Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Signal transduction wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Paracrine signalling wikipedia , lookup
Ribosomally synthesized and post-translationally modified peptides wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Peptide synthesis wikipedia , lookup
Gene expression wikipedia , lookup
Magnesium transporter wikipedia , lookup
Metalloprotein wikipedia , lookup
Homology modeling wikipedia , lookup
Interactome wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Point mutation wikipedia , lookup
Protein purification wikipedia , lookup
Expression vector wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Western blot wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Biosynthesis wikipedia , lookup
Amino acid synthesis wikipedia , lookup
Genetic code wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Biochemistry wikipedia , lookup
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
145
GENETIC METHODS OF
POLYMER SYNTHESIS
Introduction
Polymers have found enormous technological significance in a variety of areas
including plastics, resins, fibers, and biomaterials. Both the chemical nature of
the polymer and its structure are critical in determining its properties; the importance of polymer structure in dictating function has, for many decades, fueled
the development of synthetic strategies for creating well-defined macromolecules.
During the past 15 years, the application of biosynthetic strategies has gained
increasing prominence as a polymer synthetic method, owing to the fact that the
biosynthesis of macromolecules offers precise control of monomer sequence and
macromolecular structure. This control is the source of the information storage,
hierarchical assembly, mechanical strength, and enzymatic properties of natural
macromolecules such as nucleic acids and proteins, which serve as an inspiration
to the polymer chemist. Genetic methods of polymer synthesis have therefore been
used to create protein-based polymeric materials inspired by natural molecules
such as collagen, silk, and elastin, as well as materials comprising amino acid
sequences with no natural counterpart. The macromolecules produced by these
strategies exhibit novel and well-controlled properties, and genetically directed
synthetic methods promise the contribution of additional advanced materials in
the future.
Properties of Synthetic Polymers
Synthetic polymers, used here to describe polymers produced by chemical (ie, nonbiological) methods, have enjoyed great technological success since the beginning
of their development in the early 20th century. A nearly limitless set of monomers
can be polymerized by a vast variety of chemical transformations, which has contributed to the successful application of polymers in disparate applications, such
as textiles, high temperature materials, and biomaterials. The properties of polymers are controlled by their chemical composition and by structural parameters
such as polymer chain length, stereochemistry, monomer sequence, and topology.
Because chemical polymerizations are statistical processes, synthetic polymers
exist as a population of different molecules of varying structure, rather than as a
collection of identical molecules. The properties of a synthetic polymer can therefore only be characterized with respect to the average properties of the chain
Encyclopedia of Polymer Science and Technology. Copyright John Wiley & Sons, Inc. All rights reserved.
146
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
population and the distribution around this average. Such heterogeneity has not
adversely affected the use of these macromolecules in bulk application, but does
have profound consequences when attempting to engineer polymer properties on
the nanometer length scale. In this regime, important parameters such as functional group placement and macromolecular assembly are directly affected by
variations in molecular weight, sequence, stereochemistry, and topology.
There have been many advances in synthetic polymer chemistry that have
increased the control of polymer structure. These advances began most notably
in the 1950s with the implementation of Ziegler–Natta polymerizations (1,2) and
living polymerizations (3–5), which permitted, for the first time, control over architectural parameters such as stereochemistry and molecular weight, respectively.
The development of these methods was critical to developing an understanding of
structure–property relationships in polymeric materials and for the production of
useful plastics, fibers, and elastomers. Since that time, a variety of novel chemical strategies for controlling polymerizations have continued to evolve, including
the use of organometallic catalysts to control stereochemistry in olefinic polymers; atom-transfer radical polymerizations that permit living free-radical polymerization; living N-carboxyanhydride polymerizations; and living rutheniumcatalyzed ring opening metathesis polymerizations (2,6–15). These methods have
been extremely important for producing stereoregular materials, synthetic macromolecules with unusual self-assembly properties and phase separation behavior,
polymers with triggered and controlled response, and functionalized polymers
with controlled molecular weight. Despite these advances, precise control of functional group placement remains elusive in polymers produced by chemical methods, which can limit their utility in applications where precise molecular recognition, catalysis, and assembly is desired. Because genetically directed methods
of polymer synthesis offer precise control over molecular weight, stereochemistry,
and sequence, they have been increasingly applied over the last 15 years toward
the synthesis of exactly defined protein based materials that have potential uses
in materials, biomedical, and nanotechnology applications.
Biosynthesis of Proteins
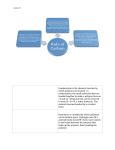
The control over protein synthesis in living organisms is derived from the templated nature of the genetically directed polymerization (Fig. 1). A DNA message
encoding an amino acid sequence—naturally derived or artificial—is transcribed
(converted) to a messenger RNA (mRNA) sequence. This sequence is translated
(decoded) by transfer RNA (tRNA) molecules, which deliver amino acids to the
ribosome. At the ribosome, the corresponding amino acid sequence is linked together enzymatically to form the protein. The tRNA molecules carry the appropriate amino acid to its exactly specified location along the protein chain via codon–
anticodon pairing, and are charged with the appropriate amino acid by a highly
selective class of enzymes, the aminoacyl tRNA synthetases. These enzymes exhibit an extremely small error frequency in amino acid incorporation—fewer than
one in 10,000 errors (16)—so the amino acid sequence is preserved with great fidelity during protein biosynthesis; this permits the exact control of the placement
of over 20 different amino acids along the protein backbone. This primary sequence
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
147
Fig. 1. Schematic of protein biosynthesis.
directs the formation of regular secondary structure (helical, coil, or sheet) and
mediates the 3-D structure, assembly, and molecular recognition that are the hallmark of biological materials and the source of their remarkable properties. The
strict sequence control, then, allows the synthesis of a remarkably diverse set of
materials from a single set of building blocks. For example, the range of properties
of Silk (qv), Collagen (qv), and elastin, which are each synthesized from the same
set of 20 natural amino acids, arises from differences in their primary sequences
and consequently different conformations and assembly.
