliminated. The new streamlined two-stage olefination process was applied to a diverse group of hindered ketones (Table 9). Benzophenone 115, adamantanone 142, and pinacolone 143 produced alkenes 130, 137, and 139 in excellent yields (entries 1-3). Dienonate 145 was obtained from verbenone 144 in nearly quantitative yield (entry 4). The ethoxyacetylide addition to camphor 146 and 3,3,5,5-tetramethylcyclohexanone 147 failed to go to completion resulting in slightly lower yields for alkenoates 148 and 149. However, the yields were still quite reasonable (entries 5 and 6). As illustrated in Table 9, we developed an atom-economical olefination strategy. The limitation in the method is the (E/Z)-selectivity (entries 3-6). The effects of the reaction temperature on the (E/Z)-selectivity of the rearrangement of ethoxyacetylene 138 can be seen in Table 10. The selectivity shifted from favoring the (Z)-isomer to favoring the (E)-isomer by lowering the reaction temperature to 78 oC (entries 4 and 5). The effect of the temperature was found to be independent of reaction time (entries 3 and 5). While temperature could be used to influence the formation of the major isomer, all attempts to obtain a single isomer by varying the temperature were unsuccessful. Exposure of the purified (E/Z)-mixture of α,βunsaturated ester 139 to the normal reaction conditions resulted in no observable change in the (E/Z)-ratio. This suggests that isomerization under the reaction conditions is not a contributing factor to the (E/Z)-selectivity. Ultimately, temperature control alone was insufficient to control selectivity. 125 Table 9: Olefination of Hindered Ketonesa R2 R1 1. BuLi, H O G Entry 2. AuCl3 (5 mol %) EtOH (5 equiv), CH2Cl2 Substrate Ph R3 R1 KK Yield (%) Ph O 115 Ph O 2 3 O Product Ph 1 R2 OEt, THF CO2Et CO2Et O 143 O 137 99 139 99b 96c 144 CO2Et 145 68% (84%)c,d 5 146 O 6 CO2Et 147 O CO2Et 148 149 a d 91 CO2Et 142 4 130 73 (85)d See Chapter 7 for details. bCa. 4:3 ratio of olefin isomers. c Ca. 2:1 ratio of olefin isomers. 10 mol % of AuCl3 employed. Value in parentheses is the calculated yield based on recovered ketone 126 Table 10: Temperature Effect on (E/Z)-Selectivitya AuCl3 (5 mol %) EtOH (5 equiv), OH OEt O CH2Cl2 OEt 138 Entry 1 2 3 4c 5 139 o Time 5 min 5 min 24 h 20 min 17 h Temperature C 25 0 25 78 78 to 30 E/Z Ratiob 3:4 2:3 2:3 2:1 2:1 Yield (%) 93 99 99 84 99 a Typical procedure: Gold(III) chloride added to a solution of propargyl alcohol 138 (1 equiv), EtOH (5 equiv), and CH2Cl2 at the indicated temperature. b Determined by 1H NMR c 20 mol % of AuCl3 was used To identify conditions that afford the enoate products stereoselectively, we examined the rearrangement of secondary propargyl alcohols (prepared by addition of lithium ethoxyacetylide to the corresponding aldehydes). The dampened reactivity of secondary propargyl alcohols, compared to that of the tertiary alcohols which ionize more easily, along with the greater steric distinction between the aliphatic substituent and the hydrogen were hoped to make stereocontrol more facile. A qualitative screening of gold catalysts, additives, and solvents was conducted, and the results can be seen in Figure 67. catalyst (10 mol%) additive (10 equiv) OH 150 OEt O OEt solvent, r.t 151 P.AuCl catalyst: AuCl AgSbF6 > AuCl3, AuCl, Ph3 >> AgSbF6 additive: EtOH >> CF3CH2OH, AcOH, PhOH, morpholine, none solvent: THFCH2Cl2 > THF, CH2Cl2, EtOH, H2O Figure 67: Qualitative Screening for Optimal Conditions 127 Of the protic additives screened, which were believed to assist in the formal 1,3-hydroxy migration, ethanol was the most effective. A mixed solvent system of 1:1 THF and CH2Cl2 proved superior to the use of either solvent independently. Although all of the gold catalysts screened performed well, a mixed AuCl/AgSbF6 system provided the best results. Similar to previous results, a 5 mol % catalyst loading was found to be optimal. The new rearrangement conditions were then tested on a series of representative secondary alcohols (Table 11). Neopentyl alcohol 150 yielded the non-enolizable enoate 151 with almost complete selectivity (entry 1a). Alkyl-substituted alcohols 152, 154, and 156 gave enoates 153, 155, and 157 (entries 2a-4a) without any elimination to the enynes. Addition of camphorsulfonic acid (CSA) to the reaction mixture improved the selectivity of most reactions (entries 1b-6b). Table 11: Meyer-Schuster Reaction of Ethoxyalkynyl Carbinolsa 5 mol% AuCl/AgSbF6 10 equiv EtOH OH O R LL Entry 1aa R THFCH2Cl2 (1:1) rt, 3060 min OEt Substrate OEt MM Yield (%, E/Z) Product OH O 1bb 91 (97:3) OEt 93 (E only) OEt 150 151 a O OH 2a 2bb OEt OEt O OH b 3b 5 90 (86:14) 155 154 4aa 91 (63:37) OEt 5 OEt 82 (99:1) 153 152 3aa 80 (94:6) O OH 4bb 75 (57:43) OEt OEt 157 156 128 78 (70:30) Table 11: Continued 5 mol% AuCl/AgSbF6 10 equiv EtOH OH R LL OEt THFCH2Cl2 (1:1) rt, 3060 min Entry Substrate 5aa OH O R OEt MM O 72 (40:60) OEt b 5b OEt 70 (40:60) 159 158 6aa Yield (%, E/Z) Product O OH 6bb 90 (47:53) OEt OEt 92 (91:9) 133 132 a Solutions of AgSbF6 (5 mol %) and AuCl (5 mol %) in 1:1 THFCH2Cl2 added sequentially to ethoxyalkynl carbinol (1.0 equiv) and EtOH (10 equiv) in 1:1 THFCH2Cl2. No camphorsulfonic acid (CSA) was included unless otherwise indicated. b 1.0 Equiv CSA. The addition of camphorsulfonic acid (CSA) not only improved the selectivity of the reaction but also accelerated the reaction, while addition of 2,6-di-tert-butyl)-4-methylpyridine (DTBMP, an acid scavenger) inhibited the reaction. These results suggest that exchangeable protons play a significant role in the gold-catalyzed rearrangement. To test whether the gold catalyst has an active role in the reaction mechanism, or if the reaction is simply catalyzed by acid generated under the reaction conditions, a series of control experiments were conducted. Propargyl alcohol 138 was exposed to a variety of acidic conditions (Figure 68). Use of 20 mol % of TsOH·H2O in place of the gold catalyst resulted in a 70:15 mole ratio of 138 and 139, along with unidentified product, after 18 hours (Eq 38). Aqueous acidic conditions were also tested. The use of 1 equivalent of AcOH in a 1:1 THFH2O mixture resulted in no reaction, while the use of 50 mol % HCl resulted in incomplete conversion after 1 hour. The use of strong acids like HCl is much more likely to promote undesired side reactions and can only be used with acid-stable compounds. Therefore, although acid may be generated under the reaction conditions, there seems to be an important role for the gold catalyst. 129 OH O "acid" OEt OEt 138 Eq (38) 139 TsOH (20 mol %) EtOH (5 equiv), 138 CH2Cl2 Eq (39) 138 138 + 70% 1 equiv. AcOH 139 15% no reaction THF:CH2Cl2 1:1 50 mol% HCl Eq (40) 138 THF:CH2Cl2 1:1 1h 138 33% + 139 67% Figure 68: Gold Salts vs. Acid Catalysts The exact role of the gold catalyst in the reaction mechanism is still unknown. One possible mechanistic sequence for the MeyerSchuster rearrangement of 150 to 151 can be seen in Figure 69. Coordination between the cationic gold catalyst and the electron-rich system of the alkyne allows for -addition of ethanol to generate 1,1-diethoxyallene 161, after immediate expulsion of water. The formation of allene 161 is consistent with the lack of βhydroxy ester byproducts in any of the reactions, which is in sharp contrast to the acidic hydrolysis of ethoxyalkynyl carbinols.176,177 The gold catalyst may or may not promote subsequent reincorporation of water to produce 162. The stereoselectivity of the reaction is presumably set in this step. Control experiments have shown that there is no isomerization of the (Z)-enoates to the (E)-enoates under the reaction conditions. Therefore, we believe that the non-thermodynamic product distribution is the result of kinetic control. Collapse of 162 yields α,β-unsaturated ester 151. 130 OH O [Au+], EtOH OEt OEt 151 150 [Au+] H EtOH O Au Au+ + EtOH H2O • OEt 160 OEt OEt 161 OH OEt OEt H2O [Au+] H 162 Figure 69: Hypothesis on the MeyerSchuster Reaction Pathway The mechanism depicted in Figure 69 suggests that there is incorporation of an external alcohol into the α,β-unsaturated ester, based on competitive collapse of tetrahedral intermediate 162. Alcohol 150 was subjected to the standard reaction conditions with replacement of ethanol with n-propanol, following the assumption that ethanol and n-propanol would be ejected from the tetrahedral intermediate at a similar rate. α,β-Unsaturated ethyl (151) and n-propyl (163) esters were obtained in an approximately 1:1 ratio (Figure 70). Exposure of enoate ester 151 to the same reaction conditions yielded no evidence of transesterification. Therefore, n-propanol incorporation must occur before product formation, which is consistent with our mechanistic hypothesis. OH 5 mol % AuCl/AgSbF6 10 equiv n-PrOH OEt 150 THFCH2Cl2 (1:1) r.t. 30 min O O 151 50% Figure 70: Incorporation of External Alcohol Additive 131 OPr OEt 163 50% Our research to this point has mainly focused on the use of cationic gold catalysts to achieve the MeyerSchuster reaction. The use of cationic gold and other precious metal catalysts offer many advantages in synthesis. 184 The ability of these catalyst to engage in redox chemistry, through facile interconversion between oxidation states, has contributed to their use in a variety of applications. However, when selectively using these catalysts for their -Lewis acidity, their ability to engage in redox chemistry is unnecessary and often makes interpretation of the reaction mechanism difficult. Another limitation of the precious metal catalysts, which is often ignored or confined to a footnote, is their high cost. This makes low catalyst loading and high turnover essential for developing a cost effective method. Working under the hypothesis that the MeyerSchuster rearrangement of ethoxyalkynyl carbinols by late transition metal-catalysts stems from simple Lewis acid/base interactions, we became interested in identifying other Lewis acids that could provide similar or superior activity. Table 12 provides a summary of our catalyst screenings, which focused primarily (though not exclusively) on soft transition metal salts. In an open reaction vessel, ethoxyacetylene 154 was dissolved in methylenechloride and treated with reagent-grade ethanol (ca. 510 equiv) and 1 mol % of various Lewis acid catalysts (and one protic acid, camphorsulfonic acid, entry 1) at room temperature. Reactions were allowed to proceed for up to 24 hours or until TLC analysis showed consumption of starting material. The lone protic acid (CSA) is listed first; the rest of the table is arranged by the typical cost of the catalysts (per mole), from lowest to highest. From this general screening emerged three top choices: copper(II) triflate, indium(III) chloride, and scandium(III) triflate, and all further studies were conducted with these catalysts. Of these, indium(III) chloride was the least reactive; the copper and scandium triflates were comparable in reactivity. All three are air-stable powders and are convenient to handle and use. It should be noted that many of the catalysts (most notably HfCl 4 and PtCl4) that provided trace conversion with 1 mol % catalyst loading could achieve complete conversion with increased catalyst loading. Even with the higher catalyst loading, HfCl 4 was competitive with the aforementioned catalysts in terms of cost. 132 Table 12: Screening of Alternative Catalysts OH 1 mol% catalyst C5H11 154 OEt CH2Cl2/EtOH CO2Et C5H11 155 Entry Catalyst Time (h) Conversion E/Z Ratioa 1 CSA 24 0 _ 2 NiCl2·xH2O 24 0 _ 3 CuI 24 0 _ 4 Cu(OAc)2 24 Trace n.d. 5 CeCl3·7H2O 24 0 _ 6 HfCl4 24 Trace n.d. 7 ZrCl4 24 Trace n.d. 8 Cu(C5H4F3O2)b 24 0 _ 9 Hg(CF3CO2)2 24 Trace n.d. 10 NaReO4 24 0 _ 11 Cu(OTf)2 1.5 100% 89:11 12 InCl3 24 100% 84:16 13 Ag(OTf)2 0.3 100% 43:57 14 Pd(OAc)2 24 <100%c 63:37 15 Sc(OTf)3 1.2 100% 90:10 16 PtCl4 24 Trace 17 AuCl3 24 a As observed by 1H NMR. b Copper(II) trifluoroacetylacetonate detected by TLC analysis but not by 1H NMR after workup. n.d c <100% c >20:1 Ethoxyacetylene 197 could be In an attempt to gain more insight on these Lewis acid-catalyzed MeyerSchuster reactions, the effects of various additives were explored. Two acidic and two basic additives were chosen: 1 mol % CSA, 1.0 equivalent acetic acid (AcOH), 1 mol % 2,6-di-tert-butyl-4methylpyridine (DTBMP), and 1.0 equivalent magnesium oxide (MgO). 