Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Human genome wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Non-coding DNA wikipedia , lookup
Genetic engineering wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Copy-number variation wikipedia , lookup
Oncogenomics wikipedia , lookup
Ridge (biology) wikipedia , lookup
Transposable element wikipedia , lookup
Genomic library wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Gene therapy wikipedia , lookup
History of genetic engineering wikipedia , lookup
Public health genomics wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Gene nomenclature wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Metagenomics wikipedia , lookup
Genome editing wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
Maximum parsimony (phylogenetics) wikipedia , lookup
Genomic imprinting wikipedia , lookup
Gene desert wikipedia , lookup
Genome (book) wikipedia , lookup
Minimal genome wikipedia , lookup
Gene expression profiling wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Pathogenomics wikipedia , lookup
Designer baby wikipedia , lookup
Microevolution wikipedia , lookup
Gene expression programming wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Genome evolution wikipedia , lookup
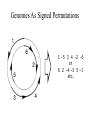
GRAPPA: Large-scale whole genome phylogenies based upon gene order evolution Tandy Warnow, UT-Austin Department of Computer Sciences Institute for Cellular and Molecular Biology Program in Evolution, Ecology, and Behavior Center for Computational Biology and Bioinformatics Whole-Genome Phylogenetics A A C D X B E Y E Z C F B D W F Genomes As Signed Permutations 1 –5 3 4 -2 -6 or 6 2 -4 –3 5 –1 etc. Genomes Evolve by Rearrangements 1 2 3 4 5 6 7 9 10 1 2 3 –8 9 –7 -8 4 –6 –7 5 –5 –6 6 -4 –5 7 –4 8 9 10 • Inversion (Reversal) • Transposition • Inverted Transposition 8 Genome Rearrangement Has A Huge State Space • DNA sequences : 4 states per site • Signed circular genomes with n genes: 2 n 1 (n 1)! states, 1 site • Circular genomes (1 site) – with 37 genes: – with 120 genes: 2.56 10 3.70 10 52 states 232 states Why use gene orders? • “Rare genomic changes”: huge state space and relative infrequency of events (compared to site substitutions) could make the inference of deep evolution easier, or more accurate. • Our research shows this is true, but accurate analysis of gene order data is computationally very intensive! Phylogeny reconstruction from gene orders • Distance-based reconstruction: estimate pairwise distances, and apply methods like NeighborJoining or Weighbor • “Maximum Parsimony”: find tree with the minimum length (inversions, transpositions, or other edit distances) • Maximum Likelihood: find tree and parameters of evolution most likely to generate the observed data This talk • Distance-based methods: we show how to estimate genomic distances appropriately for the Generalized Nadeau-Taylor model • Parsimony-style methods: we can find very good solutions to NP-hard problems (inversion and breakpoint phylogeny) quite quickly • Validation of these approaches on real and simulated data Distance-based methods Genomic Distance Estimators • Standard: – Breakpoint distance – (Minimum) Inversion distance • Our estimators: We attempt to estimate the actual number of events (the “true evolutionary distance”): – EDE [Moret et al, ISMB’01] – Approx-IEBP [Wang and Warnow, STOC’01] – Exact-IEBP [Wang, WABI’01] Breakpoint Distance • Breakpoint distance=5 1 2 3 4 5 6 7 8 9 10 1 –3 –2 4 5 9 6 7 8 10 Minimum Inversion Distance • Inversion distance=3 1 2 3 4 8 9 10 1 2 3 –8 –7 –6 –5 –4 9 10 1 8 –3 –2 –7 –6 –5 –4 9 10 1 8 –3 9 10 7 5 6 7 2 –6 –5 –4 Measured Distance vs. Actual Number of Events Breakpoint Distance Inversion Distance 120 genes, inversion-only evolution Generalized Nadeau-Taylor Model • Three types of events: – Inversions – Transpositions – Inverted Transpositions • Events of the same type are equiprobable • Probability of the three types have fixed ratio: Inv : Trp : Inv.Trp = (1-a-b):a:b Estimating True Evolutionary Distances for Genomes Given fixed probabilities for each type of event, we estimate the expected breakpoint distance after k random events, or the expected inversion distance after k random events. Inverting these functions gives us a better estimate of true evolutionary distances. • Approx-IEBP [Wang and Warnow 2001] • Exact-IEBP [Wang 2001] • EDE [Moret et al, ISMB 2001] (inversion based) Estimating True Evolutionary Distances for Genomes (cont.) Estimating the expected Inversion