The mechanical properties of these natural proteins, which can be
comparable to and sometimes exceed those of high performance synthetic materials (Table 1), coupled with their potential biological activity, have been a
motivation for the synthesis of polymers via genetic methods. Capturing these
properties in genetically engineered artificial protein polymers offers the possibility of biologically derived performance materials and other protein materials
that can be engineered with properties not currently found in the natural protein
repertoire (eg, novel folding motifs and assemblies). Furthermore, the exact molecular weight control and precise specification of amino acid position of genetically
directed methods provides materials that carry sufficient information to exactly
control their folding and assembly. Because proteins assume thermodynamically
stable structures in solution, the 3-D placement of functional groups on the atomic
Table 1. Representative Properties of Several Natural Proteins and Synthetic Fibersa
Material
B. mori silk
N. clavipes silk
Collagen (mammalian tendon)
Elastin (bovine ligament)b
Polylactic acid, 50,000–300,000 MW
Bone
Kevlar (49 fiber)
Synthetic rubber
a Adapted
b From
UTS, MPa
Modulus, GPa
% Strain at break
740
875–972
120
2
28–50
160
3600
50
10
11–13
1.2
0.0011
1.2–3.0
20
130
0.001
20
17–18
13
150
2–6
3
2.7
850
from Ref. 17, with permission from Elsevier.
Ref. 18.
148
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
length scale, with assembly controlling placement on longer length scales, is possible; the advantages of this assembly include the purposeful control of the mechanical, chemical, and biological properties of the artificial protein polymer chain. The
variety of amino acid sequences that can be constructed and produced in cellular
hosts via protein engineering methods provides access to a wide variety of protein
polymers with diverse properties. Additionally, the incorporation of non-natural
amino acids in these polymers has also significantly broadened the versatility of
in vivo protein synthesis as a route to producing polymeric materials.
Genetically Directed Synthetic Strategies
Recombinant DNA methods have been traditionally used in site-directed mutagenesis studies designed to probe protein folding or enzymatic activity. The ease with
which genetic sequences can be constructed has, however, led to the increased use
of these methods for the synthesis of proteins with repetitive sequences. Naturally
occurring materials with desired properties can be produced in organisms such as
bacteria and yeast, simply by transferring the DNA encoding the natural protein
into the appropriate expression host. Alternatively, short, repetitive amino acid
sequences that are found in naturally occurring proteins can be linked together to
form an artificial protein (one that does not exist in nature). Finally, since the folding of certain amino acid motifs is understood, amino acid sequences not found in
nature can be designed from scratch (de novo design) to produce artificial proteins
with desired chemical, biological, and/or physical behavior.
The production of naturally occurring proteins, such as silk, collagen, or
elastin, is motivated by their excellent mechanical properties. In one approach for
producing these proteins, the cDNA encoding the natural protein is isolated from
an organism that produces the protein of interest and is cloned into an expression
plasmid that carries the necessary genetic information to direct the expression
host to produce both RNA and protein. While this approach guarantees that the
polymer sequence will match that of the natural protein, difficulties with gene
stability and protein synthesis can be encountered when the genes are extremely
long and repetitive and when the coding preferences of the organism from which
the DNA is isolated are different than those of the expression host. In many
cases, it is not possible or necessary to construct a single amino acid sequence
that encodes the full-length protein, and so shortened DNA sequences from the
natural protein can be used.
Since many natural structural proteins comprise highly repetitive amino
acid sequences, strategies to produce artificial repetitive proteins with properties
analogous to those of the natural proteins have also been developed. The artificial
repetitive proteins are either based on naturally occurring amino acid sequences
or on sequences identified via de novo design. The overall strategy for gene design
and expression is summarized in Figure 2. Synthetic genes encoding the artificial proteins are produced via standard molecular biological protocols; the genes
for short amino acid sequences (30–40 amino acids, 90–120 base pairs) are first
produced by solid-phase chemical synthesis and are then ligated into circular plasmid DNA, which serves as a source of the DNA “monomer” for future experiments.
The initial solid-phase synthesis of the short genes permits use of DNA sequences
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
149
Fig. 2. Protein engineering strategy for the genetically directed synthesis of protein
polymers.
that reflect the natural coding preferences of the organism in which the protein
will be expressed and allows reduction of repetition in codon usage. It also permits
the inclusion of amino acids with desired chemical functionality, or the insertion
of biologically active sequences, to capture a variety of desired materials properties in the resulting protein. After the sequence of the gene in the plasmid in
confirmed, the monomer is isolated from the plasmid and multimerized enzymatically. The multimerization proceeds in a head-to-tail orientation to form genes
that can have lengths of up to 5000 base pairs (approximately 1675 amino acids);
these genes are ultimately ligated into an expression plasmid (vide infra).
There are several different strategies (random, iterative, and recursive) that
have emerged for the construction of multimers, as outlined in Figure 3, although
the use of one strategy is not exclusive of another; ie, a combination of these approaches can be used in the construction of a single artificial gene (19). In random
approaches, as the general one described above, DNA monomers are oligomerized
in a single step, creating a population of oligomers of different lengths. In iterative approaches, a DNA segment is oligomerized in a series of uniform steps, each
step adding one DNA segment to the growing oligomer. In recursive approaches,
the DNA monomers are joined sequentially, with the length of the ligated segments doubling at each step. Random approaches offer the advantage of rapid
creation of a library of repetitive genes of different lengths in a single step. Although this method does not guarantee the synthesis of a specific gene of desired
length, it has been the most widely adopted of the multimerization strategies,
owing to its simplicity and the fact that ligation conditions can be controlled
to increase the probability of obtaining multimers of a given molecular weight
range. Iterative and recursive approaches offer the advantage of production of
genes with a specific and predetermined molecular weight, which is desirable
if the effects of sequential changes in molecular weight or the impact of a precise and alternating block copolymer architecture are of interest. These methods requires repetitive cloning steps, the exact number depending on the desired
150
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
Fig. 3. Multimerization strategies employed for the construction of artificial genes.