133 The effect of the additives on the reaction rate (qualitatively) and the stereoselectivity (quantitatively) of the reaction are summarized in Table 13. Table 13: Effect of Additives 1 mol% catalyst <additive> OH C5H11 154 OEt CO2Et C5H11 CH2Cl2/EtOH 155 Entry Catalyst Additive Amount Time (h) E/Z Ratioa 1 InCl3 None _ 24 84:16 2 InCl3 CSA 1 mol % 6 67:33 3 InCl3 DTBMP 1 mol % 24 75:25 4 InCl3 AcOH 1.0 equiv 24 91:9 5 InCl3 MgO 1.0 equiv 24 78:22 6 Cu(OTf)2 None _ 1.5 89:11 7 Cu(OTf)2 CSA 1 mol % 1.5 86:14 8 Cu(OTf)2 DTBMP 1 mol % 24 76:24 9 Cu(OTf)2 AcOH 1.0 equiv <1 89:11 10 Cu(OTf)2 MgO 1.0 equiv 24b 76:24 11 Sc(OTf)3 None _ 1.2 90:10 12 Sc(OTf)3 CSA 1 mol % <1 92:8 13 Sc(OTf)3 DTBMP 1 mol % 4 91:9 14 Sc(OTf)3 AcOH 1.0 equiv <1 92:8 15 Sc(OTf)3 MgO 1.0 equiv 24c n.d. a As observed by 1H NMR b Conversion (60%) 134 c Conversion (30%) Lewis acid and protic acids catalyze the MeyerSchuster reaction, so one would expect acidic additives to accelerate the reaction and basic additives to quench or retard the reaction. This hypothesis is supported by the results presented in Table 13. However, the fact that basic additives retard but do not quench the reaction suggests that protic acid, though helpful, is not required for catalytic activity. Therefore, one can choose between a short reaction time (e.g., entries 9 or 14) and reaction conditions that are presumably free of protic acid (e.g., entry 5). Since the Sc(OTf)3 and Cu(OTf)2 were more reactive and gave better (E)-selectivity than the InCl3 catalyst in the absence of additives, they were used in the two-step olefination of aldehydes and ketones. As expected, ethoxyacetylene addition to both ketones and aldehydes proceeded smoothly and with high isolated yields (Table 14). Table 14:Ethoxy Acetylene Addition O R1 R2 G a H OEt n-BuLi, THF 78 oC to rt R1 R2 OH JJ OEt Entry Substrate R1 R2 Yield (%)a 1 164 n-C7H15 H > 99 2 165 Cyclohexyl H 76 3 166 t-Bu H 97 4 143 t-Bu Me 83 5 131 Ph H 92 Isolated yield The resulting secondary and tertiary propargyl alcohols were treated with either 1 mol % of Cu(OTf)2 or Sc(OTf)3 to complete the two-step olefination (Table 15). Use of EtOH as a cosolvent instead of an additive provided superior selectivity (entries 1-4). Aliphatic substitution at the propargyl position was universally tolerated. In the case of aliphatic di-substituted alkenes, (E)-selectivity increased as branching increased (entries 1-8). Stereoselection eroded when tri135 substituted alkenes were formed, or an aromatic group was introduced at the propargyl position (entries 9-12). The two catalyst systems gave similar results; however, scandium (III) triflate performed slightly better. Scandium(III) triflate also has the advantage of being recoverable and reusable after aqueous work upwithout loss of activity.185 Table 15: Scandium and Copper Rearrangements R1 R2 OH OEt Entry Substrate 1a 2a R1 CO2Et R1 CH2Cl2/EtOH (4:1) R2 Product Cu(OTf)2 154 Sc(OTf)2 n-Heptyl 155 H Yield (%)b E/Z Ratioc 53 91:9 63 E only 3 Cu(OTf)2 64 97:3 4 Sc(OTf)2 70 E only 71 E only 75 E only 94 E only 97 E only 86 57:43 89 58:42 90 76:24 93 77:23 5 156 6 7 9 150 12 Cyclohexyl 157 H Cu(OTf)2 tert-Butyl 151 H Sc(OTf)2 138 10 11 Cu(OTf)2 Sc(OTf)2 8 a Catalyst R2 1 mol% catalyst Cu(OTf)2 tert-Butyl 139 Me Sc(OTf)2 132 Cu(OTf)2 Phenyl 133 H Sc(OTf)2 EtOH (5.0 equiv) in CH2Cl2 b Isolated yield c 1 As observed by H NMR Up until this point, our substrates had been limited to those containing aliphatic and aryl substituents. To test the functional group compatibility of this method, N-Boc-serine methyl ester 167 was converted to into the tert-butyldimethylsilyl (TBS) ether 168. It was then included 136 in the reaction mixture during the conversion of 156 to 157 (Figure 71). Ester 157 was isolated in 75 % yield, and the TBS protected N-Boc-serine methyl ester was recovered in near quantitative yield. The high recovery of 168 indicates that our current MeyerSchuster conditions are compatible with typical alkyl esters, amine carbamates, and silyl ethers. OH CO2Et OEt Boc H N 156 1 mol% Sc(OTf)3, 1 h O rt, CH2Cl2/EtOH (4:1) OMe 157, 75% yield Boc H N OR O OMe OTBS R = H: 167 R = TBS: 168 168, 99% recovery Figure 71: Functional Group Compatability In our earlier gold(III) chloride catalyst work, we developed a two-stage olefination method that avoided the need to purify the propargyl alcohol intermediate. The use of the crude propargyl alcohol allowed for higher yields over the two steps and greatly reduced the time needed to complete the overall olefination. The use of 1 mol % scandium(III) triflate in place of 5 mol % of the more expensive gold(III) chloride had no negative effect and yields were generally excellent (Table 16). A series of trisubstituted olefins were prepared (entries 1-7) in good to excellent yield, although control of stereochemistry was limited (entries 1-2, 7). Entries 1-4 illustrate the ability of this method to olefinate hindered ketones in excellent yields. Preparation of 169 is of particular interest since we were unable to prepare it through HornerWadsworthEmmons reaction (refer to Chapter 3). 137 the use of the Table 16: Two-Stage Olefination of Ketones and Aldehydes O R1 1.Li R2 R2 OEt 2.MeyerSchustera G Entry Substrate 1 ()-menthone 58 CO2Et R1 KK Product CO2Et Yield (%)b E/Z Ratioc 98 n.d.e 97 58:42 99 _ 96 _ 60 _ 78 _ 93 39:61 80 E only 169 2 verbenone 144 CO2Et 3 145 benzophenone 115 CO2Et 4 adamantanone 142 130 CO2Et 137 CO2Et 5 4-tert-butyl cyclohexanone 170 171 CO2Et 6 cycloheptanone 172 7 acetophenone 174 173 CO2Et 175 8 CO2Et pivaldehyde 166 151 138 Table 16: Continued O R1 1.Li R2 R2 OEt 2.MeyerSchustera G Entry CO2Et R1 KK Substrate Product Yield (%)b E/Z Ratioc 94 89:11 94 89:11 97 36:61d 80 E only CO2Et 9 ()-perillaldehyde 176 177 CO2Et 10 4-methoxybenzaldehyde 178 11 2-furaldehyde 180 179 O O CO2Et 181 F 12 pentafluorobenzaldehyde 182 F CO2Et F 183 F F a b 1 mol % Sc(OTf)3, CH2Cl2/EtOH (4:1) Isolated yield c Determined by 1H NMR unless otherwise stated d E and Z isomers were isolated separately e Unable to determine the E/Z ratio by 1H NMR The method also proved efficient for the preparation of disubstituted olefins (entries 812). Excellent (E)-selectivity was achieved for aliphatic substituents (entry 8); however, selectivity diminished when aryl (entries 10-12) or vinyl (entry 9) substituents were introduced (exception is entry 12). Both electron-rich (entries 10 and 11) and electron-deficient (entry 12) aryl substituents at the propargyl position were tolerated. In our earlier studies using gold and silver salts to catalyze the MeyerSchuster reaction, we suggested a mechanism in which the alcoholic additive becomes incorporated into 50 % of the product via an intermediate 1,1-diethoxy-allene 161 (refer to Figure 69). This hypothesis was supported by the observation that the ethyl and propyl esters were obtained in a roughly 1:1 ratio when n-propanol was used as the additive. This experiment was repeated 139 with the use of the scandium(III) triflate catalyst. Instead of obtaining the expected 1:1 ratio of ethyl and propyl esters, we obtained a ratio of 25:75 (as estimated by 1H NMR, Eq 38 Figure 72). In other words, there was only a 25 % retention of the original ethoxy group. Based on our previous mechanism, the ethoxy group should be retained in at least 50 % of the product, even in the presence of excess n-propanol. The possibility of trans-esterification was eliminated by a control experiment in which pure ethyl ester 151 was subjected to the reaction conditions. There was no evidence of the propyl ester product, even after extended reaction times (Eq 39, Figure 72). 1 mol% Sc(OTf)3 5 equiv n-PrOH OH Eq (38) CH2Cl2 OEt O O OEt OPr 151 150 163 25 : 75 ratio! O OEt Eq (39) 1 mol% Sc(OTf)3 5 equiv n-PrOH 151 CH2Cl2, 24 h O OPr 163 not observed Figure 72: Test for trans-Esterification These observations led us to suggest a slightly modified mechanism for the scandium(III) triflate catalyst (Figure 73). Based on the consistent lack of β-hydroxy ester byproducts, the scandium(III) triflate-catalyzed MeyerSchuster reaction most likely also proceeds via intermediate 1,1-diethoxy-allene NN. Subsequent addition of a second equivalent of ethanol would give rise to tetrahedral intermediate OO, which can hydrolyze via intermediate PP to reach the α,β-unsaturated ester MM. Invoking ortho-ester intermediate OO can account for up to a 67 % incorporation of the alcohol additive; however, we observed a 75 % incorporation. One possible explanation would be dynamic alcohol exchange reactions of ortho-ester OO. 140 OH 1 mol % Sc(OTf)3 R LL CO2Et R CH2Cl2/EtOH OEt MM EtOH H2O R NN • EtOH EtOH OEt OEt R OEt OEt OEt OO OH OEt OEt H2O EtOH R PP Figure 73: Revised Mechanistic Hypothesis for Scandium(III)Salts A series of experiments were conducted with varying amounts of n-PrOH. In every case, there was almost complete statistical incorporation of the n-propanol additive (Table 17). These results provide support for a hemi-labile intermediate OO, that can undergo partial equilibrium before proceeding irreversibly to the α,β-unsaturated ester MM. The equilibrium can also account for the high (E)-selectivity observed in the case of disubstituted olefins with the scandium catalyst, by allowing for the thermodynamic establishment of the olefin geometry at the stage of the ortho-ester. We have developed an efficient, two-step, atom-economical method for the olefination of aldehydes and ketones. The first step was acetylide addition to a carbonyl substrate. Acetylide addition is relatively insensitive to steric congestion, which allowed for the use of hindered ketones as substrates. The second step was the conversion of the resulting propargyl alcohol to the desired α,β-enone by the use of MeyerSchuster reaction. The MeyerSchuster reaction was found to proceed more readily and with lower catalyst loading when the propargyl alcohol contained an oxygen-activated alkyne. 