distance: EDE [Moret, Wang, Warnow, Wyman 2001] – Closed-form formula based upon an empirical estimation of the expected inversion distance after k random events (based upon 120 genes and inversion only, but robust to errors in the model) . – Polynomial time, fastest of the three. Absolute Difference •120 genes •Inversion only evolution (Similar relative performance under other models) Accuracy of Neighbor Joining Using Distance Estimators •120 genes •All three event types equiprobable •10, 20, 40, 80, and 160 genomes •Similar relative performance under other models Summary of Distance-based Reconstruction Methods • Statistically-based estimation of genomic distances improves NJ analyses. • Our IEBP estimators assume knowledge of the probabilities of each type of event, but are robust to model violations. • EDE is based upon an inversion-only evolutionary model, but is robust. • Best performing method: Weighbor(EDE); second best is NJ(EDE); both are robust to model violations. • Worst performing is NJ(BP). • Accuracy is very good, except when very close to saturation. Maximum Parsimony on Rearranged Genomes (MPRG) • The leaves are rearranged genomes. • Find the tree that minimizes the total number of rearrangement events A A B 3 6 E C 2 B D C 3 4 Total length = 18 F D Optimization problems for gene order phylogeny • Breakpoint phylogeny: find the phylogeny which minimizes the total number of breakpoints (NP-hard, even to find the median of three genomes) • Inversion phylogeny: find the phylogeny which minimizes the sum of inversion distances on the edges (NP-hard, even to find the median of three genomes) Inversion and Breakpoint phylogenies • When the data are close to saturated, even Weighbor(EDE) analyses are insufficiently accurate. In these cases, our initial investigations suggest that the inversion and breakpoint phylogeny approaches may be superior. • Problem: finding the best trees is enormously hard, since even the “point estimation” problem is hard (worse than estimating branch lengths in ML). Local optimum MP score Global optimum Phylogenetic trees GRAPPA (Genome Rearrangement Analysis under Parsimony and other Phylogenetic Algorithms) http://www.cs.unm.edu/~moret/GRAPPA/ • Heuristics for NP-hard optimization problems • Fast polynomial time distance-based methods • Contributors: U. New Mexico,U. Texas at Austin, Universitá di Bologna, Italy • Freely available in source code at this site. • Project leader: Bernard Moret (UNM) ([email protected]) Benchmark gene order dataset: Campanulaceae • 12 genomes + 1 outgroup (Tobacco), 105 gene segments • NP-hard optimization problems: breakpoint and inversion phylogenies (techniques score every tree) 1997: BPAnalysis (Blanchette and Sankoff): 200 years (est.) Benchmark gene order dataset: Campanulaceae • 12 genomes + 1 outgroup (Tobacco), 105 gene segments • NP-hard optimization problems: breakpoint and inversion phylogenies (techniques score every tree) 1997: BPAnalysis (Blanchette and Sankoff): 200 years (est.) 2000: Using GRAPPA v1.1 on the 512-processor Los Lobos Supercluster machine: 2 minutes (200,000-fold speedup per processor) Benchmark gene order dataset: Campanulaceae • 12 genomes + 1 outgroup (Tobacco), 105 gene segments • NP-hard optimization problems: breakpoint and inversion phylogenies (techniques score every tree) 1997: BPAnalysis (Blanchette and Sankoff): 200 years (est.) 2000: Using GRAPPA v1.1 on the 512-processor Los Lobos Supercluster machine: 2 minutes (200,000-fold speedup per processor) 2003: Using latest version of GRAPPA: 2 minutes on a single processor (1-billion-fold speedup per processor) Summary • Weighbor(EDE) and NJ(EDE) are highly accurate polynomial time distance-based reconstructions, except when datasets are close to saturated • GRAPPA (inversion phylogeny or breakpoint phylogeny) produces highly accurate estimates of trees, and even of ancestral gene orders, acceptably fast Limitations and ongoing research • Current methods limited to single chromosomes with equal gene content (or very small amounts of deletions and duplications) -- we are working on developing reliable techniques for genomes with unequal gene content Acknowledgements • Funding: The David and Lucile Packard Foundation, and The National Science Foundation. • Collaborators: Bernard Moret (UNM), David Bader (UNM), Bob Jansen (UT), Linda Raubeson (CWU) • Students: Li-San Wang (now postdoc of Junhyong Kim at Penn), Jijun Tang (UNM), and others at UNM Phylolab, U. Texas Please visit us at http://www.cs.utexas.edu/users/phylo/