Adapted from Ref. 19, with permission from Elsevier.
length of the gene. The different approaches are used depending on the requirements of a specific macromolecular design, and demonstrate the flexibility of genetic methods of polymer synthesis for the production of varied types of polymer
architectures.
Once the appropriate gene library is constructed, individual genes are incorporated into an expression plasmid, which is then incorporated into an expression
host, most commonly E. coli. Because only one type of plasmid is maintained per
cell, the plasmid DNA of individual bacterial colonies can be screened to identify
and isolate a specific synthetic gene of desired length. The chosen synthetic gene
is expressed from an expression plasmid (Fig. 2), which directs the synthesis of
mRNA and protein in the cell. The expression plasmid also contains a promoter
that regulates protein production and that can be controlled by inexpensive external means (eg, addition of chemicals, change in temperature). During protein
expression, host cells are grown to a desired density, and then protein synthesis
is initiated by simple addition of chemicals or change in temperature. Protein
polymers can be produced at high levels in host cells; protein generally accumulates intracellularly (although it can also be transported out of the cell) and
is then isolated from the cellular proteins via cell lysis, washing, precipitation,
and chromatographic methods. The proteins are analyzed via amino acid analysis, chromatographic methods, mass spectrometry, NMR, and immunochemical
analysis to confirm their identity; the pure product is absolutely homogeneous
in both molecular weight and sequence. A variety of protein polymers based on
silk, collagen, elastin, mussel adhesive proteins, and viral spike proteins have
been produced by these methods, and there are also reports of the production of
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
151
hybrid polymeric materials comprising synthetic polymers coupled to genetically
engineered motifs. There are limited reports of the synthesis of the mussel adhesive and viral spike protein polymers (20,21) and the hybrid polymers (22–24).
The vast majority of materials have been produced from silk, collagen, elastin,
and artificial amino acid sequences (Table 2); research in the design, synthesis,
and characterization of these materials will therefore by presented below.
Silk-Like Protein Polymers
Naturally occurring silk (qv) is produced by a variety of organisms such as the
silkworm (eg, Bombyx mori), spider (eg, Nephila clavipes), and scorpion, and the
toughness and mechanical properties of the silk fiber has interested materials
scientists for its use as a lightweight, high performance fiber, and in composite applications. Of all the silks, those from B. mori and from the dragline of
N. clavipes have been the most extensively studied. The silk from the silkworm
B. mori has been cultivated for centuries and used as a textile fiber. Spider silks,
in contrast, are a family of proteins, in which there are multiple types of silk; for
example, N. clavipes has seven different types of silk, each of which is tailored
for a specific function. All silks have a highly repetitive amino acid sequence,
which is characteristic of the fibrous proteins (eg, silk, collagen, and elastin) that
serve a mechanical or structural function; globular proteins that serve catalytic
or molecular recognition functions do not contain such repetitive sequences. The
exact nature of the repetitive amino acid sequence depends on the type of silk, but
includes alanine- and glycine-rich regions that have a high propensity for β-sheet
formation via intrachain hydrogen bonding, interrupted by less crystalline or
amorphous domains. The crystalline region from B. mori fibroin, for example, has
the sequence [GAGAGSGAAG(SGAGAG)8 Y], and it is estimated to occur approximately 70 times within the protein (101). The repeated motifs from spider silk
are less conserved, but a 13 amino acid repeat, (YGGLGSQGAGRGG), based on
cDNA sequences, has been identified (102); this consensus region is generally followed by a short polyalanine sequence. The novel mechanical properties of silks
are believed to arise from the folding of the protein into small β-sheet crystallites that are dispersed in a noncrystalline matrix (103); shear forces during fiber
spinning permit processing and alignment of the crystallites to produce excellent
fiber properties. The dragline of N. clavipes, for example, dissipates energy over a
large surface area and balances stiffness (modulus of 10–50 GPa), strength (tensile strength of near 1.0 GPa), and extensibility (elongation to break of 10–30%)
(104) (Table 1). These mechanical properties compare will to those of synthetic
high performance materials, with a high tensile strength that approaches that of
Kevlar, a toughness 3 times that of aramid fibers, and a strength 5 times that of
steel by weight.