141 Table 17: Incorporation of External Alcohol Additive 1 mol% Sc(OTf)3 <n-propanol> OH 150 a CO2R CH2Cl2 OEt R = Et, 151 R = nPr, 163 Entry PrOH (equiv) 150:PrOH 151:163a 1 1 50:50 55:45 2 2 33:67 42:58 3 3 25:75 38:62 4 5 17:83 25:75 5 10 9:91 22:78 6 100 1:99 16:84 As observed by 1H NMR Gold(III) chloride was the first catalyst to provide consistent results. The relatively expensive gold catalyst required a 5 mol % catalyst loading and showed little stereochemical control. The lack of stereochemical control was partially addressed by the use of a Au(I)/Ag(I) catalyst system. Inclusion of 1.0 equivalent of CSA allowed for the preparation of a variety of disubstituted olefins with high (E)-selectivity. A screening of various Lewis acids revealed that the less expensive Sc(OTf)3 could be substituted for the precious metal catalysts without sacrificing yield. Not only did the use of Sc(OTf)3 allow for the catalyst loading to be lowered to 1 mol %, it also proved significantly more adept in controlling the stereochemistry of the rearrangement. Future plans are to test scandium(III) triflate’s ability to catalyze the MeyerSchuster rearrangement of propargyl alcohols derived from aliphatic alkynes, which have proven to be poor substrates for the gold catalysts. We would also like to determine whether the electronic nature of the alkyne effects the (E/Z)-selectivity of the reaction, for both the gold and scandium-based catalysts. Finally, further mechanistic experiments will be conducted to identify the exact role of gold and scandium salts in the reaction mechanism. 142 In conclusion, we outlined and discussed a strategy for the olefination of aldehydes and ketones using the MeyerSchuster rearrangement of ethoxyalkynyl carbinols. The method appears to be limited only by the ability to access the requisite propargyl alcohols via ethoxyacetylide addition to carbonyls. This method is likely to find widespread application in organic synthesis, particularly for its unique ability to complete the olefination of hindered ketones in excellent yield. 143 CHAPTER 7 EXPERIMENTAL: MEYERSCHUSTER GENERAL EXPERIMENTAL PROCEDURES: 1 H-NMR and 13 C-NMR spectra were recorded on 300 MHz spectrometer using CDCl3 as the deuterated solvent. The chemical shifts () are reported in parts per million (ppm) relative to the residual CHCl3 peak (7.26 ppm for 1H NMR, 77.0 ppm for 13 C NMR). The coupling constants (J) were reported in Hertz (Hz). IR spectra were recorded on an FTIR spectrometer on NaCl discs. Mass spectra were recorded using chemical ionization (CI) or electron ionization (EI) techniques. Yields refer to isolated material judged to be ≥95 % pure by 1H NMR spectroscopy following silica gel chromatography. All chemicals were used as received unless otherwise stated. Tetrahydrofuran (THF) was purified by passing through a column of activated alumina. The n-BuLi solutions were titrated with menthol dissolved in tetrahydrofuran using 1,10-phenanthroline as the indicator. The purifications were chromatography using silica gel F-254 (230-499 mesh particle size). 144 perfomed by flash OH 113 (2S, 5R)-1-Hex-1-ynyl-2-isopropyl-5-methyl-cyclohexanol [113] To a THF solution (5 mL) of 1-hexyne (0.44 mL, 3.94 mmol) was added n-BuLi (1.29 mL, 2.96 mmol, 2.29 M) dropwise over 5 min at –78 °C under argon atmosphere. The solution was allowed to warm to 0 °C over 1 h and held at 0 °C for an additional 30 min. The solution was then recooled to –78 °C and ()-menthone (0.34 mL, 1.97 mmol) was added in one portion. The solution was allowed to warm to room temperature over 1 h and held at room temperature for an additional 3 h. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO 4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 20/1 – 10/1) to give (2S, 5R-1-hex-1-ynyl-2-isopropyl-5-methylcyclohexanol (113) in 96 % yield (0.45 g) as a clear colorless oil. H NMR (300 MHz, CDCl3): 0.83-0.96 (m, 12H), 1.23-1.57 (m, 10H), 1.65-1.78 (m, 2H), 1.92 1 (dt, J = 13.6, 3.7 Hz, 1H), 2.20 (t, J = 7.2 Hz, 2H), 2.35-2.45 (m, 1H); 13 C NMR (75 MHz, CDCl3): 13.5, 18.3, 18.6, 20.5, 21.8, 21.9, 23.9, 27.3, 28.2, 30.9, 34.9, 50.5, 50.9, 71.8, 83.7, 85.0 For full characterization refer to Spino, C.; Beaulieu, C. J. Am. Chem. Soc. 1998, 120(45), 1183211833. 145 146 147 HO 117 1,1,3-Triphenyl-2-propyne-1-ol [117] To a THF solution (6 mL) of phenylacetylene (0.85 mL, 7.69 mmol) was added n-BuLi (3.3 mL, 6.59 mmol, 2.0 M) dropwise over 5 min at –78 °C under argon atmosphere. The solution was allowed to warm to 0 °C over 1 hr and held at 0 °C for an additional 30 min. The solution was then recooled to –78 °C and benzophenone (1.0 g, 5.52 mmol) was added in one portion. The solution was allowed to warm to room temperature over 1 h and held at room temperature for an additional 3 hr. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO 4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 20/1 – 10/1) to give 1,1,3-triphenyl-2-propyne-1-ol (117) in 100 % yield (1.57 g) as a white crystalline solid. H NMR (300 MHz, CDCl3): 2.85 (s, 1H), 7.28-7.42 (m, 9H), 7.48-7.53 (m, 2H), 7.64-7.73 (m, 1 3H); For full characterization refer to Sanz, R.; Martinez, A.; Alvarez-Gutierrez, J. M.; Rodriguez, F. Eur. J. Org. Chem. 2006, 6, 13831386. 148 149 OH 118 1-Phenyl-hept-2-yn-1-ol [118] To a THF solution (6 mL) of 1-hexyne (0.76 mL, 6.59 mmol) was added n-BuLi (2.47 mL, 5.65 mmol, 2.29 M) dropwise over 5 min at –78 °C under argon atmosphere. The solution was allowed to warm to 0 °C over 1 h and held at 0 °C for an additional 30 min. The solution was then recooled to –78 °C and benzaldehyde (0.48 mL, 4.71 mmol) was added in one portion. The solution was allowed to warm to room temperature over 1 hr and held at room temperature for an additional 3 hr. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 20/1 – 10/1) to give 1-phenyl-hept-2-yn-1-ol (118) in 84 % yield (0.75 g) as a clear yellow oil. H NMR (300 MHz, CDCl3): 0.92 (t, J = 7.2 Hz, 3H), 1.32-1.61 (m, 4H), 2.08 (dd, J = 6.1, 3.1 1 Hz, 1H), 2.28 (td, J = 6.9, 1.9 Hz, 2H), 5.46 (d, J = 6.0 Hz, 1H), 7.29-7.46 (m, 3H), 7.55 (d, J = 7.2 Hz, 2H); For full characterization refer to Fuerstner, A.; Shi, N. J. Am. Chem. Soc. 1996, 118(49), 1234912357. 150 151 OH 126 3-Ethyl-1-phenyl-1-pentyn-3-ol [126] To a THF solution (6 mL) of phenylacetylene (1.55 mL, 14.1 mmol) was added n-BuLi (4.6 mL, 10.6 mmol, 2.29 M) dropwise over 5 min at –78 °C under argon atmosphere. The solution was allowed to warm to 0 °C over 1 hr and held at 0 °C for an additional 30 min. The solution was then recooled to –78 °C and 3-pentanone (0.75 mL, 7.1 mmol) was added in one portion. The solution was allowed to warm to room temperature over 1 hr and held at room temperature for an additional 3 hr. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO 4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 20/1 – 10/1) to give 3-ethyl-1-phenyl-1-pentyn-3-ol (126) in 100 % yield (1.33 g). light yellow oil; 1H NMR (300 MHz, CDCl3) 1.11 (t, J = 7.4 Hz, 6H), 1.77 (q d, J = 7.4, 4.5 Hz, 4H), 1.94 (br s, 1H), 7.28-7.33 (m, 3H), 7.40-7.44 (m, 2H). For full characterization refer to Galli, C. et al. J. Org. Chem. 1994, 59, 67866795. 152 153 Typical procedure for solvent screening with AuCl 3, Table 4 To a MeCN solution (1 mL) of 1,1,3-triphenyl-2-propyne-1-ol 117 (23.3 mg, 0.82 mmol) in a 1 dram vial under Ar was added AuCl3 (1.2 mg, 0.004 mmol) at room temperature. After 24 hr, the reaction mixture was filtered through a plug of SiO2 with the aid of an ethyl acetate/hexane mixture (1/7). The filtrate was concentrated and resulting residue was subjected to 1H NMR. Conversion to 122 was 5 %. Typical procedure for screening of water equivalence, Table 5 To a MeCN solution (1 mL) of 1,1,3-triphenyl-2-propyne-1-ol 117 (23.3 mg, 0.82 mmol) in a 1 dram vial under Ar was added water (44 L, 2.46 mmol) followed by AuCl3 (1.2 mg, 0.004 mmol). Reaction mixture was heated to 60 oC in an oil bath for 24 hr, then allowed to cool to room temperature. Reaction mixture was concentrated and the resulting residue was subjected to 1H NMR. Qualitative ratio of 122 to the unidentified by-product was determined. Typical procedure for AuCl3 catalyst loading in CH3CN, Table 6 To a MeCN solution (1 mL) of of 1,1,3-triphenyl-2-propyne-1-ol 117 (23.3 mg, 0.82 mmol) in a 1 dram vial under Ar was added water (44 L, 2.46 mmol) followed by AuCl3 (2.4 mg, 0.008 mmol). Reaction mixture was heated to 60 oC in an oil bath for 2 hr, then allowed to cool to room temperature. Reaction mixture was concentrated and the resulting residue was subjected to 1H NMR. Qualitative ratio of 122 to the unidentified by-product was determined. Typical procedure for preparation of α,β-unsaturated ketones 122 and 125 and enyne 127 in MeCN, Figure 63 To a MeCN solution (8 mL) of 1,1,3-triphenyl-2-propyne-1-ol 117 (102 mg, 0.36 mmol) in a 2neck, 25 mL round bottom flask was added water (19 L, 1.08 mmol) followed by AuCl3 (22 mg, 0.073 mmol). Reaction was heated to 60 oC for 18 hr, then allowed to cool to room temperature. Reaction mixture was concentrated and the resulting residue was purified by flash chromatography (hexanes/ethyl acetate = 100/1) to give 122 in 90 % yield (92 mg). 154 Ph O Ph Ph 122 1,3,3-triphenyl-propenone [122]: yellow oil; 1H NMR (300 MHz, CDCl3) 7.12 (s, 1H), 7.167.21 (m, 2H), 7.25-7.28 (m, 3H), 7.35-7.4 (m, 7H), 7.46-7.51 (m, 1H), 7.91 (d, J = 7.1 Hz, 2H); For full characterization refer to Gurdeep et al. J. Chem. Soc. Perkin Trans. I. 1985, 12891294. O 125 (E)-1-Phenyl-hept-1-en-3-one [125] H NMR (300 MHz, CDCl3) 0.77-1.02 (m, 3H), 1.20-1.47 (m, 2H), 1.57-1.78 (m, 2H), 2.67 (t, 1 J = 7.4 Hz, 2H), 6.75 (d, J = 16.2 Hz, 1H), 7.35-7.47 (m, 3H), 7.49-7.66 (m, 3H); For full characterization refer to Concellon, J.; Rodriguez-Solla, H.; Mejica, C. Tetrahedron 2006, 62(14), 32923300. 127 3-Ethyl-pent-3-en-1-ynyl benzene [127] H NMR (300 MHz, CDCl3) 1.14 (t, J = 7.5 Hz, 3H), 1.91 (dt, J = 6.8, 1.1 Hz, 3H), 2.18-2.27 1 (m, 2H), 5.81 (qt, J = 6.8, 1.2 Hz, 1H), 7.27-7.39 (m, 3H), 7.40-7.50 (m, 2H); For full characterization refer to Mayr, H.; Halberstadt-Kausch, I. Chem. Ber. 1982, 115(11), 34793515. 155 156 157 158 Typical procedure for the preparation of α,β-unsaturated ketones 122, 125, and 128 in CH2Cl2, Figure 64 To a CH2Cl2 solution (8 mL) of 3-ethyl-1-phenyl-1-pentyn-3-ol 126 (0.10 g, 0.54 mmol) in an open flask was added 95 % ethanol (0.15 mL, 2.7 mmol) followed by AuCl 3 (33 mg, 0.11 mmol). After 18 hr the reaction mixture was diluted with diethyl ether (10 mL) and concentrated. The resulting residue was purified by flash chromatography (hexanes/ethyl acetate = 100/1) to give 3-ethyl-1-phenyl-pent-2-en-1-one 128 in 31 % yield (31.8 mg). Typical procedure for the preparation of ethoxy propargylic alcohols 129, 132, 134, 136, 138, 150, 152, 154, 156, 158 To a THF solution (7 mL) of ethyl ethynyl ether (0.7 g, ca. 40% by weight in hexanes, ca. 9 mmol was added n-BuLi (1.5 mL, 3.4 mmol, 2.3 M) dropwise over 5 min at –78 °C under argon atmosphere. The solution was allowed to warm to 0 °C over 1 hr and held at 0 °C for an additional 30 min. The solution was then recooled to –78 °C and pinacolone (0.30 mL, 2.4 mmol) was added in one portion. The solution was allowed to warm to room temperature over 1 hr and held at room temperature for an additional 3 hr. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. The residue was purified using silica gel column chromatography (hexanes/ethyl acetate = 20/1 – 7/1) to give 1-ethoxy-3methyl-3-tert-butyl-1-propyn-3-ol (138) in 83 % yield (0.34g). Ph Ph OH 129 OEt 3-ethoxy-1,1-diphenyl-prop-2-yn-1-ol (129): yellow oil; 1H NMR (300 MHz, CDCl3) 1.41 (t, J = 7.1 Hz, 3H), 2.64 (s, 1H), 4.18 (q, J = 7.1 Hz, 2H), 7.21-7.34 (m, 6H), 7.60-7.63 (m, 4H); C NMR (75 MHz, CDCl3) 14.4, 42.2, 74.4, 74.8, 95.9, 125.9, 127.2, 128.0, 146.1; IR (neat) 13 3543, 3444, 3060, 2982, 2263, 1450, 1170, 1004, 640 cm-1; HRMS (EI) Calcd for C17H16O2 (M+) 252.1150. Found 252.1153. 159 OH 132 OEt 3-Ethoxy-1-phenyl-prop-2-yn-1-ol [132] H NMR (300 MHz, CDCl3) 1.39 (t, J = 7.1 Hz, 3H), 2.04 (d, J = 5.9 Hz, 1H), 4.15 (q, J = 7.1 1 Hz, 2H), 5.51 (d, J = 6.0 Hz, 1H), 7.31-7.40 (m, 3H), 7.48-7.59 (m, 2H); 13 C NMR (75 MHz, CDCl3) 14.4, 38.8, 64.6, 74.8, 95.4, 126.5, 128.0, 128.5, 129.2; IR (neat) 3401, 2981, 2226, 1718, 1450 cm-1; HRMS (EI) calcd for C11H12O2 (M+) 176.0834. Found 176.0837. Consistent with data from Raucher, S.; Bray, B. J. Org. Chem. 1987, 52(11), 23322333. OH OEt 134 4-tert-butyl-1-ethoxyethynyl-cyclohexanol [134]: white solid; 1H NMR (300 MHz, CDCl3) 0.87 (s, 9H), 0.98 (t t, J = 11.9, 3.5 Hz, 1H), 1.38 (t, J = 7.1 Hz, 3H), 1.26-1.55 (m, 4H), 1.71 (br d, J = 12.8 Hz, 2H), 1.89 (s, 1H), 1.94 (br d, J = 10.6 Hz, 2H), 4.09 (q, J = 7.1 Hz, 2H); 13 C NMR (75 MHz, CDCl3) 14.3, 25.0, 27.6, 32.2, 41.1, 41.3, 47.1, 69.6, 74.4, 94.0; IR (neat) 3390, 2944, 2865, 2257, 1479, 1444, 1393, 1365, 1178, 1059, 900, 834 cm-1; HRMS (CI) Calcd for C14H24O2 (M+H+) 225.1855. Found 225.1856. OH OEt 136 2-ethoxyethynyl-adamantan-2-ol [136]: clear, colorless oil; 1H NMR (300 MHz, CDCl3) 1.37 (t, J = 7.1 Hz, 3H), 1.53 (br s, 1H), 1.57 (br s, 1H), 1.68-1.80 (m, 7H), 1.88 (br s, 2H), 2.14 (br t, J = 12.6 Hz, 4H), 4.09 (q, J = 7.1 Hz, 2H); C NMR (75 MHz, CDCl3) 14.6, 27.1, 27.3, 32.0, 13 35.9, 38.0, 39.8, 43.2, 72.7, 74.6, 93.9; IR (neat) 3426, 2902, 2854, 2666, 2259, 1712, 1450, 1247, 1010, 916, 866 cm-1; HRMS (CI) Calcd for C14H20O2 (M+H+) 221.1542. Found 221.1538. 160 OH OEt 138 1-ethoxy-3-methyl-3-tert-butyl-1-propyn-3-ol [138]: clear, colorless oil; 1H NMR (300 MHz, CDCl3) 1.03 (s, 9H), 1.37 (t, J = 7.1 Hz, 3H), 1.41 (s, 3H), 1.71 (s, 1H), 4.08 (q, J = 7.1 Hz, 2H); C NMR (75 MHz, CDCl3) 14.3, 25.2, 25.6, 38.4, 41.9, 73.9, 74.2, 92.8; IR (neat) 3479, 13 2971, 2873, 2261, 1481, 1392, 1369, 1219, 1094, 1007, 908, 878 cm-1; HRMS (CI) Calcd for C10H19O2 ([M+H]+) 171.1385. Found 171.1390. OH OEt 150 1-Ethoxy-4,4-dimethyl-pent-1-yn-3-ol [150]: 1H NMR (300 MHz, CDCl3) 0.97 (s, 9H), 1.38 (t, J = 7.1 Hz, 3H), 4.03 (d, J = 6.0 Hz, 1H), 4.10 (q, J = 7.1 Hz, 2H); 13C NMR (75 MHz, CDCl3) 14.2, 25.2, 35.8, 38.0, 71.0, 74.3, 94.0; IR (neat) 3431, 2956, 2714, 2264, 1629 cm-1; HRMS (EI) calcd for C9H16O2 (M+) 156.1150. Found 156.1103. OH OEt 152 1-Ethoxy-5-phenyl-pent-1-yn-3-ol [152]: 1H NMR (300 MHz, CDCl3) 1.38 (t, J = 7.1 Hz, 3H), 1.66 (d, J = 5.3 Hz, 1H), 1.97-2.03 (m, 2H), 2.78 (t, J = 7.9 Hz, 2H), 4.11 (q, J = 7.1 Hz, 2H), 4.41 (ap. q, J = 5.3 Hz, 1H), 7.16-7.31 (m, 5H); 13 C NMR (75 MHz, CDCl3) 14.3, 31.6, 38.8, 40.1, 61.7, 74.6, 94.0, 125.8, 128.3, 128.4, 141.6; IR (neat) 3400, 3084, 3061, 2931, 2862, 2262, 1722, 1603, 1496 cm-1; HRMS (EI) calcd for C13H16O2 (M+) 204.1145. Found 204.1150. 161 OH OEt 154 1-Ethoxy-dec-1-yn-3-ol [154]: 1H NMR (300 MHz, CDCl3) 0.86-0.90 (m, 3H), 1.21-1.46 (m, 10H), 1.37 (t, J = 7.1 Hz, 3H), 1.56-1.70 (m, 3H), 4.09 (q, J = 7.1 Hz, 2H), 4.39 (q, J = 6.3 Hz, 1H); 13 C NMR (75 MHz, CDCl3) 14.0, 14.2, 22.5, 25.3, 29.2, 29.2, 31.7, 38.6, 39.6, 62.3, 74.4, 93.5; IR (neat) 3381, 2927, 2263, 1722, 1467 cm-1; HRMS (CI) calcd for C12H22O2 (M+H+) 199.1698. Found 199.1692. OH OEt 156 1-Cyclohexyl-3-ethoxy-prop-2-yn-1-ol [156]: 1H NMR (300 MHz, CDCl3) 0.83-1.3 (m, 6H), 1.38 (t, J = 7.1 Hz, 3H), 1.57-1.84 (m, 6H), 4.10 (q, J = 7.1 Hz, 2H), 4.18 (t, J = 5.7 Hz, 1H); C NMR (75 MHz, CDCl3) 14.3, 25.9, 25.9, 26.4, 28.1, 28.6, 38.3, 44.6, 67.0, 74.5, 94.3; IR 13 (neat) 3411, 2980, 2460, 1719, 1450 cm-1; HRMS (CI) calcd for C11H18O2 (M+H+) 183.1385. Found 183.1390. OH OEt 158 1-Cyclohex-1-enyl-3-ethoxy-prop-2-yn-1-ol [158]: 1H NMR (300 MHz, CDCl3) 1.38 (t, J = 7.1 Hz, 3H), 1.55-1.65 (m, 8H), 2.07 (br s, 1H), 4.12 (q, J =7.1 Hz, 2H), 4.77 (d, J = 5.5 Hz, 1H), 5.85 (s, 1H); 13 C NMR 14.3, 22.3, 22.5, 24.0, 24.9, 38.0, 66.7, 74.6, 94.6, 123.8, 138.0, 143.8; IR (neat) 3392, 2980, 2929, 2858, 2837, 2658, 2837, 2659, 2455, 2263, 1716 cm -1; HRMS (EI) calcd for C11H16O2 (M+) 180.1151. Found 180.1150. 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 Typical procedure for preparation of α,β-unsaturated esters 130, 133, 135, 137, 139, 141 using AuCl3 (Table 7 and Table 8) To a CH2Cl2 solution (8 mL) of 1-ethoxy-3-methyl-3-tert-butyl-1-propyn-3-ol (138) in an open flask was added 95 % ethanol (0.16 mL, 2.9 mmol) followed by AuCl3 (35 mg, 0.12 mmol, fine powder). After 5 min, the reaction mixture was filtered through a plug of SiO2 with the aid of an ethyl acetate/hexane mixture (1/7). The filtrate was concentrated and purified using silica gel column chromatography (hexanes/ethyl acetate = 50/1) to give 3,4,4-trimethyl-pent-2-enoic acid ethyl ester (139) in 86% yield (85 mg). Ph O Ph OEt 130 Ethyl 3,3-diphenylpropenoate [130]: yellow oil; 1H NMR (300 MHz, CDCl3) 2.01 (t, J = 7.1 Hz, 3H), 4.95 (q, J = 7.1 Hz, 2H), 8.09-8.12 (m, 2H), 8.18-8.30 (m, 8H); For full characterization refer toLe Tadic-Biadatti et al. J. Org. Chem. 1997, 62, 559563 and Rathke, M. et al. Synth. Commun. 1990, 20, 869875. O OEt 133 Ethyl cinnamate [133]: yellow oil; 1H NMR (300 MHz, CDCl3, E-Isomer) 1.33 (t, J = 7.1 Hz, 3H), 4.26 (q, J = 7.1 Hz, 2H), 6.44 (d, J = 16.0 Hz, 1H), 7.35-7.53 (m, 5H), 7.69 (d, J = 16.0 Hz, 1H); Z-Isomer 1.24 (t, J = 7.1 Hz, 3H), 4.18 (q, J = 7.1 Hz, 2H), 5.95 (d, J = 12.6 Hz, 1H), 6.95 (d, J = 12.6 Hz, 1H), 7.33-7.38 (m, 3H), 7.57-7.59 (m, 2H); For full characterization refer to Wadsworth, E. J. Am. Chem. Soc. 1961, 83, 1733. 182 OEt O 135 4-tert-butylcyclohexylidene-acetic acid ethyl ester [135]: clear, colorless oil; 1H NMR (300 MHz, CDCl3) 0.84 (s, 9H), 1.04-1.28 (m, 3H), 1.26 (t, J = 7.1 Hz, 3H), 1.76-1.95 (m, 3H), 2.14 (t d, J = 13.5, 3.9 Hz, 1H), 2.27-2.32 (m, 1H), 3.86 (d q, J = 13.7, 2.7, 1H), 4.12 (q, J = 7.1 Hz, 2H), 5.58 (s, 1H); C NMR (75 MHz, CDCl3) 14.3, 27.5, 28.4, 29.2, 29.5, 32.4, 37.9, 47.8, 13 59.4, 112.7, 163.5, 166.9; IR (neat) 3412, 2948, 2867, 1716, 1648, 1478, 1365, 1247, 1143, 1041, 862 cm-1; HRMS (CI) Calcd for C14H24O2 (M+H+) 225.1855. Found 225.1849. OEt O 137 adamantan-2-ylidene-acetic acid ethyl ester [137]: clear, colorless oil: 1H NMR (300 MHz, CDCl3) 1.27 (t, J = 7.1 Hz, 3H), 1.86 (br s, 6H), 1.93-1.96 (m, 6H), 2.43 (br s, 1H), 4.07 (br s, 1H), 4.13 (q, J = 7.1 Hz, 2H), 5.58 (s, 1H); For full characterization refer to L. Griaud et al. Tetrahedron, 1998, 54, 1189911906. O OEt 139 (E/Z)-3,4,4-trimethyl-pent-2-enoic acid ethyl ester [139]: clear, colorless oil: 1H NMR (300 MHz, CDCl3, E-isomer) 1.10 (s, 9H), 1.28 (t, J = 7.1 Hz, 3H), 2.16 (br d, J = 1.1 Hz, 3H), 4.14 (q, J = 7.1 Hz, 2H), 5.74 (q, J = 1.1 Hz, 1H); 1H NMR (300 MHz, CDCl3, Z-isomer) 1.20 (s, 9H), 1.28 (t, J = 7.1 Hz, 3H), 1.84 (br d, J = 1.3 Hz, 3H), 4.14 (q, J = 7.1 Hz, 2H), 5.63 (q, J = 1.3 Hz, 1H); 13C NMR (75 MHz, CDCl3, (E/Z)-mixture) 14.1, 14.3, 15.1, 23.9, 28.5, 29.0, 36.4, 37.9, 59.4, 60.0, 112.9, 116.6, 158.5, 167.2, 167.5, 167.9; IR (neat, (E/Z)-mixture) 2970, 2873, 1719, 1634, 1466, 1372, 1262, 1182, 1123, 1054, 868 cm-1; HRMS (EI) Calcd for C10H18O2 (M+) 170.1307. Found 170.1306. 183 O OEt 141 3-butyl-hept-2-enoic acid ethyl ester [141]: clear, colorless oil: 1H NMR (300 MHz, CDCl3) 0.89-0.94 (m, 6H), 1.27 (t, J = 7.1 Hz, 3H), 1.28-1.47 (m, 8H), 2.13 (br t, J = 7.6 Hz, 2H), 2.59 (br t, J = 7.6 Hz, 2H), 4.13 (q, J = 7.1 Hz, 2H), 5.61 (s, 1H); C NMR (75 MHz, CDCl3) 13.88, 13 13.92, 14.3, 22.4, 23.0, 29.8, 30.8, 31.9, 38.1, 59.3, 115.0, 164.8, 166.6; IR (neat) 2958, 2931, 2872, 1716, 1642, 1466, 1378, 1190, 1147, 1039, 862 cm-1; HRMS (CI) Calcd for C13H24O2 (M+H+) 213.1855. Found 213.1857. 184 185 186 187 188 189 190 191 192 193 Typical two-step procedure for preparation of α,β-unsaturated esters 130, 137, 139, 145, 147, 149 using AuCl3 (Table 9) To a THF solution (2.6 mL) of ethyl ethynyl ether (0.13g, ca. 40 % by weight in hexanes, ca. 2 mmol) was added n-BuLi (0.40 mL, 0.75 mmol, 2.0 M) dropwise over 5 min at –78 °C under argon atmosphere. The solution was allowed to warm to 0 °C over 1 hr and held at 0 °C for an additional 30 min. The solution was then recooled to –78 °C and 2-adamantanone (75 mg, 0.5 mmol) was added in one portion. The solution was allowed to warm to room temperature over 1 hr and held at room temperature for an additional 3 hr. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated. To the concentrated mixture in an open flask was added 6 mL of CH 2Cl2, followed by 95 % ethanol (0.14 mL, 2.5 mmol) and AuCl3 (7.6 mg, .025 mmol, fine powder). [NOTE: 15.2 mg, 0.05 mmol of AuCl3 was used for the preparation of 147 and 149.] After 30 min, the reaction mixture was filtered through a plug of SiO2 with the aid of an ethyl acetate/hexane mixture (1/7). The filtrate was concentrated and purified using silica gel column chromatography (hexanes/ethyl acetate = 50/1) to give adamantan-2- ylidene-acetic acid ethyl ester (137) in 99 % yield (109 mg) over two steps. CO2Et 145 (4,6,6-trimethyl-bicyclo[3.1.1]hept-3-en-(2E/Z)-ylidene)-acetic acid ester [145]: clear oil; 1H NMR (300 MHz, CDCl3, E-isomer) 0.86 (s, 3H), 1.28 (t, J = 7.1 Hz, 3H), 1.40 (s, 3H), 1.68 (d, J = 7.9 Hz, 1H), 1.90 (d, J = 1.5 Hz, 3H), 2.20-2.45 (m, 1H), 2.53-2.63 (m, 2H), 4.08-4.20 (m, 2H), 5.32 (s, 1H), 7.13 (s, 1H); 1H NMR (300 MHz, CDCl3, Z-isomer) 0.84 (s, 3H), 1.26 (t, J = 7.1 Hz, 3H), 1.44 (s, 3H), 1.58 (d, J = 8.8 Hz, 1H), 1.86 (d, J = 1.4, 3H), 2.20-2.45 (m, 1H), 2.53-2.63 (m, 2H), 4.08-4.20 (m, 2H), 5.46 (s, 1H), 5.77 (s, 1H); 13C NMR (75 MHz, CDCl3, E/Z mixture) 14.3, 14.4, 21.7, 21.8, 23.2, 23.6, 26.4, 26.5, 37.5, 38.1, 45.3, 47.8, 48.2, 49.0, 49.1, 53.1, 59.2, 59.3, 107.6, 110.0, 117.6, 121.6, 156.9, 158.0, 159.6, 161.2, 166.8, 167.4; IR (neat) 194 2979, 2930, 2870, 1708, 1622, 1466, 1443, 1380, 1370, 1226, 1164, 1040, 874, 705 cm -1; HRMS (EI) Calcd for C14H20O2 (M+) 220.1463. Found 220.1462. CO2Et 147 (1,7,7-trimethyl-bicyclo[2.2.1]hept-(2E/Z)-ylidene)-acetic acid ethyl ester [147]: clear, colorless oil; 1H NMR (300 MHz, CDCl3) diagnostic resonance signals for E and Z isomers appeared at 5.58 (br t, J = 2.3 Hz, 1H), 5.64 (br t, J = 2.0 hz, 1H); 13 C NMR (75 MHz, CDCl3, E/Z mixture) 12.7, 13.2, 14.5, 14.6, 19.0, 19.1, 19.8, 20.2, 27.5, 27.8, 34.0, 34.4, 38.6, 40.6, 44.0, 44.6, 48.2, 50.0, 53.9, 54.0, 59.6, 60.0, 108.2, 112.2, 166.9, 167.6, 175.6; IR (neat, E/Z mixture) 2955, 2874, 1714, 1652, 1450, 1368, 1306, 1288, 1200, 1177, 1097, 1048, 873, 850 cm-1; HRMS (CI) Calcd for C14H22O2 (M+H+) 223.1698. Found 223.1695. CO2Et 149 (3,3,5,5-tetramethyl-cyclohexylidene)-acetic acid ethyl ester [149]: clear, colorless oil; 1H NMR (300 MHz, CDCl3) 0.95 (s, 6H), 0.97 (s, 6H), 1.28 (t, J = 7.1 Hz, 3H), 1.33 (s, 2H), 1.95 (s, 2H), 2.61 (s, 2H), 4.15 (q, J = 7.1 Hz, 2H), 5.69 (s, 1H); C NMR (75 MHz, CDCl3) 14.3, 13 30.8, 30.9, 34.8, 34.9, 42.2, 51.1, 52.5, 59.4, 115.8, 160.4, 166.6; IR (neat) 2953, 2896, 1716, 1646, 1461, 1376, 1229, 1154, 1043, 862 cm-1; HRMS (EI) Calcd for C14H24O2 (M+) 224.1776. Found 224.1771. 