Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Epigenetics of diabetes Type 2 wikipedia , lookup
Medical genetics wikipedia , lookup
History of genetic engineering wikipedia , lookup
Gene expression programming wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Koinophilia wikipedia , lookup
Ridge (biology) wikipedia , lookup
Heritability of IQ wikipedia , lookup
Minimal genome wikipedia , lookup
Genomic imprinting wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Saethre–Chotzen syndrome wikipedia , lookup
Population genetics wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Genome evolution wikipedia , lookup
Public health genomics wikipedia , lookup
Gene expression profiling wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Designer baby wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Genome (book) wikipedia , lookup
Oncogenomics wikipedia , lookup
Frameshift mutation wikipedia , lookup
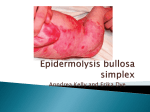
Centre for Arab Genomic Studies A Division of Sheikh Hamdan Award for Medical Sciences The Catalogue for Transmission Genetics in Arabs CTGA Database Epidermolysis Bullosa Letalis Alternative Names Epidermolysis Bullosa, Junctional, HerlitzPearson Type Herlitz-Pearson Type Epidermolysis Bullosa Epidermolysis Bullosa, Junctional, Herlitz Type Epiligrin WHO International Classification of Diseases Congenital malformations, deformations and chromosomal abnormalities junctional epidermolysis bullosa characterized by localized to generalized blistering, nail dystrophy, scarring alopecia, and mucosal involvement; and the Herlitz type of junctional epidermolysis bullosa, associated with extensive skin and mucosal blistering, nail dystrophy, exuberant granulation tissue, enamel defects, and a high perinatal mortality resulting from overwhelming infections and respiratory complications. The junctional epidermolysis bullosa subtype, generalized atrophic benign epidermolysis bullosa, is characterized by life-long blistering, resulting in cutaneous atrophy, diffuse scarring alopecia, pigmentary changes, and nail and tooth abnormalities. OMIM Number 226700 Mode of Inheritance Autosomal recessive Gene Map Locus 18q11.2, 1q32, 1q25-q31 Description Epidermolysis bullosa is a group of inherited disorders of the epithelial basement membrane zone that manifest with blistering of the skin and mucous membranes after minor trauma. The disease appears to be one of the most frequent monogenic causes of infant mortality among Arabs. The disease is traditionally classified into three groups according to the level of cleavage within the skin: Epidermolysis bullosa simplex results from separation of the skin above the basement membrane, junctional epidermolysis bullosa is caused by blister formation within the basement membrane, and in the dystrophic form of epidermolysis bullosa, blisters appear below the basement membrane. To date, all cases of junctional epidermolysis bullosa have been shown to be transmitted in an autosomal recessive fashion. Clinical and genetic heterogeneity are characteristic of junctional epidermolysis bullosa. Two major clinical variants of this epidermolysis bullosa subtype have been described: the moderately severe non-Herlitz Molecular Genetics The genetic basis of all major clinical variants of epidermolysis bullosa has been delineated. Specific mutations have been demonstrated in 10 different genes expressed within the dermoepidermal adhesion zone, with at least six different genes being involved in the pathogenesis of junctional epidermolysis bullosa. Herlitz junctional epidermolysis bullosa has been shown to be associated with mutations in one of the 3 laminin-5 genes (LAMB3, LAMA3, LAMC2), resulting in premature termination of protein translation, or reduced levels of mRNA transcripts resulting from non-sense–mediated mRNA decay. NonHerlitz junctional epidermolysis bullosa cases have also been assigned to mutations within the laminin-5 genes. The current mutation detection strategy followed in the United States and Europe entails analysis of patient samples for the recurrent mutations in LAMB3 followed by sequential screening of LAMB3, LAMC2, and finally LAMA3. Copyright © Centre for Arab Genomic Studies 1 The observation of reduced type XVII collagen/bullous pemphigoid antigen 2 expression in the skin of generalized atrophic benign epidermolysis bullosa patients led to the identification of causative mutations within the gene encoding this protein. Other clinical cases of generalized atrophic benign epidermolysis bullosa were shown to be caused by mutations affecting laminin-5-encoding genes. Epidemiology in the Arab World Saudi Arabia Nakano et al. (2002) identified two consanguineous families originating from Saudi Arabia with non-Herlitz junctional epidermolysis bullosa. Affected members of both families were analyzed for mutations in LAMB3, LAMA3, and LAMC2 genes. Both patients turned out to be homozygous for the Q46X mutation in the LAMC2 gene. Nakano et al. (2002) further investigated the genealogic relationship between the two unrelated families and found out that both originated from a Bedouin tribe of the Empty Quarter that is now split between Saudi Arabia and the United Arab Emirates. Sudan Nakano et al. (2002) identified a consanguineous family originating from Sudan with generalized atrophic benign epidermolysis bullosa. The affected member of the family presented with milia over his face. Immunofluorescence studies performed on frozen skin sections indicated lack of staining for type XVII collagen. United Arab Emirates Nakano et al. (2002) identified a consanguineous family originating from the United Arab Emirates with non-Herlitz junctional epidermolysis bullosa. The affected child in the family was analyzed for mutations in LAMB3, LAMA3, and LAMC2 genes. The child turned out to be heterozygote for two mutations, Q1083X and 1296insA, in the LAMB3 gene. The Q1083X mutation has been found in Haifa in Palestine. Interestingly, the two grandmothers of the affected child originated from that village. Nakano et al. (2002) quoted the presence of two different mutations in the affected child to emphasize the fact that the presence of consanguinity does not preclude compound heterozygosity. Yemen Nakano et al. (2002) identified a consanguineous family originating from Yemen with non-Herlitz junctional epidermolysis bullosa. The affected child in the family was analyzed for mutations in LAMB3, LAMA3, and LAMC2 genes. The child turned out to be homozygous for the 1942delG aberration in the LAMB3 gene. References Nakano A, Lestringant GG, Paperna T, Bergman R, Gershoni R, Frossard P, Kanaan M, Meneguzzi G, Richard G, Pfendner E, Uitto J, Pulkkinen L, Sprecher E. Junctional epidermolysis bullosa in the Middle East: clinical and genetic studies in a series of consanguineous families. J Am Acad Dermatol. 2002; 46(4):510-6. Contributors Ghazi: 3.10.2004 Copyright © Centre for Arab Genomic Studies 2