Protein engineering strategies have been widely employed for the production of silk-like proteins, both in research investigations aimed at understanding
the molecular basis for the excellent mechanical properties of silk, as well as in
commercial applications that employ protein engineering methods as a source
of specialty materials (or potentially materials for bulk fiber applications). Protein engineering of silk is motivated in large part by the difficulty of isolating
152
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
Table 2. Representative Consensus Sequences Used for Genetically Synthesized
Protein Polymersa
Protein
Silk-like proteins
(GAGAGS)x
[(GAGAGS)9 GAAGY)x
[(GAGAGS)n -(GAAVTGRGDSPASAAGY)m ]x
[(GAGAGS)n -(GAAPGASIKVAVSAGPSAGY)]x
[GGAGSGYGGGYGHGYGSDGG(GAGAGS)3 ]2,4,6
[(GXG)n -(A)m ]x
Natural protein
model
B. mori silk
B. mori silk
B. mori
silk/fibronectin
B. mori
silk/laminin
S. c. ricini/B. mori
silks
N. clavipes
dragline silk
[(GPGGYGPGQQ)n -(A)m ]x
N. clavipes
dragline silk
[GLGGQGGGAGQGGYG]x
N. clavipes
dragline silk
N. clavipes
dragline silk
N. clavipes
dragline silk
N. clavipes
dragline silk
N. clavipes
dragline silk
N. clavipes
dragline silk
N. clavipes
flagelliform silk
N. clavipes
flagelliform silk
[SGRGGLGGQGAGA10 GGAGQGGYGGLGSQGT]x
[SGRGGLGGQGAGA5 GGAGQYGGLGSQG]x
[SGRGGLGGQGAGMA5 MGGAGQYGGLGSQG]x
[SGRGYSLGGQGAGA5 GGAGQYGGLGSQG]x
[SGPGGYGPGQQT]x
[(GPGGSGPGGY)2 -GPGGK]11
[{(AEAEAKAK)2
AG(GPGQQ)6 GS}9 (AEAEAKAK)2 AG(GPGQQ)]
[A18 TS(GVGAGYGAGAGYGVGAGYGAGVGYGAGAGY)TS]4
Partial cDNA construct
Partial cDNA construct
Collagen, cDNA constructs
proα1
proα1 and proα2
S. cynthia ricini/B.
mori silk
N. clavipes
dragline silk
A. diadematus silk
Human type I, III
collagen
Human type I
collagen
Expression host
Ref.
E. coli
E. coli
E. coli
26
26,61
27,61
E. coli
61
E. coli
28
E. coli,
B. subtilis,
P. pastoris
E. coli,
B. subtilis,
P. pastoris
E. coli
29–33
E. coli
34
E. coli
41–43
E. coli
41–43
E. coli
41,44
E. coli
34
E. coli
46
E. coli
47
E. coli
45
E. coli,
mammalian,
transgenic
plants
Mammalian,
transgenic
animals
25,38,40
P. pastoris,
S. frugiperda
P. pastoris
29–33
35
36,37
38,39
50
48,55
49
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
153
Table 2. (Continued)
Protein
proα1
proα2
proα1
Collagen-like proteins
(GESGREGAPGAEGSPGRDGSPGAKGDRGET)6
(GAPGAPGAPGPVGPAGKSGDRGETGPAGPP)8,10
Partial cDNA
GPE[{(GPQ)(GPE)4 }2 ]x
GXP-GXQ-rich random polymers
(GPP)32
[GAP(GPP)4 ]x
[(GAP(GPP)4 )2 GLPGPKGDRGDAGPKGADGSPGPAGPAGPVGSP]n
(GAPGAPGSQGAPGLQ)52
Elastin-like proteins
(VPGXG)x
[(VPGVG)n (VPGXG)m ]x
[(VPGVG)n (VPGAG)m (VPGGG)p ]x b
[GVGVP GVG(F/V)P
GXG(F/V)P-(GVGVP)3 ]n
[GVGVP GVG(F/V)P
GXG(F/V)P-GVGVP GVGFP
G(V/F)GFP]n
[GVGVP GVG(F/K)P
GEGFP-GVGVP GVG(F/V)P
G(F/K)GVP]n
[GVGIP GFGEP GEGFP
GVGVP-GFGFP GFGIP]n
(GVGIP)x , (AVGVP)x
ABc
BABc
Natural protein
model
Expression host
Ref.
Human type I
collagen
Human type I
collagen
Human type I
collagen
H. polymorpha
54
Transgenic
mouse
Transgenic
mouse
51
Human type I
collagen
Human type I
collagen
Mouse type I, rat
type III
collagens
Bovine collagen
Collagen-like
Collagen-like
Collagen-like
Collagen-like, Cell
binding
B. brevis
57
B. brevis
57
P. pastoris
53
S. cerevisiae
P. pastoris
E. coli
E. coli
E. coli
56
58
59,60
61
61
Collagen-like
E. coli
61–63
Mammalian
elastin
E. coli, yeast,
transgenic
plants
E. coli
61,64–74
E. coli
19,68,76
E. coli
75
E. coli
75
Mammalian
elastin
E. coli
75
Mammalian
elastin
Mammalian
elastin
Mammalian
elastin
Mammalian
elastin
E. coli
75
E. coli
75
E. coli
77–79
E. coli
77–79
Mammalian
elastin
Mammalian
elastin
Mammalian
elastin
Mammalian
elastin
52
67,73,80,81
154
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
Table 2. (Continued)
Protein
(GVG(V/I)P)10 GVGVPGRGDSP(GVG(V/I)P)10
[LD-CS5d -(GVPGI)x ]y
[LD-CS5-G(VPGIG)20 VP]5
RKTMG[LD-CS5G(VPGIG)20,25 VP]5,3,1 LEKAAKLE
[LD-CS5-G
((VPGIG)2 VPGKG(VPGIG)2 )4 VP]3
Silk-elastin-like proteins
[(GVGVP)n (GAGAGS)m ]x
[(GVGVP)n (GAGAGS)m (GAAVTGRGDSPASAAGY)(GAGAGS)p ]x
[(GVGVP)n (GEGVP)(GVGVP)m (GAGAGS)p ]x
[(GAGAGS)n (GVGVP)m (GKGVP)(GVGVP)p ]x
Artificial proteins
[(AG)n PEG]x
[(AG)n EG]x
[(GA)3 GX]x
ED(E17 D)x EE, benzylated
Helix-[(AG)3 PEG]10 -Helixe
Helix-[(AG)3 PEG]10
Helix
[(AAAQ)x (AAAE)(AAAQ)x ]y
[GKGSAQA]x
[AKPSYPPTYK]x
[LSVQTSAPLTVSDGK]16
[SGLDFDNNALRIKLG]26
[QLSLRVSEPLDTSHGV]64
Natural protein
model
Expression host
Ref.