195 196 197 198 199 200 201 Typical Procedure for the Gold-Catalyzed Meyer–Schuster Rearrangement of Ethoxyalkynyl Carbinols, 133, 151, 153, 155, 157, 159 (Table 11). A mixture of AuCl (7.4 mg, 0.032 mmol) in 1:1 CH2Cl2–THF (5 mL) was prepared and allowed to stir for 20 min to give a homogeneous solution with an insoluble residue. A separate 25 mL roundbottomed flask under argon was charged with 150 (100 mg, 0.64 mmol), EtOH (95 %, 0.36 mL, 6.4 mmol) and a solution of AgSbF6 (11 mg, 0.032 mmol) in 1:1 CH2Cl2–THF (5 mL). The mixture of AuCl in 1:1 CH2Cl2–THF (5 mL) was then added dropwise. After 40 min, the reaction mixture was filtered through a plug of SiO 2 with the aid of 1:7 Et2O–hexane. The filtrate was concentrated and purified using silica gel column chromatography (Et2O–hexane, 1:50) to give ethyl 4,4-dimethylpent-2-enoate (151); yield 0.091 g (91 %, 97:3 E/Z ratio). Typical Procedure for the Gold-Catalyzed Meyer–Schuster Rearrangement of Ethoxyalkynyl Carbinols in the Presence of Camphorsulfonic Acid (CSA) (Table 11) A mixture of AuCl (7.4 mg, 0.032 mmol) in 1:1 CH2Cl2–THF (5 mL) was prepared and allowed to stir for 20 min to give a homogeneous solution with an insoluble residue. A separate 25 mL roundbottomed flask under argon was charged with 150 (100 mg, 0.64 mmol), EtOH (95 %, 0.36 mL, 6.4 mmol), CSA (0.15 g, 0.64 mmol) and a solution of AgSbF6 (11 mg, 0.032 mmol) in 1:1 CH2Cl2–THF (5 mL). The mixture of AuCl in 1:1 CH 2Cl2–THF (5 mL) was then added dropwise. After 40 min, the reaction mixture was filtered through a plug of SiO 2 with the aid of 1:7 Et2O–hexane. The filtrate was concentrated and purified using silica gel column chromatography (Et2O–hexane, 1:50) to give ethyl (E)-4,4-dimethylpent-2- enoate (151); yield 0.093 g (93 %). O OEt 151 Ethyl (E)-4,4-dimethyl-pent-2-enoate [151] H NMR (300 MHz, CDCl3) 1.08 (s, 9H), 1.29 (t, J = 7.1 Hz, 3H), 4.19 (q, J = 7.1 Hz, 2H), 1 5.73 (d, J = 15.9 Hz, 1H), 6.97 (d, J = 15.9 Hz, 1H); For full characterization refer to Comasseto, J. et. al. Tetrahedron 2005, 61, 23192326. 202 O OEt 153 Ethyl 5-phenyl-pent-2-enoate [153] H NMR (300 MHz, CDCl3) E-isomer 1.21 (t, J = 7.1 Hz, 3H), 2.38-2.50 (m, 2H), 2.70 (t, J = 1 7.1 Hz, 2H), 4.11 (q, J = 7.1 Hz, 2H), 5.77 (d, J = 15.6 Hz, 1H), 6.93 (dt, J = 15.6, 6.8 Hz, 1H), 7.06-7.27 (m, 5H); Z isomer 1.28 (t, J = 7.1 Hz, 3H), 2.77 (t, J = 7.1 Hz, 2H), 2.94-3.03 (m, 2H), 4.16 (q, J = 7.1 Hz, 2H), 5.78 (d, J = 11.2, 1H), 6.23 (dt, J =11.2, 7.3 Hz, 1H), 7.15-7.33 (m, 5H); For full characterization refer to Yamada, T. et. al. J. Am. Chem. Soc. 129, 129(43), 1290212903. O OEt 155 (E)-Dec-2-enoic acid ethyl ester [155]; H NMR (300 MHz, CDCl3) 0.86-0.90 (m, 3H), 1.26-1.31 (m, 8H), 1.28 (t, J = 7.1 Hz, 3H), 1 1.42-1.47 (m, 2H), 2.19 (ddd, J = 14.6, 7.1, 1.2 Hz, 2H), 4.18 (q, J = 7.1 Hz, 2H), 5.80 (br d, J = 15.6 Hz, 1H), 6.96 (dt, J = 15.6, 7.0 Hz, 1H); For full characterization refer to Concellón, J. M.; Concellón, C.; Méjica, C. J. Org. Chem. 2005, 70, 61116113. O OEt 157 (E/Z)-3-Cyclohexyl-acrylic acid ethyl ester [157]: H NMR (300 MHz, CDCl3) E-Isomer 1.12-1.31 (m, 5H), 1.29 (t, J = 7.1 Hz, 3H), 1.64-1.77 1 (m, 5H), 2.04-2.17 (m, 1H), 4.18 (q, J = 7.1 Hz, 2H), 5.76 (dd, J = 15.8, 1.4 Hz, 1H), 6.91 (dd, J = 15.8, 6.8 Hz); Z-Isomer 1.12-1.31 (m, 5H), 1.29 (t, J =7.1 Hz, 3H), 1.64-1.77 (m, 5H), 3.233.34 (m, 1H), 4.17 (q, J = 7.1 Hz, 2H), 5.65 (dd, J = 11.5, 1.0 Hz, 1H), 6.02 (dd, J = 11.5, 9.8 Hz, 1H). For full characterization refer to Concellón, J. M.; Concellón, C.; Méjica, C. J. Org. Chem. 2005, 70, 61116113. 203 O OEt 159 (E/Z)-3-(1-Cyclohexen-1-yl)-2-propenoic acid ethyl ester [159]: H NMR (300 MHz, CDCl3) E-Isomer 1.29 (t, J = 7.1 Hz, 3H), 1.65-1.73 (m, 4H), 2.11-2.16 1 (m, 2H), 2.20-2.25 (m, 2H), 4.20 (q, J = 7.1 Hz, 2H), 5.76 (d, J = 16.4 Hz, 1H), 6.16 (t, J = 4.1 Hz, 1H), 7.28 (d, J = 15.7 Hz, 1H); Z-Isomer 1.28 (t, J = 7.1 Hz, 3H), 1.57-1.63 (m, 4H), 2.172.2 (m, 2H), 2.24-2.26 (m, 2H), 4.16 (q, J = 7.1 Hz, 2H), 5.58 (d, J = 12.7 Hz, 1H), 6.00 (t, J = 3.8 Hz, 1H), 6.31 (dd, J =12.7, 0.9 Hz, 1H); For full characterization refer to Stille, J. K. et. al. J. Am. Chem. Soc. 1987, 109, 813817. 204 205 206 207 208 209 Typical procedure for the preparation of -unsaturated esters 133, 139, 151, 155, 157 using Sc(OTf)3, Table 15. To a 4:1 v/v CH2Cl2/ethanol solution (10 mL) of 1-ethoxydec-1-yn-3-ol (154, 0.10 g, 0.51 mmol) in an open flask was added Sc(OTf)3 (2.5 mg, 0.005 mmol). Progress of the reaction was monitored by TLC analysis. After 1 hr, the reaction mixture was concentrated under reduced pressure and purified using silica gel column chromatography (hexanes/ethyl acetate, 50:1) to give ethyl (E)-dec-2-enoate (155) in 70 % yield (70 mg). Typical two-step procedure for the preparation of -unsaturated esters 130, 137, 145, 151, 169, 171, 173, 175, 177, 179, 181, 183 using Sc(OTf) 3, Table 16. To a THF solution (2.6 mL) of ethyl ethynyl ether (0.13 g, ca. 40 % by weight in hexanes, ca. 2 mmol) was added n-BuLi (0.40 mL, 0.75 mmol, 2.0 M) dropwise over 5 min at 78 oC under argon atmosphere. The solution was allowed to warm to 0 oC over 1 hr and held at 0 oC for an additional 30 min. The solution was then recooled to 78 oC and 2-adamantanone (142, 75 mg, 0.50 mmol) was added in one portion. The solution was allowed to warm to room temperature over 1 hr and held at room temperature for an additional 3 hr. Saturated aqueous NH4Cl solution was added to quench the reaction and the mixture was extracted with ethyl acetate. The organic layer was washed sequentially with water, saturated aqueous sodium bicarbonate, and brine. The organic layer was dried over MgSO4, filtered, and concentrated under reduced pressure. To the concentrated mixture in an open flask were added CH2Cl2 (8 mL), absolute ethanol (2 mL), and Sc(OTf)3 (2.5 mg, 0.005 mmol). After 6 hr, the reaction mixture was concentrated under reduced pressure and purified using silica gel column chromatography (hexanes/ethyl acetate, 50:1) to give adamantan-2-ylidene-acetic acid ethyl ester (137) in 96 % yield over two steps (106 mg). 210 CO2Et 169 (2-Isopropyl-5-methyl-cyclohexylidiene)-acetic acid ethyl ester [169]: H NMR (300 MHz, CDCl3, E/Z-mixture, diagnostic peaks) 2.55 (ddd, J = 12.9, 5.5, 1.5 Hz), 1 3.14 (dd, J = 12.9, 4.3 Hz), 3.48-3.52 (m), 4.13 (q, J = 7.1 Hz), 4.14 (q, J = 7.1 Hz), 5.63 (br s); C NMR (75 MHz, CDCl3, E/Z-mixture) 14.3, 18.1, 19.5, 20.5, 20.8, 21.8, 23.4, 26.1, 26.8, 13 27.0, 27.6, 30.4, 31.6, 33.6, 33.9, 35.4, 36.1, 40.0, 43.5, 50.8, 52.6, 55.9, 59.3, 59.4, 113.3, 116.3, 164.8, 167.1. O OEt 173 Cycloheptyliden-acetic acid ethyl ester [173]: H NMR (300 MHz, CDCl3) 1.27 (t, J = 7.1 Hz, 3H), 1.51-1.56 (m, 4H), 1.60-1.69 (m, 4H), 1 2.37 (dt, J = 1.3 Hz, 2H), 2.87 (dt, J = 1.3 Hz, 2H), 4.13 (q, J =7.1 Hz, 2H), 5.66 (quint, J = 1.2 Hz, 1H); For full characterization refer to Friese, A. et al. J. Med. Chem. 2002, 45, 15351542. O O 175 (E/Z)-Ethyl-β-methyl cinnamate [175]: H NMR (300 MHz, CDCl3) E-Isomer 1.32 (t, J = 7.1 Hz, 3H), 2.58 (d, J = 1.2 Hz, 3H), 4.22 1 (q, J = 7.1 Hz, 2H), 6.14 (q, J = 1.3 Hz, 1H), 7.1-7.5 (m, 5H); Z-Isomer 1.08 (t, J = 7.1 Hz, 3H), 2.18 (d, J = 1.4 Hz, 3H), 4.00 (q, J = 7.1 Hz, 2H), 5.91 (q, J =1.4 Hz, 1H), 7.1-7.5 (m, 5H); For full characterization refer to Tsuda, T. et. al. J. Org. Chem. 1988, 53, 607610. 211 O O 177 (E/Z)-3-(4-Isopropenyl-cyclohex-1-enyl)-acrylic acid ethyl ester [177]: Diagnostic peaks 1H NMR (300 MHz, CDCl3) E-Isomer 5.77 (d, J = 15.8 Hz, 1H), 6.18 (br s, 1H), 7.30 (d, J = 15.8 Hz, 1H); Z-Isomer 5.61 (d, J = 12.6 Hz, 1H), 6.02 (br s, 1H), 6.33 (d, J = 12.6 Hz, 1H). O O 179 O (E/Z)-Ethyl-3-(4-methoxyphenyl)-2-propenoate [179]: H NMR (300 MHz, CDCl3) E-Isomer 1.33 (t, J =7.1 Hz, 3H), 3.84 (s, 3H), 4.25 (q, J = 7.1 Hz, 1 2H), 6.31 (d, J = 16.0 Hz, 1H), 6.90 (d, J =8.6 Hz, 2H), 7.48 (d, J = 8.6 Hz, 2H), 7.64 (d, J = 16.0 Hz, 1H); For full characterization refer to Aggarwal, V. et. al. J. Am. Chem. Soc. 2003, 125, 60346035. Z-Isomer 1.28 (t, J = 7.1 Hz, 3H), 3.82 (s, 3H), 4.19 (q, J = 7.1 Hz, 2H), 5.83 (d, J =12.7, 1H), 6.84 (d, J =12.7, 1H), 6.88 (d, J = 8.5 Hz, 2H), 7.69 (d, J = 8.7 Hz, 2H); For full characterization refer to Mueller, A. et . al. Org. Lett. 2007, 9, 53275329. O CO2Et 181 (E/Z)-Ethyl-3-(2-furyl)-propenoate [181]: H NMR (300 MHz, CDCl3) E-Isomer 1.32 (t, J = 7.1 Hz, 3H), 4.24 (t, J = 7.1 Hz, 2H), 6.31 (d, 1 J = 15.8 Hz, 1H), 6.46 (dd, J = 3.4, 1.8 Hz, 1H), 6.60 (d, J = 3.4 Hz, 1H), 7.44 (d, J = 15.8 Hz, 1H), 7.46 (d, J =6.5 Hz, 1H); For full characterization refer to Cristau, H. J. et. al. Tetrahedron 1998, 54, 15071522. Z-Isomer 1.32 (t, J = 7.1 Hz, 3H), 4.22 (q, J = 7.1 Hz, 2H), 5.74 (d, J = 12.9 Hz, 1H), 6.50 (dd, J = 3.5, 1.7 Hz, 1H), 6.79 (d, J =12.9 Hz, 1H), 7.47 (d, J =1.6 Hz, 1H), 7.67 (d, J = 3.5 Hz, 1H); For full characterization refer to Reetz, M. T. et. al. Eur. J. Org. Chem. 2003, 34853496. 212 F O F O F F 183 F (E)-3-Pentafluorophenyl-acrylic acid ethyl ester [183]: H NMR (300 MHz, CDCl3) 1.35 (t, J = 7.1 Hz, 3H), 4.29 (q, J = 7.1 Hz, 2H), 6.74 (d, J = 16.5 1 Hz, 1H), 7.64 (d, J = 16.5 Hz, 1H); For full characterization refer to Shen, Y. et. al. Syn. Comm. 1991, 21, 14031408. 213 214 215 216 217 218 219 220 221 222 223 LIST OF REFERENCES 1. Sachs, J.; Malaney, P. The economic and social burden of malaria. Nature 2002, 415, 680685. 2. Report No. Who/HTM/GMP/2008.1 (World Health Organization, Geneva 2008). 3. Luzzatto, L. Genetics of red cells and susceptibility to malaria. Blood 1979, 54, 961976 4. Hill, A. V.; Elvin, J.; Willis, A. C.; Aidoo, M.; Allsopp, C. E. M.; Gotch, F. M.; Gao, X. M.; Takiguchis, M.; Greenwood, B. M.; Townsend, A. R. M.; McMichael, A. J.; Whittle, H. C. Molecular analysis of the association of HLA-B53 and resistance to severe malaria. Nature 1992, 360, 434443. 5. Anderson, R. M.; May, R. Infectious Diseases of Humans: Dynamics and Control; Oxford Univ. Press, Tokyo, 1991. 6. Colluzzi, M. The clay feet of the malaria giant and its African roots: hypothesis and inferences about origin, spread and control of Plasmodium falciparum. Parassitologia 1999, 41, 277283. 7. Gallup, J.; Sachs, J. The economic burden of malaria. Am. J.Trop. Med. Hyg. 2001, 64(1,2), 8596. 8. Yamada, T. Casual relationships between infant mortality and fertility in developed and less developed countries. South. Econ. 1985, 52, 364371. 9. Galloway, P. R.; Lee, R. D.; Hammel, E. A. From Death to Birth: Mortality Decline and Reproductive Change; Montgomery, M. R. & Cohen, B.,Eds.; National Academy Press, Washington DC, 1988; pp 182226 10. Handa, S. The impact of education, income, and mortality on fertility in Jamaica. World Dev. 2000, 28, 173186. 224 11. Rosenweig, M.; Schultz, T. P. Consumer demand and household production: the relationship between fertility and child mortality. Am. Econ. Rev. 1983, 73, 3843. 12. Ridley, R. G. Medical need, scientific opportunity and the drive for antimalarial drugs. Nature, 2002, 415, 686693. 13. Meshnick, S.; Dobson, M. in Antimalarial Chemotherapy, Mechanisms of Action, Resistance, and New Directions in Drug Discovery; Rosenthal, P., Ed.; Humana, Totowa, New Jersey, 2001; pp 1526. 14. Wellems, T.; Plowe, C. Chloroquine-resistant malaria. J. Infect. Dis. 2001, 184, 770776. 15. Robert, A.; Dechy-Cabaret, O.; Cazelles, J.; Meunier, B. From Mechanistic Studies on Artemisinin Derivatives to New Modular Antimalarial Drugs. Acc. Chem. Res. 2002, 35,167174. 16. Francis, S.; Sullivan, D. J.; Goldberg, D. Hemoglobin metabolism in the malaria parasite. Plasmodium falciparum. Annu. Rev. Microbiol. 1997, 51, 97123. 17. Ridley, R.; Dorn, A.; Vippagunta, S.; Vennerstrom, J. Haematin (haem) polymerization and its inhibition by quinoline antimalarials. Ann. Trop. Med. Parasitol. 1997, 91, 559566. 18. Sugioka, Y.; Suzuki, M. The chemical basis for the ferriprotoporphyrin IX-chloroquine complex induced lipid peroxidation. Biochim. Biophys. Acta 1991, 1074, 1924. 19. Ginsburg, H.; Krugliak, M. Chloroquine—some open questions on its antimalarial mode of action and resistance. Drug Resist. Update 1999, 2, 6370. 20. Price, R. Artemisinin drugs: novel antimalarial agents. Exp. Opin. Invest. Drugs 2000, 9, 18151827. 21. Haynes. R. Artemisinin and derivatives: the future for malaria treatment? Curr. Opin. Infect. Dis. 2001, 14, 719726. 22. White, N. J. Antimalarial drug resistance and combination chemotherapy. Phil. Trans. R. Soc. Lond. B. 1999, 354, 739749. 225 23. Klayman, D. L. Qinghaosu (artemisinin): an antimalarial drug from China. Science 1985, 228, 10491055. 24. Price, R.; Van Vugt, M. Phaipun, L.; Luxemberger, C.; Simpson, T.; White, N. J.; Nosten, F. Adverse effects in patients with acute falciparum malaria treated with artemisinin derivatives. Am. J. Trop. Med. Hyg. 1999, 60, 547555. 25. Duc, D. D.; de Vries, P. J.; Khanh, N. X.; Binh, L. N.; Kager, P. A.; van Boxtel, C. J. The pharmacokinetics of a single dose of artemisinin in healthy Vietnamese subjects. Am. J. Trop. Med. Hyg. 1994, 51, 785790. 26. Bhakuni, R. S.; Jain, D. C. Sharma, R. P. In Artemisia; Wright, C. W., Ed.; Taylor & Francis: London, 2002; pp 211248. 27. Dhingra, V.; Rao, K. V.; Narasu, M. L. Current status of artemisinin and its derivatives as antimalarial drugs. Life Sci. 2000, 66, 279300. 28. Haynes, R. K.; Fugmann, B.; Stetter, J.; Riekmann, K.; Heilmann, H. D.; Chan, H. W.; Cheung, M. K.; Lam, W. L.; Wong, H. N.; Croft, S. L.; Vivas, L.; Rattray, L.; Stewart, L.; Peters, W.; Robinson, B. L.; Edstein, M. D.; Kotecka, B.; Kyle, D. E.; Beckermann, B.; Gerisch, M.; Radtke, M.; Schmuck, G.; Steinke, W.; Wolborn, U.; Schmeer, K.; Romer, A. Artemisone – A highly active antimalarial drug of the artemisinin class. Angew. Chem. Int. Ed. 2006, 45, 20822088. 29. Hsu, E. The history of quig hao in the Chinese material medica. Trans. Royal Soc. Trop. Med. Hyg. 2006, 100, 505508. 30. Liu, J. M.; Ni, M. Y.; Fan, J. F.; Tu, Y. Y.; Wu, Z. H.; Wu, Y. L.; Chou, W. S. Structure and reaction of arteannuin. Acta Chim. Sin. 1979, 37, 129143. 31. Lapkin, A. A.; Plucinski, P. K.; Cutler, M. Comparative assessment of technologies for extraction of artemisinin. J. Nat. Prod. 2006, 69, 16531664. 32. Rodrigues, R. A. F.; Foglio, M. A.; Junior, S. B.; Santos, A. d. S.; Rehder, V. L. Otimização do processo de extração e isolamento do antimalárico artemisinina a partir de Artemisia annua L. Quim. Nova 2006, 29, 368372. 33. Kohler, M.; Haerdi, W.; Christen, P.; Veuthey, J. –L. Extraction of artemisinin and artemisinic acid from Artemisia annua L. using supercritical carbon dioxide. J. Chromatogr., A 1997, 785, 353360. 226 34. Quispe-Condori, S.; Sánchez, D.; Foglio, M. A.; Rosa, P. T. V.; Zetzi, C.; Brunner, G.; Meireles, M. A. A. Global yield isotherms and kinetic of artemisinin extraction from Artemisia annua L leaves using supercritical carbon dioxide. J. Supercrit. Fluids 2005, 36, 4048. 35. Enserink M. Source of new hope against malaria is in short suppy. Science, 2005, 307, 33. 36. Lenihan, J. R.; Tsuruta, H.; Diola, D.; Renninger, N. S.; Regentin, R. Developing an industrial artemisinic acid fermentation process to support the cost-effective production of antimalarial artemisinin-based combination therapies. Biotechnol. Prog. 2008, 24, 10261032. 37. Zeng, Q.; Qiu, F.; Yuan, L. Production of artemisinin by genetically-modified microbes. Biotechnol. Lett. 2008, 30, 581592. 38. Acton, N.; Roth, R. J. On the conversion of dihydroartemisinic acid into artemisinin. J. Org. Chem. 1992, 36103614. 39. Ro, D. –K.; Paradise, E. M.; Ouellet, M.; Fisher, K. J.; Newman, K. L.; Ndungu, J. M.; Ho, K. A.; Eachus, R. A.; Ham, T. S.; Kirby, J.; Chang, M. C. Y.; Withers, S. T.; Shiba, Y.; Sarpong, R.; Keasling, J. D. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940943. 40. Dietrich, J. A.; Yoshikuni, Y.; Fisher, K. J.; Woolard, F. X.; Ockey, D.; McPhee, D. J.; Renninger, N. S.; Chang, M. C. Y.; Baker, D.; Keasling, J. D. A novel semibiosynthetic route for artemisinin production using engineered substrate-promiscuous P450. ACS Chem. Biol. 2009, 4(4), 261267. 41. Chang, M. C. Y.; Eachus, R. A.; Trieu, W.; Ro, D. –K.; Keasling, J. D. Engineering Eschericha coli for production of functionalized terpenoids using plant P450s. Nat. Chem. Biol. 2007, 3, 274277. 42. Nair, M. S. R.; Basile, D. V. Bioconversion of arteannuin B to artemisinin. J. Nat. Prod. 1993, 56(9), 15591566. 43. Jung, M.; Yao, Y.; ElSholy, H. N.; McChesney, J. D. One-step stereospecific synthesis of ()-arteannuin B. J. Nat. Prod. 1987, 50, 972973. 227 44. Lansbury, P. T.; Novak, D. M. Total synthesis of (±)arteannuin B. Tetrahedron Lett. 1986, 27, 39673970. 45. Dhingra, V.; Narasu, M. L. Purification and characterization of an enzyme involved in biochemical transformation of arteannuin B to artemisinin from Artemisia annua. Biochem. Biophys. Res. Commun. 2001, 281, 558561. 46. Schmid, G.; Hofheinz, W. Total synthesis of qinghaosu. J. Am. Chem. Soc. 1983, 105, 624625. 47. Xu, X, X.; Zhu, J.; Huang, D. –Z.; Zhou, W. –S. Total synthesis of arteannuin and deoxyarteannuin. Tetrahedron 1986, 42(3), 819828. 48. Avery, M. A.; Jennings-White, C.; Chong, W. K. M. The total synthesis of (+)artemisinin and (+)9desmethylartemisinin. Tetrahedron Lett. 1987, 28(4), 46294632. 49. Avery, M. A.; Jennings-White, C.; Chong, W. K. M. Simplified analogues of the antimalarial artemisinin: synthesis of 6,9desmethylartemisinin. J. Org. Chem. 1989, 54, 17921795. 50. Ravindranathan, T.; Kumar, M. A.; Menon, R. B.; Hiremath, S. V. Stereroselective synthesis of artemisinin. Tetrahedron Lett. 1990, 31(5), 755758. 51. Avery, M. A.; Chong, W. K. M.; Jennings-White, C. Stereoselective total synthesis of (+)artemisinin, the antimalarial constituent of Artemisia annua L. J. Am. Chem. Soc. 1992, 114, 974979. 52. Liu, H. –J.; Yeh, W. –L.; Chew, S. Y. A total synthesis of the antimalarial natural product (+)qinghaosu. Tetrahedron Lett. 1993, 34(28), 44354438. 53. Yadav, J. S.; Babu, R. S.; Sabitha, G. Stereoselective total synthesis of (+)artemisinin. Tetrahedron Lett. 2003, 44, 387389. 54. Stork, G.; Jung, M. E.; Colvin, E.; Noel, Y. Synthetic routes to halomethyl vinylsilanes. J. Am. Chem. Soc. 1974, 96, 36823684. 55. Roth, R. J.; Acton, N. A simple conversion of artemisinic acid into artemisinin. J. Nat. Prod. 1989, 52(5), 11831185. 228 56. Constantino, M. G.; Beltrame, M., Jr.; da Silva, G. V. J. A novel asymmetric total synthesis of (+)-artemisinin. Syn. Comm. 1996, 26(2), 321329. 57. Reetz, M. T. Dyoptropic rearrangements and related exchange processes. In Advances in Organometallic Chemistry, Stone, F. G. A., West, R.; Eds.; Academic Press: New York, NY, 1977; Vol. 16, pp 3365. 58. Mulzer, J.; Hoyer, K.; Müller-Fahrnow, A. Relative migratory aptitude of substituents and stereochemistry of dyotropic ring enlargements of β-lactones. Angew. Chem. Int. Ed. Engl. 1997, 36, 14761478. 59. Cathgart, R. C.; Bovenkamp, J. W.; Moir, R. Y.; Bannard, R. A. B.; Casselman, A. A. Migration of hydroxyl in secondary systems during solvolysis in acid solutions. Can. J. Chem. 1977, 55, 37743785. 60. Caggiano, L.; Davies, J.; Fox, D. J.; Moody, D. C.; Warren, S. A novel silica catalyzed stereoselective cyclic carbamate and carbonate rearrangement. Chem. Commun. 2003, 16501651. 61. Brown, G. D. Two new compounds from Artemisia annua. J. Nat. Prod. 1992, 55, 17561760. 62. Wallaart, T. E.; Pras, N.; Quax, W. J. Isolation and identification of dihydroartemisinic acid hydropperoxide from Artemisia annua: a novel biosynthetic precursor of artemisinin. J. Nat. Prod. 1999, 62, 11601162. 63. Sy, L. –K.; Brown, G. D. The mechanism of spontaneous autoxidation of dihydroartemisinic acid. Tetrahedron 2002, 58, 897908. 64. Nowak, D. M.; Lansbury, P. T. Synthesis of (+)-artemisinin and (+)-deoxoartemisinin from arteannuin B and arteannuic acid. Tetrahedron 1998, 54, 319336. 65. Davidson, B. S.; Plavcan, K. A.; Meinwald, J. Intramolecular Diels-Alder approach to cadinane and amorphane sesquiterpenes. J. Org. Chem. 1990, 55, 39123917. 66. Haynes, R. K.; King, G. R.; Vonwiller, S. C. Preparation of a bicyclic analogue of qinghao (artemisinic) acid via a lewis acid catalyzed ionic Diels-Alder reaction involving a hydroxyl diene and cyclic enone and facile conversion into (±)-6,9desmethylqinghaosu. J. Org. Chem. 1994, 59, 47434748. 229 67. Avery, M. A.; Fan, P.; Karle, J. M.; Bonk, J. D.; Miller, R.; Goins, K. D. Structureactivity relationships of the antimalarial agent artemisinin. 3. Total synthesis of (+)-13carbaartemisinin and related tetra- and tricyclic structures. J. Med. Chem. 1996, 39, 18851897. 68. Fry, A. J.; Little, R. D.; Leonetti, J. A general mechanistic scheme for intramolecular electrochemical hydrocyclizations. Mechanism and electroreductive cyclization of .omega.-keto.alpha.,.beta.-unsaturated esters. J. Org. Chem. 1994, 59, 50175026. 69. Schwaebe, M.; Little, R. D. Asymmetric reductive cyclization. Total synthesis of ()C10-desmethyl arteannuin B. J. Org. Chem. 1996, 61, 32403244. 70. Dreiding, A. S.; Goldberg, O.; Deja, I.; Rey, M. The synthesis of stereoisomers of arteannuin B. Helv. Chim. Acta 1980, 63, 24552468. 71. Wu, H.; Moeller, K. D. Anodic coupling reactions: A sequential cyclization route to the arteannuin ring skeleton. Org. Lett. 2007, 9(22), 45994602. 72. Barriault, L.; Deon, D. H. Total synthesis of (+)-arteannuin M using the tandem oxyCope/ene reaction. Org. Lett. 2001, 3(12), 19251927. 73. Gawley, R. E. The Robinson annelation and related reactions. Synthesis 1976, 777794. 74. Jung, M. E. A review of annulations. Tetrahedron 1976, 32, 331. 75. Stork, G.; Jung, M. E.; Noel, Y. Synthetic routes to halomethyl vinylsilanes. J. Am. Chem. Soc. 1974, 96(11), 36843688. 76. Sato, F.; Ishikawa, H.; Watanabe, H.; Miyake, T.; Sato, M.; Specific hydromagnesiation of prop-2-ynylic alcohols. A simple and specific route to terpenoids. J. Chem. Soc., Chem. Comm. 1981, 718720. 