Elastin/cellbinding
domain
Fibronectin/elastin
Fibronectin/elastin
Fibronectin/elastin
E. coli
75
E. coli
E. coli
E. coli
66
88
88
Fibronectin/elastin
E. coli
89
Elastin/B. mori
silk
Elastin/B. mori
silk/fibronectin
E. coli
61,82,87
E. coli
61,82
Elastin/B. mori
silk
B. mori silk/elastin
E. coli
83,84
E. coli
82,85–87
E. coli
E. coli
100
90
E. coli
91,98,99
E. coli
E. coli
E. coli
E. coli
E. coli
E. coli
E. coli
92–94
95
95
95
96
97
20
E. coli
E. coli
E. coli
21
21
21
Random coil
Silk mimetic
sequence
Silk mimetic
sequence
Helical
Leucine zipper
Leucine zipper
Leucine zipper
Helical
Random coil
Mussel adhesive
mimetic
Viral spike protein
Viral spike protein
Viral spike protein
a One-letter abbreviations of the amino acids are employed except as noted in the table. The letter X
indicates positions in which several different amino acids have been encoded. Only general consensus
repeats are given; minor linker regions, fusion tags, random sequences of amino acids, and minor
substitutions in isolated repeats are deleted for simplification.
b The A and G residues are dispersed randomly throughout the sequence to reduce gene repetition.
cA
blocks:
[VPGEG(IPGAG)4 ]14 ,
[(APGGVPGGAPGG)2 ]x ,
[VPGVG(IPGVGVPGVG)2 ]19 ,
[VPGEG(VPGVG)4 ]30 , [VPGEG(VPGVG)4 ]48 , [(VPGMG)5 ]x ; B blocks: [VPAVG(IPAVG)4 ]16 ,
[VPGFG(IPGVG)4 ]14 .
d CS5 = GEEIQIGHIPREDVDYHLYP; CS5 is a cell-binding domain from fibronectin.
e Helix = SGDLENEVAQLEREVRSLEDEAAELEQKVSRLKNEIEDLKAE.
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
155
these proteins in large quantities from the predatory spider, which cannot be cultivated like the silkworm. Despite advances in recombinant DNA methods, no
complete cDNA or genomic DNA sequence of a natural silk protein has been successfully utilized for protein expression. The difficulties arise primarily because
of the extreme length and repetitiveness of these genes, and also potentially because of the poor codon correspondence between the animal and the expression
host, which places an unbalanced demand on aminoacyl-tRNA pools during bacterial protein expression. As a result of these difficulties, genetic deletions and/or
premature termination during protein synthesis are often observed. However,
the relatively simple, repetitive amino acid sequences that comprise silk proteins
can be easily used to construct artificial silk-like proteins, and this approach has
addressed the instability and poor production of protein from the expression plasmids prepared from the natural cDNA. Indeed, these approaches have been used
to produce silk-like protein polymers containing nearly 1000 amino acids from
bacterial expression systems, and somewhat longer polymers in yeast, indicating
the flexibility of the method. Limitations in predicting protein polymer structure
and function from the properties of the repetitive amino acid sequences can occur, but have not caused serious problems in the design of silk-like artificial proteins; the repetition of consensus sequences of natural silks provides protein-based
polymers in which the conformational properties of the original protein are well
preserved.
The instability of long cDNA sequences has limited their lengths to less than
2.5 kb. Recently, a 1.5-kb partial cDNA fragment of N. clavipes dragline silk has
been cloned and the 43-kDa protein has been expressed from E. coli (25). However,
most work has focused on the bacterial expression of silk-like proteins from synthetic genes, as mentioned above. Earliest reports of this approach date back to
the work of Cappello and co-workers in 1990 (26). In these initial studies, a variety
of different silk-like protein sequences with the repetitive sequence (GAGAGS)n
were constructed, (where G is glycine, A is alanine, and S is serine) on the basis
of the B. mori silk fibroin. The 108-bp monomer, flanked by Ban I restriction sites
which permit only head-to-tail ligation of monomers, was multimerized enzymatically, and the multimers were fractionated and ligated into an expression system
controlled by a strong, temperature-sensitive promoter that permits induction of
protein expression by simply raising temperature to above 40◦ C. The (GAGAGS)n
sequence proved intractable because of the formation of very stable and insoluble
β-sheet structures, and so more complex sequences with improved solubility have
been produced via introduction of noncrystalline regions dispersed between the
β-sheet crystalline regions. For example, artificial proteins designed to contain
both the crystalline segments of silk fibroin and the cell adhesion domain of fibronectin have also been synthesized by Cappello and co-workers, and have been
shown via wide-angle X-ray scattering and molecular simulations to crystallize
according to the models for Silk I (27). More recently, a sequence that alternates
the (GAGAGS)4 repeat from B. mori with a (VPGVG)8 repeat from elastin has
been produced, and the protein maintains excellent materials properties based on
the combination of crystallinity and elasticity imparted by the two repetitive domains of the 832-amino acid artificial protein (105). Copolymers of the crystalline
region of B. mori silk fibroins with the unordered glycine-rich region of Samia
156
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
cynthia ricini have also been produced to improve the tractability of the B. mori
crystalline domain (28).
Similar success has been observed for the expression of repetitive artificial
proteins based on spider silk amino acid sequences. Genes for partial amino acid
sequences from N. clavipes have been generated, and repetitive genes of these
sequences have been produced via recursive multimerization strategies. Proteins
comprising 8 or 16 repeats of a poly(alanine) β-sheet hard segment with either
interspersed (GXG) variants or (GPGGY) and (GPGQQ) alternating pentamers
as the soft segment have been expressed in both E. coli and Bacillus subtilis
by Fahnestock and co-workers (29–31). When expressed in E. coli under control of the strong promoter of the bacteriophage T7, approximately 10–30% of
the soluble cell protein is the expression target sequence, although some truncated products are formed in these expression systems. The proteins are easily
purified via chromatographic methods or via solubility differences between the
artificial repetitive protein and other cellular proteins (31,32). When expressed in
the yeast Pichia pastoris, from genes employing the codon preferences for Pichia
(33), truncated products are eliminated and protein yields are increased, with
silk-like proteins comprising 10% of the total protein (yields of 1 g/L). A chromosomal integration strategy is used to establish the genes in Pichia by homologous
recombination, and expression is controlled by the strong promoter AOX1, which
is induced by addition of small amounts of methanol. Fusing the target protein to
secretion signals and prosequences of Saccharomyces cerevisiae permits efficient
secretion of the target protein from the cell and into the extracellular medium,
although at much lower yields. The advantages of this system include the fact
that P. pastoris is a useful host for large-scale fermentation, and that extracellular localization of an expression protein target has many potential advantages for
low cost production (simpler recovery, purification, and the potential for expression from immobilized cells). Circular dichroism (CD) characterization of these
proteins in dilute aqueous solution indicates that the protein polymer adopts
a random coil conformation, with increasing β-sheet content upon drying, consistent with the reported behavior of natural silk proteins upon spinning into
fibers.
The production of other synthetic genes based on the amino acid sequences
of dragline silk from N. clavipes has also been achieved by Kaplan and co-workers.
From the partial cDNA sequences of this silk, two consensus repeats (NCMAG1
and NCMAG2, abbreviated for N. clavipes major ampullate gland) have been
identified: (GGAGQGGYGGLGSQGAGRGGLGGQGAG), followed by a polyalanine region, and (GPGGYGPGQQGPGGYAPGQQPSGPGS), also followed by a
polyalanine region. Repetitive proteins with sequences based on NCMAG1 and
NCMAG2 have been produced from E. coli via expression of artificial repetitive
genes from a pQE9 expression vector under control of a bacteriophage T5 promoter (34); these proteins contain a hexahistidine fusion to facilitate purification
via metal chelate affinity chromatography. Yields of approximately 15 mg/L have
been observed, although the yield of the silk-like repetitive proteins decreases
with increasing length. Characterization via CD confirms the expected presence
of β-sheet structure in these proteins. Additional silk-like proteins, also modeled
after the dragline silk of N. clavipes, have been produced in E. coli by other research groups (35,106); in general, these proteins have molecular weights ranging
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
157
from 10 to 160 kDa (molecular weights of natural spider silk proteins can reach
740 kDa), with yields of purified protein ranging from 2 to 20 mg/L.
In addition to expression from bacterial and yeast expression hosts, silk
proteins have also been expressed from transgenic hosts, motivated in part by
the low expression yields obtained in microorganisms. Tobacco and potato plants
have been successfully utilized as transgenic hosts for producing silk-like proteins
based on dragline silk (36,37). In these investigations, at least 2% of the total soluble protein in the endoplasmic reticulum of the tobacco and potato leaves and
potato tubers is reported to be silk. In addition, partial cDNA clones of the silk
protein from Araneus diadematus and from N. clavipes have been expressed in
mammalian cells [baby hamster kidney (BHK) cells] to yield several grams of
protein that is recovered in soluble form by ammonium sulfate precipitation. The
25–50-mg/L yields provide an ultimate yield of more than 12 g of material purified from conditioned culture media (38). For production of larger amounts of these
recombinant silk-like proteins, expression from goat milk may prove a viable alternative, and is currently under development; transgenic goats that express silk
proteins in their milk have been produced by Nexia Biotechnologies, Inc., and
methods to purify these proteins have been developed (39,107–109).
Additional areas of active research have been to mimic the fiber spinning
conditions of the spider and to control β-sheet assembly in order to produce artificial silk protein materials that have mechanical properties similar to those of the
natural silk protein fibers. In general, the spinning of recombinant, silk-like artificial proteins requires dissolution in harsh solvents such as hexafluoroisopropanol,
formic acid, or 9 M lithium bromide. For example, fibers have been spun from solutions of these proteins in hexafluoroisopropanol with quenching in methanol and
wet drawing. Such fibers exhibit the properties [tenacities of 2 g/denier (gpd) and
8–10% elongation] of traditional textile fibers, but do not match the properties
of natural dragline silk (110). Silk proteins produced from bacterial sources have
also been spun into fibers from more benign aqueous processing conditions that
better mimic those of the spider. In 2002, Arcidiacono and co-workers reported
the aqueous solution spinning of silk sequences derived from N. clavipes dragline
silk and expressed from E. coli (40). The proteins are produced via expression
of the C-terminal cDNA of N. clavipes dragline (42.9 kDa), or via expression of
the consensus sequences NCMAG1 and NCMAG2 of N. clavipes (55 kDa). The
key to the successful spinning of these proteins from aqueous solution has been
to eliminate the drying of the protein at any stage of purification and processing,
by increasing protein concentration via removal of water during purification, with
processing in dilute denaturing buffer (160 mM–1 M urea). The content of β-sheet
structure in the fibers increases with time, as expected, and fiber diameters range
from 10 to 60 µm in diameter, depending on the weight percentage of the spin
dope and the identity of the protein. Fibers are insoluble in water and birefringent, indicating the formation of oriented fibers, although no mechanical property
measurements have been reported. The A. diadematus-derived protein produced
in mammalian cells (38) has also been processed in a similar manner—first concentrated into an aqueous spin dope exceeding 23% (w/v) protein, coagulated in
methanol/water, and exposed to wet draw conditions. The fibers isolated via these
methods exhibit a tenacity of 2.26 gpd and 55–130% extensibility, which yield a
toughness equivalent to that of silk, although the properties do not exactly mimic
158
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
natural silk fibers, which have a tenacity of 7–11 gpd and an extensibility of 30%
(vide supra).