77. Sato, F.; Watanabe, H.; Tanaka, Y.; Sato, M. Hydromagnesiation of 3trimethylsilylprop-2-yn-1-ol. A simple route to (E)-3-trimethylsilylalk-2-en-1-ols. J. Chem. Soc., Chem. Comm. 1982, 11261127. 78. Miller, R. B.; Al-Hassan, M. I. The tetrahydropyranyl ether of (E)-3-bromotrimethylsilyl-2-propen-1-ol, a single synthon for the (E)-β-formlvinyl anion and cation. Tetrahedron Lett. 1983, 24(20), 20552058. 230 79. Denmark, S.; Habermas, K. L.; Hite, G. A. 19. Silicon-directed Nazarov cyclization. Substituent and heteroatom effects on the reaction. Helv. Chim. Acta 1988, 71, 168194. 80. Zweifel, G.; Lewis, W. Stereoselective synthesis of ((E)-and (Z)-1-Halo-1alkenyl)silanes from alkynes. J. Org. Chem. 1978, 43(14), 27392744. 81. Singletary, J. A.; Lam, H.; Dudley, G. B. A succinct method for preparing the StorkJung vinylsilane Robinson annulations reagent. J. Org. Chem. 2005, 70, 739741. 82. Schulte-Elte, K. H.; Ohloff, G. 21. Über eine aussergewöhnliche stereospezifität bei der hydroborierung der diastereomeren (1R)-isopulegole mit diboran. Helv. Chim. Acta 1967, 50, 153. 83. Mancuso, A. J.; Swern, D. Activated dimethyl sulfoxide: useful reagents for synthesis. Synthesis 1981, 165185. 84. Nakashima, K.; Imoto, M.; Sono, M.; Tori, M.; Nagashima, F.; Asakawa, Y. Total synthesis of ()-(7S, 10R)-calamenene and ()-(7S, 10R)-2-hydroxycalamenene by use of a ring-closing metathesis reaction. A comparison of the cis- and trans-isomers. Molecules 2002, 7, 517527. 85. Halterman, R. L.; Crow, L. D. Preparation of chiral annulated indenes derived from nopinone, verbenone and menthone. Tetrahedron Lett. 2003, 44, 29072909. 86. Avery, M. A.; Gao, F.; Chong, W. K. M.; Hendrickson, T. F.; Inman, W. D.; Crews, P. Synthesis, conformational analysis, and antimalarial activity of tricyclic analogs of artemisinin. Tetrahedron 1994, 50, 957972. 87. Morita, Y.; Suzuki, M.; Noyori, R. An organozinc aid in alkylation and acylation of lithium enolates. J. Org. Chem. 1989, 54, 17851787. 88. Imamoto, T.; Takiyama, N.; Nakamura, K.; Hatajima, T.; Kamiya, Y. Reactions of carbonyl compounds with Grignard reagents in the presence of cerium chloride. J. Am. Chem. Soc. 1989, 111, 43924398. 89. Handbook of Metathesis; Grubbs, R. H., Ed.; Wiley-VCH: New York, 2003. 231 90. Hoye, T. R.; Jeffrey, C. S.; Tennakoon, M. A.; Wang, J.; Zhao, H. Relay ring-closing metathesis (RRCM): A strategy for directing metal movement throughout olefin metathesis sequences. J. Am. Chem. Soc. 2004, 126, 1021010211. 91. Dudley, G. B.; Engel, D. A.; Ghiviriga, I.; Lam, H.; Poon, K. W. C.; Singletary, J. A. Synthesis of (+)-dihydro-epi-deoxyarteannuin B. Org. Lett. 2007, 9(15), 28392842. 92. Majumder, U.; Rainier, J. D. Olefinic-ester cyclization using Takai-Utimoto reduced titanium alkylidenes. Tetrahedron Lett. 2005, 46, 72097211. 93. Schollkopf, U. Carbonyl-olefin transformation with (C6H5)3P=CH2.-Wittig reaction. Angew. Chem. 1959, 71, 260273. 94. Boutagy, J.; Thomas, R. Olefin synthesis with organic phosphonate carbanions. Chem. Rev. 1974, 74, 8799. 95. Patai, S.; Pappoport, Z. The Chemisty of Enones, Wiley: Chichester, 1989 96. Kϋrti, L.; Czakó, B. Strategic Applications of Named Reactions in Organic Synthesis; Elsevier: New York, NY 2003; pp 89 and references cited. 97. Maryanoff, B. E.; Reitz, A. B. The Witting olefination reaction and modifications involving phosphoryl-stablized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863927. 98. Kelly, S. E. Alkene Synthesis; Trost, B. M., Fleming, I., Eds.; Comprehensive Organic Synthesis; Pergamon: Oxford, 1991; Vol.1, pp 729817. 99. Trost, B. M. The atom economy—a search for synthetic efficiency. Science 1991, 254, 14711477. 100. Shindo, M. Ynolates as functional carbanions. Synthesis, 2003, 22752288. 101. Fuks, R.; Viehe, H. G. Heterosubstituierte acetylene, XXV C2-kettenverlängerung von aldehyden und ketonen mit inaminen: die synthese substituierter acrylamide. Chem. Ber. 1970, 103, 564572. 232 102. Hsung, R. P.; Zificsak, C. A.; Wei, L. –L.; Douglas, C. J.; Xiong, H.; Mulder, J. A. Lewis acid promoted hetero [2+2] cycloaddition reactions of aldehydes with 10propynyl-9(10H)-acridone. A highly stereoselective synthesis of acrylic acid derivatives and 1,3-dienes using an electron deficient variant of ynamine. Org. Lett. 1999, 1, 12371240. 103. Hsung, R. P., Ed. Chemistry of electon-deficient ynamines and ynamides (Tetrahedron Symposium-in-Print Number 118). Tetrahedron 2006, 62, 37713938. 104. You, L.; Al-Rashid, Z. F.; Figueroa, R.; Ghosh, S. K.; Li, G.; Lu, T.; Hsung, R. P. A two-carbon homologation of aldehydes and ketones using ynamides. Synlett 2007, 16561662. 105. Rupe, H.; Kambli, E. Ungesättigte aldehydes aus acetylen-alkoholen. Helv. Chim. Acta 1922, 9, 672. 106. Ma, D.; Lu, X. Isomerization of propargylic alcohols catalyzed by an iridium complex. Tetrahedron Lett. 1989, 30(16), 21092122. 107. Lu, X.; Ji, J.; Ma, D.; Shen, W. Facile synthesis of 1,4-diketones via palladium complex catalyzed isomerization of alkynediols. J. Org. Chem. 1991, 56(20), 57745778. 108. Lu, X.; Ji, J.; Guo, C.; Shen, W. Isomerization of alkynemono-ols catalyzed by palladium(0) complex and diols. J. Organomet. Chem. 1992, 428, 259266. 109. Salah, M. K. E.; Pellicciari, R. Rhodium-catalyzed redox isomerization of hydroxyl alkynes to trans keto and hydroxy vinyl esters. A short stereoselective synthesis of dipeptide isoesters. Tetrahedron Lett. 1995, 36(25), 44974500. 110. Tanaka, K.; Shoji, T. Cationic rhodium(I)/BINAP complex-catalyzed isomerization of secondary propargylic alcohols to α,β-enones. Org. Lett. 2005, 7(16), 35613563. 111. Trost, B. M.; Livingston, R. C. Two-metal catalyst system for redox isomerization of propargyl alcohols to enals and enones. J. Am. Chem. Soc. 1995, 117(37), 95869587. 112. Trost, B. M.; Livingston, R. C.; An atom-economic and selective rutheniumcatalyzed redox isomerization of propargylic alcohols. An efficient strategy for the synthesis of leukotrienes. J. Am. Chem. Soc. 2008, 130(36), 1197011978. 233 113. Edens, M.; Boerner, D.; Chase, C. R.; Nass, D.; Schiavelli, M. Mechanism of the Meyer-Schuster rearrangement. J. Org. Chem. 1977, 42(10), 34023408. 114. Andres, J.; Cardenas, R.; Silla, E. Tapia, O. A theoretical study of the MeyerSchuster reaction mechanism: minimum-energy profile and properties of transitionstate structure. J. Am. Chem. Soc. 1988, 110, 666674. 115. Tapia, O.; Lluch, J. M.; Cardenas, R.; Andres, J. Theoretical study of salvation effects of chemical reactions. A combined quantum chemical/Monte Carlo study of the Meyer-Schuster reaction mechanism in water. J. Am. Chem. Soc. 1989, 111(3), 829835. 116. Meyer, K. H.; Schuster, K. Umlagerung tertiärer äthinyl-carbinole in ungesättigte ketone. Chem. Ber. 1922, 55, 819. 117. Swaminathan, S.; Narayanan, K. V. The rearrangements. Chem. Rev. 1971, 71, 429438. Rupe and Meyer-Schuster 118. Charbardes, P.; Querou, Y., French Patent 1554805 (Jan. 24, 1969); C. A. 72, 43923 (1970). 119. Pauling H. α,β-Ungesättigte carbonylverbindungen durch umlagerung von acetylencarbinolen. Chimia 1973, 7, 383386. 120. Pauling, H.; Andrews, D. A.; Hindley, N. C. The rearrangement of α-acetylenic alcohols to α,β-unsaturated carbonyl compounds by silylvanadate catalysts. Helv. Chem. Acta 1976, 59, 12331243. 121. Olson, G. L.; Morgan, K. D.; Saucy, G. A new vanadium (V)-catalyzed rearrangement of 17-ethynyl-17-hydroxy steroids to 17(20)-en-21-al compounds key intermediates in the synthesis of the corticosteroid side chain. Synthesis 1976, 2526. 122. Olson, G. L.;Cheunĝ, H. C.; Morgan, K. D.; Borer, R.; Saucy, G. 59. A stereospecific synthesis of vitamin A from 2,2,6-trimethyl-cyclohexanone. Helv. Chim. Acta 1976, 59, 567585. 123. Mercier, C.; Chabardes, P. Organometallic chemistry in industrial vitamin A and vitamin E synthesis. Pure & Appl. Chem. 1994, 66(7), 15091518. 234 124. Erman, M. B.; Aul’chenko, I. S.; Kheifits, L. A.; Dulova, V. G.; Novikov, J. N.; Vol’pin, M. E. The rearrangement of tertiary propargyl alcohols to α,β-unsaturated aldehydes in the presence of polymeric organosilyl vanadates. Tetrahedron Lett. 1976, 34, 29812984. 125. Lorber, C. Y.; Osborn, J. A. Cis-dioxomolybdenum(VI) complexes as new catalysts for the Meyer-Schuster rearrangement. Tetrahedron Lett. 1996, 37(6), 853856. 126. Chabardes, P.; Kuntz, E.; Varagnat, J. Use of oxo-metallic derivatives in isomerization. Tetrahedron 1977, 33, 17751783. 127. Trost, B. M.; Chung, C. K. Vanadium-catalyzed addition of propargyl alcohols and imines. J. Am. Chem. Soc. 2006, 128(32), 1035810359. 128. Chabardes, P. Ti/Cu and Ti/Ag catalysts for the isomerization of α-acetylenic alcohols into α,β-ethylenic carbonyl derivatives. Tetrahedron Lett. 1988, 29(48), 62536256. 129. Choudary, B. M.; Prasad, A. D.; Valli, V. L. K. Shape-selective isomerization of αacetylenic alcohols to α,β-ethylenic carbonyl compounds by vanadium-pillared montomorillonite catalyst. Tetrahedron Lett. 1990, 31, 75217522. 130. Narasaka, K.; Kusama, H.; Hayashi, Y. Rearrangement of allylic and propargylic alcohols catalyzed by the combined use of tetrabutylammonium perrhenate(VII) and p-toluenesulfonic acid. Chem. Lett. 1991, 14131416. 131. Sherry, B. D.; Radosevich, A. T.; Toste, F. D. A mild C-O bond formation catalyzed by a rhenium-oxo complex. J. Am. Chem. Soc. 2003, 125, 60766077. 132. Stefanoni, M.; Luparia, M.; Porta, A.; Zanoni, G.; Vidari, G. A simple and versatile Re-catalyzed Meyer-Schuster rearrangement of propargylic alcohols to α,βunsaturated carbonyl compounds. Chem. Eur. J. 2009, 15, 39403944. 133. Vidari performed control experiments in which pure (Z)-isomer was fully converted to the (E)-isomer under the standard reaction conditions. 134. Egi, M.; Yamaguchi, Y.; Fujiwara, N.; Akai, S. MoAu combo catalysis for rapid 1,3-rearrangement of propargyl alcohols into α,β-unsaturated carbonyl compounds. Org. Lett. 2008, 10(9), 18671870. 235 135. Muzart, J. Gold-catalyzed reactions of alcohols: isomerization, inter- and intramolecular reactions leading to CC and Cheteroatom bonds. Tetrahedron 2008, 64, 58155849. 136. Jiménez-Núñez, E.; Echavarren, A. M. Molecular diversity through gold catalysis with alkynes. Chem. Commun. 2007, 333346. 137. Fϋrstner, A.; Davies, P. W. Catalytic carbophilic activation: catalysis by platinum and gold -acids. Angew. Chem. Int. Ed. 2007, 46, 34103449. 138. Fukuda, Y.; Utimoto, K. Efficient transformation of methyl propargyl ethers into α,β-unsaturated ketones. Bull. Chem. Soc. Jpn. 1991, 64, 20132015. 139. Pindur, U.; Schall, T. Proton acid-induced rearrangements of -alkynylestradiol methyl ethers. 1993, 10991103. Lieb. Ann. Chem. 1993, 10, 10991103 140. Zhang, L. Tandem Au-catalyzed 3,3-rearrangement-[2 + 2] cycloadditions of propargylic esters: expeditious access to highly functionalized 2,3-indoline-fused cyclobutanes. J. Am. Chem. Soc. 2005, 127, 1680416805. 141. Oh, C. H.; Kim, A.; Park, W.; Park, D. I.; Kim, N. Synthesis of 2-acyl-3arylnapthalenes by dual catalysts of sodium tetrachloroaurate toward alkynepropargylic acetates. Synlett 2006, 27812784. 142. Buzas, A.; Gagosz, F. J. Gold(I) catalyzed isomerization of 5-en-2-yn-1-yl acetates: An efficient access to acetoxy bicycle[3.1.0]hexanes and 2-cycloalken-1ones. J. Am. Chem. Soc. 2006, 128, 1261412615. 143. Wang, S.; Zhang, L. A gold-catalyzed unique cycloisomerization of 1,5-enynes: Efficient formation of 1-carboxycyclohexa-1,4-dienes and carboxyarenes. J. Am. Chem. Soc. 2006, 128, 1427414275. 144. Lemiere, G.; Gandon, V.; Cariou, K.; Fukuyama, T.; Dhimane, A. –L.; Fensterbank, L.; Malacria, M. Tandem gold(I)-catalyzed cyclization/electrophilic cyclopropanation of vinyl allenes. Org. Lett. 2007, 9, 22072209. 145. Yeom, H. S.; Yoon, S. J.; Shin, S. Au(I)-catalyzed tandem [3,3]-sigmatropic rearrangement-cycloisomerization cascade as a route to spirocyclic furans. Tetrahedron Lett. 2007, 48, 48174820. 236 146. Correa, A.; Marion, N.; Fensterbank, L.; Malacria, M.; Nolan, S. P.; Cavallo, L. Golden carousel in catalysis: the cationic gold/propargylic ester cycle. Angew. Chem. Int. Ed. 2008, 47, 718721. 147. Mamane, V.; Gress, T.; Krause, H.; Fürstner, A. Platinum- and Gold-catalyzed cycloisomerization reactions of hydroxylated enynes. J. Am. Chem. Soc. 2004, 126, 86548655. 148. Shi, X.; Gorin, D. J.; Toste, F. D. Synthesis of 2-cyclopentenones by gold(I)catalyzed Rautenstrauch rearrangement. J. Am. Chem. Soc. 2005, 127, 58025803. 149. Johansson, M. J.; Gorin, D. J.; Staben, S. T.; Toste, R. D. Gold(I)-catalyzed stereoselective olefin cyclopropanation. J. Am. Chem. Soc. 2005, 127, 1800218003. 150. Gorin, D. J.; Dubé, P.; Toste, F. D. Synthesis of benzonorcaradienes by gold(I)catalyzed [4+3] annulations. J. Am. Chem. Soc. 2006, 128, 1448014481. 151. Sugawara, Y.; Yamada, W.; Yoshida, S.; Ikeno, T.; Yamada, T. Carbon dioxidemediated catalytic rearrangement of propargyl alcohols into α,β-unsaturated ketones. J. Am. Chem. Soc. 2007, 129, 1290212903. 152. Yu, M.; Li, G.; Wang, S.; Zhang, L. Gold-catalyzed efficient formation of α,βunsaturated ketones from propargylic acetates. Adv. Synth. Catal. 2007, 349, 871875. 153. When 2-butanone was used as a solvent there was always competitive elimination. The use of acetonitrile as the solvent was believed to lessen the reactivity of Au(PPh3)NTf2 through solvent coordination with the catalyst. 154. Wang, S.; Zhang, L. Gold-catalyzed efficient formation of alkenyl enol esters/carbonates from trimethylsilylmethyl-substituted propargyl esters/carbonates. Org. Lett. 2006, 8, 45854587. 155. Wang, S.; Zhang, L. A highly efficient preparative method of α-ylidene-βdiketones via AuIII-catalyzed acyl migration of propargylic esters. J. Am. Chem. Soc. 2006, 128, 84148415. 156. Zhang, L.; Wang, S. Efficient synthesis of cyclopentenones from enynyl acetates via tandem Au(I)-catalyzed 3,3-rearrangement and the Nazarov reaction. J. Am. Chem. Soc. 2006, 128, 14421443. 237 157. Zhang, L. Tandem Au-catalyzed 3,3-rearrangement-[2 + 2] cycloadditions of propargylic esters: expeditious access to highly functionalized 2,3-indoline-fused cyclobutanes. J. Am. Chem. Soc. 2005, 127, 1680416805. 158. For Cu refer to, Barluenga, J.; Riesgo, L.; Vicente, R.; López, L. A.; Tomás, M. Rearrangement of propargylic esters: metal-based stereospecific synthesis of (E)and (Z)-Knoevenagel derivatives. J. Am. Chem. Soc. 2007, 129, 77727773. 159. Marion, N.; Díez-González, S.; de Frémont, P.; Noble, A. R.; Nolan, S. P. Au Icatalyzed tandem [3,3] rearrangement-intramolecular hydroarylation: mild and efficient formation of substituted indenes. Angew. Chem. Int. Ed. 2006, 45, 36473650. 160. Zhao, J.; Hughes, C. O.; Toste, F. D. Synthesis of aromatic ketones by a transition metal-catalyzed tandem sequence. J. Am. Chem. Soc. 2006, 128, 74367437. 161. Bovonsombat, P.; McNelis, E. Formations of mixed β,β-dihaloenals from halogenated secondary alkynols. Tetrahedron Lett. 1992, 33(50), 77057708. 162. Yu, M.; Zhang, G.; Zhang, L. Gold-catalyzed efficient preparation of linear αhaloenones from propargylic acetates. Tetrahedron 2009, 65, 18461855. 163. Marion, N.; Carlqvist, P.; Gealageas, R.; de Frémont, P.; Maseras, F.; Nolan, S. P. [(NHC)AuI]-Catalyzed formation of conjugated enones and enals: an experimental and computational study. Chem. Eur. J. 2007, 13, 64376451. 164. Ramón, R. S.; Marion, N.; Nolan, S. P. [(NHC)AuCl]-catalyzed Meyer-Schuster rearrangement: scope and limitation. Tetrahedron 2009, 65, 17671773. 165. Lee, S. I.; Baek, J. Y.; Sim, S. H.; Chung, Y. K. Gold(I)-catalyzed rearrangement of propargylic alcohols to α,β-unsaturated ketones. Synthesis 2007, 14, 21072114. 166. Imagawa, H.; Asai, Y.; Takano, H.; Hamagaki, H.; Nishizawa, M. Mercuric triflatecatalyzed reaction of propargyl acetates with water leading to vinyl ketones. Org. Lett. 2006, 8(3), 447450. 238 167. Nishizawa, M.; Hirakawa, H.; Nakagawa, Y.; Yamamoto, H.; Namba, K.; Imagawa, H. Virtually complete E-selective α,β-unsaturated ester synthesis by Hg(OTf)2-catalyzed hydration of sec-ethoxyalkynyl acetate. Org. Lett. 2007, 9(26), 55775580. 168. In addition to heteroatoms, there is a single report on a ferrocene alkyne undergoing Meyer-Schuster rearrangement (a) Banide, E. V.; Ortin, Y.; Chamiot, B.; Cassidy, A.; Niehaus, J.; Moore, A.; Seward, C. M.; Mϋller-Bunz, H.; McGlinchey, M. J. Syntheses, structures, and dimerization of ferroccenyl-and fluorenylideneallenes: pushpull multiple bonds. Organometallics 2008, 27(16), 41734182. 169. Yoshimatsu, M.; Naito, M.; Kawahigashi, M.; Shimizu, H.; Kataoka, T. MeyerSchuster rearrangement of γ-substituted propargyl alcohols: A convenient synthesis of α,β-unsaturated thioesters. J. Org. Chem. 1995, 60, 47984802. 170. Picquet, M.; Fernández, A.; Bruneau, C.; Dixneut, P. H. Efficient Rutheniumcatalysed synthesis of 3-hydroxy-1-propen-1-yl benzoates: En route to an improved isomerization of 2-propyn-1-ols into α,β-unsaturated aldehydes. Eur. J. Org. Chem. 2000, 23612366. 171. Suzuki, T.; Tokunaga, M.; Wakatsuki, Y. Efficient transformation of propargylic alcohols to α,β-unsaturated aldehydes catalyzed by ruthenium/water under neutral conditions. Tetrahedron Lett. 2002, 43, 75317533. 172. Cadierno, V.; Díez, J.; García-Garrido, S. E.; Gimeno, J. [Ru(ƞ3-2C3H4Me)(CO)(dppf)][SbF6]: a mononuclear 16e- ruthenium(II) catalyst for propargylic substitution and isomerization of HC≡CCPh2(OH). Chem. Comm. 2004, 27162717. 173. Cadierno, V.; Díez, J.; García-Garrdo, S. E.; Gimeno, J.; Nebra, N.Efficient access to conjugated dienones and diene-diones from propargylic alcohols and enolizable ketones: A tandem isomerization/condensation process catalyzed by the sixteen-electron allyl-ruthenium(II) complex [Ru(ƞ3-2-C3H4Me)(CO)(dppf)][SbF6]. Adv. Synth. Catal. 2006, 348, 21252132. 174. Cadierno, V.; Crochet, P.; Gimeno, J. Ruthenium-catalyzed isomerization of allyic and propargylic alcohols in aqueous and organic media: applications in synthesis. Synlett 2008, 8, 11051124. 175. Pelletier, S. W.; Mody, N. V. Facile synthesis of the cyclohexyl constituents of the boll weevil sex pheromone. J. Org. Chem. 1976, 41(6), 10691071. 239 176. Welch, S. C.; Hagan, C. P.; White, D. H.; Fleming, W. P.; Trotter, J. W. A stereoselective total synthesis of the antifungal mold metabolite 7α-methoxy-3a,10bdimethyl-1,2,3,3aα,7,10bβ,10cα-octahydro-4H, 9H-furo[2’,3’,4’: 4,5]naptho[2,1-c] pyran-4,10-dione. J. Am. Chem. Soc. 1977, 99(2), 549556. 177. Crich, D.; Natarajan, S.; Crich, J. Z. Synthesis of the taxol-AB-system by olefination of an A-ring C1 ketone and direct B-ring closure. Tetrahedron 1997, 53(21), 71397158. 178. Sun, C.; Lin, X.; Weinreb, S. M. Explorations on the total synthesis of the unusual marine alkaloid chartelline A. J. Org. Chem. 2006, 71(8), 31593166. 179. Engel, D. A.; Dudley, G. B. Olefination of ketones using a gold(III)-catalyzed Meyer-Schuster rearrangement. Org. Lett. 2006, 8(18), 40274029. 180. Lopez, S. S.; Engel, D. A.; Dudley, G. B. The Meyer-Schuster rearrangement of ethoxyalkynyl carbinols. Synlett 2007, 6, 949953. 181. Engel, D. A.; Lopez, S. S.; Dudley, G. B. Lewis acid-catalyzed Meyer-Schuster reactions: methodology for the olefination of aldehydes and ketones. Tetrahedron 2008, 64, 69886996. 182. Sundararaman, P.; Herz, W. Oxidative rearrangements of tertiary and some secondary allylic alcohols with chromium(VI) reagents. A new method for 1,3functional group transposition and forming mixed aldol products. J. Org. Chem. 1977, 42(5), 813819. 183. Georgy, M.; Boucard, V.; Campagne, J. –M. Gold(III)-catalyzed nucleophilic substitution of propargylic alcohols. J. Am. Chem. Soc. 2005, 127, 1418014181. 184. Gorin, D. J.; Toste, F. D. Relativistic effects in homogeneous gold catalysis. Nature 2007, 446, 395403. 185. Kobayashi, S. Sc(III) Lewis Acids; Yamamato, H., Ed.; Lewis Acids in Organic Synthesis; Wiley-VCH: New York, NY, 2000; Vol. 2, Chapter 19, pp 883910. 240 BIOGRAPHICAL SKETCH Birth Place Waukesha, Wisconsin • February 24, 1982 Educational Background Florida State University, Tallahassee, FL • August 2004 to August 2009 • Ph.D. in Organic Chemistry (anticipated completion in August 2009) • Research Advisor: Professor Gregory B. Dudley Georgia Southern University, Statesboro, GA • August 2000 to May 2004 • B.A. degree in Chemistry, with Honors - summa cum laude • Research Advisor: Professor James M. LoBue Gordon Central High School, Calhoun, GA • August 1996 to June 2000 • Valedictorian Professional Experience Florida Custom Synthesis, Inc., Tallahassee, FL • CEO, 2009–present • serving Florida’s emerging biotech industry with premium organic synthesis on demand • http://www.flsynth.com Honors and Awards • MDS Fellowship, 2004–present • Florida State University Fellowship, 2005-2006, 2007-2008 • American Institute of Chemists Foundation Outstanding Senior Award, 2004 • Georgia Southern Physical Chemistry Award, 2003 • Georgia Southern Organic Chemistry Award, 2002 • Bovey Foundation Scholarship, 2000 • Bird Scholarship, 2000 • Governors Scholarship, 2000 Teaching • Teacher’s Assistant for Organic Chemistry I, CHM 2210 241 • Teacher’s Assistant for Organic Chemistry II, CHM 2211 Research Talks: (2) ―Synthetic approaches to Artemisinin-related natural products, Didesmethylartemisinin and dihydro-epi-Deoxyarteannuin B.‖ Engel, Douglas A. Abstract of Papers, 235 th ACS National Meeting, New Orleans, LA, April 6-10, 2008, ORGN-504. (1) ―Synthetic approaches to Artemisinin-related natural products, Dihydroartemisinic Acid and Arteannuin M.‖ Engel, Douglas A. Abstract of Papers, 231 st ACS National Meeting, Atlanta, GA, March 26-30, 2006, ORGN-477. Publications: (4) Engel, D. A.; Lopez, S. S. Lewis acid-catalyzed Meyer–Schuster reactions: methodology for the olefination of aldehydes and ketones. Tetrahedron 2008, 64, 6988–6996. (invited contribution to Symposium in Print special issue) (3) Dudley, G. B.; Engel, D. A.; Ghiviriga, I.; Lam, H.; Poon, K. W. C.; Singletary, J. A. Synthesis of dihydro-epi-deoxyarteannuin B. Org. Lett. 2007, 9, 2839–2842. (2) Lopez, S. S.; Engel, D. A.; Dudley, G. B. The Meyer–Schuster rearrangement of ethoxyalkynyl carbinols. Synlett 2007, 949–953. (1) Engel, D. A.; Dudley, G. B. Olefination of ketones using a gold(III)-catalyzed Meyer–Schuster rearrangement. Org. Lett. 2006, 8, 4027–4029. 242