The difficulties in processing silk proteins is largely a result of the spontaneous assembly into β sheets of the poly(alanine) sequences of dragline silk,
especially under conditions of shear. In an effort to understand the architectural
variables that will permit purposeful control of β-sheet formation and hierarchical assembly during processing, protein engineering methods have been employed
toward the design and synthesis of silk protein sequences in which amino acids
and amino acid sequences are included for control of β-sheet formation. Incorporating sequences that can trigger β-sheet assembly, for example, as demonstrated
by Kaplan and co-workers, has been one approach to control protein properties.
In one design, methionine residues have been included to flank the (AAAAA)
units of the NCMAG1-based repetitive proteins (41–43). Under reducing conditions, these polymers readily form β-sheet structures as assessed by CD and
Fourier transform infrared (FTIR) spectroscopies; β-sheet assembly is disrupted
upon oxidation of methionine to the larger and more hydrophilic sulfoxide. These
researchers have also introduced sites of enzymatic phosphorylation that flank
the alanine-rich sequences, in order to alter the size and charge of serine residues
and control β-sheet formation (41,44). When the serine hydroxyl group in the sequence RGYSLG is phosphorylated by cAMP protein kinase, β-sheet formation is
prevented; upon dephosphorylation by alkaline phosphatase, the ability to form
β sheets is recovered. In other investigations aimed at controlling solubility and
processability of silk-like proteins, Asakura and co-workers (45) have designed
a chimeric sequence that combines the crystalline polyalanine sequence (Ala)18
of Samia cynthia ricini silk with a noncrystalline (GX)-rich sequence (GVGAGYGAGAGYGVGAGYGAGVGYGAGAGY) found in the silk fibroin of B. mori. The
introduction of the glycine-rich noncrystalline sequence in the protein results in
improved solubility of this silk-like protein and prevents β-sheet formation in the
polyalanine region of the protein, as assessed via solid-state 13 C CP/MAS NMR
studies.
The materials properties of other members of the spider silk family are also
becoming of increasing research interest. Sequences based on the flagelliform
silk protein, which produces the unique elastomeric properties of the capture spiral of spider webs, have been recently studied by Conticello and co-workers (46).
A protein containing 11 repeats of the flagelliform-derived amino acid sequence
{(GPGGSGPGGY)2 GPGGK} can be expressed in E. coli, and has been characterized by a combination of techniques including CD, FTIR, and NMR, which indicate that these molecules adopt a β-turn conformation. These results suggest that
the elastomeric properties of the flagelliform sequence may have similar origins
as those found in other β-turn-forming proteins such as elastin. The mechanical
properties of protein polymers that contain flagelliform-like amino acid sequences
can also be controlled by the inclusion of peptide motifs that are known to form
β-sheet crystallites but that are not naturally found in silk proteins.
Conticello and co-workers (47) have also produced alternating block copolymers containing a flagelliform-like sequence (GPGQQ)6 , which is derived from the
A. diadematus dragline silk fibroin, and the amphiphilic sequence (AEAEAKAK)2 .
Although the (AEAEAKAK) sequence is not a silk-derived sequence, it has been
shown to form very stable β-sheet structures in aqueous solution (111). Proteins
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
159
containing these sequences assemble into β-sheet linked membranes from concentrated aqueous solution, and membrane formation is responsive to conditions
that alter the conformation of the (AEAEAKAK) units in the polymer (47).
The application of silk-like protein polymers in many disparate applications
such as textiles, medical applications, and cosmetics will be possible, given their
demonstrated synthesis and favorable properties. One difficulty to be overcome
in the synthesis of artificial silk-like proteins from bacterial hosts is the generally low expression yields, which can be as low as 1–10 mg/L. Yeast systems offer
advantages in this regard, with potential yields of 300–1000 mg/L possible, although difficulties in purifying the protein from these expression hosts have been
encountered. While the mechanical properties of spider silk are a desirable goal, it
has remained difficult to reproduce the properties in the laboratory environment,
although continued progress is being made in this direction, owing to increased
understanding of the processing of these proteins and the ability to control the solubility of the proteins via protein engineering strategies. Improved understanding
of the impact of synthesis and processing on the resulting mechanical properties
of silk-like materials will increase the number of applications in which these polymers will find use. Coupled with the fact that silk-like proteins can also be useful
in supporting cell growth and tissue function, the proteins will find use in both
materials science and medical applications, such as in fibers, threads, membranes,
and scaffolds. The genetic tailoring of their sequence and the ability to include repeats of other structural proteins such as collagen or elastin (vide infra) has also
resulted in a variety of silk-based proteins that can be useful in varied materials
applications, including fiber reinforcement, controlled release, and scaffolds for
tissue engineering, and will continue to expand the number of uses in which these
polymers will find application.
Collagen-Like Protein Polymers
Collagen (qv) is another important fibrous structural protein that has been produced by protein engineering methods for applications in materials science and
biology. The collagens comprise a highly abundant family of extracellular matrix proteins that are found in all connective tissues. They represent 30% of the
total body proteins in mammals and are found in tendons, ligaments, cartilage,
bone, and skin. Although there are at least 20 genetically distinct types of collagen (the most abundant in mammals being types I–III), all are composed of
three helical chains that have the general amino acid sequence Gly-X-Y, in which
X is primarily proline, and Y is primarily hydroxyproline. The monomeric helical sequence first assembles into a triple helix with dimensions of 300 nm in
length and 1.5 nm in diameter, which then forms larger-scale hierarchical structures that ultimately assemble into high strength elastic fibers that form connective tissue (112). In addition to its structural role, collagen is also involved
in a variety of important biological events such as early development, cell attachment and proliferation, wound healing, and tissue remodeling. This combination of mechanical strength, higher order assembly, and biological activity of
collagen has made it a broadly used protein-based biomaterial. Collagen I, in
particular, is the most abundant structural protein in animals, and has been
160
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
widely exploited for medical use, cosmetics, therapeutics, and also as its denatured form (gelatin) in food and photographic emulsions. Although collagen is
available in large quantities from mammalian sources, extraction from animal
tissues poses the risk of contamination by viral or infectious agents, which has
become of increasing concern in recent years. Owing to this potential risk, the
simplicity of the repetitive sequence, the wide biological and materials uses of
collagen, and the ability to tailor collagen amino acid sequences, protein engineering strategies have become more prevalent for the production of recombinant
collagen.
In contrast to the production of silk-like proteins, recombinant production
of collagen-like proteins has focused primarily on the production of cDNA sequences isolated from natural sources. Fibrillar collagens have been produced in
mammalian cells (113,114), insect cells (48,49), yeast (51,115–118), and transgenic animals (51,52,118) and plants (119,120). Yields generally range from 15 to
500 mg/L, although a triple-helical, 160-kDa human type I collagen has been produced in mouse milk at 1–8-mg/mL yields (52), and a 21-kDa fragment of mouse
collagen I (from cDNA fragments) can be produced from P. pastoris at yields of
14.8 g/L of clarified broth (53).
To obtain correctly assembled collagen fibrils, most organisms employ extensive post-translational modification of procollagens, and the enzymes that mediate
these modifications have been incorporated into expression systems for collagen
production. One of the most important considerations is the hydroxylation of proline residues to 4-hydroxylproline via the action of the tetrameric (α 2 β 2 ) enzyme
prolyl 4-hydroxylase. In almost all collagens (with the exception of some marine
invertebrate collagens), hydroxylation of the proline is required for correct fibril
formation and to maintain the melting temperature of the collagen at physiologically relevant levels (approximately 40◦ C). A lack of hydroxylation reduces (and
can, under physiological conditions, eliminate) the propagation of banded fibril formation, lowers the melting transition temperature, and can render the collagen
useless for biological applications (120). Most bacterial and yeast expression hosts
do not contain the genes for prolyl 4-hydroxylase, and some insect and mammalian
expression hosts do not generate sufficient amounts of this enzyme. Coexpression
of collagen with the subunits of prolyl 4-hydroxylase has therefore been used
to permit recombinant production of functional collagen. Another consideration
in collagen expression is that the protein is generally expressed as procollagen,
which contains N- and C-terminal peptides to impart solubility. The genes for the
proteolytic enzymes N-proteinase and C-proteinase are also often included in expression hosts to allow processing of the procollagen, via removal of the N- and
C-terminal peptides, into the assembling collagen form. It has been recently reported, however, that human type I collagen proteins lacking the propeptides can
be produced from S. cerevisiae expression hosts that lack the genes for the prolyl
4-hydroxylase (117) and can still correctly fold. Generally, in expression systems
in which the proteinases are not included, procollagen can be cleaved with pepsin
post-translationally to liberate the N- and C-terminal propeptides and permit fibrillar assembly of collagen.
Vuorela and co-workers (50) have demonstrated that coexpression of subunits of human prolyl 4-hydroxylase with the proα1 (III) chains of human type
III procollagen in P. pastoris yeast expression systems permits production of
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
161
approximately 15 mg/L of properly folded homotrimeric type III procollagen that
is resistant to pepsin digestion. The yeast expression hosts also demonstrate an
expected increased level of prolyl 4-hydroxylase activity, and can be used in the
large-scale production of different types of recombinant collagen. Similar strategies have also been employed for the production of high levels of human type I
collagen in P. pastoris (121). In these investigations, the genes for the proα1 and
proα 2 chains of type I procollagen are expressed, along with the genes for both the
α and β subunits of prolyl 4-hydroxylase. The correct assembly of proα1 and proα 2
chains is observed (2:1 ratio), and high expression yields of 500 mg/L are obtained.
In contrast to these other yeast systems, properly hydroxylated, 28-kDa mouse
α1 (I) collagen fragments can be expressed from the yeast Hansenula polymorpha
without coexpression of prolyl 4-hydroxylase (54). Successful expression of the procollagen chains from insect cells has also been observed. Proα1 chains of human
type III collagen have been expressed from baculovirus vectors in S. frugiperda
Sf9 or High Five cells (48), as have wild-type and modified proα chains of human
type I collagen (55). With coexpression of human prolyl 4-hydroxylase, correctly
assembling procollagen molecules can be produced at yields of 10–20 mg/L.
Mammary gland expression has also been explored as a route to produce procollagens, although this has been more limited in scope and use to date. The general method involves the expression of genomic or cDNA transgene sequences in
conjunction with mammary-gland–specific promoters, which drives the expression
of the proteins in milk. It has been achieved for procollagen via two different approaches. In one strategy, the cDNA encoding a shortened version of recombinant
pro α 2 procollagen chains has been integrated into the mouse genome, and the procollagen has been coexpressed with the α and β subunits of prolyl 4-hydroxylase.
The expression of a thermally stable triple helical artificial α 2 (I) homotrimer at
50–200 mg/L is possible in this system (51). In another instance, transgenic mice
were generated that contained the αS1-casein mammary-gland–specific promoter
linked to a 37-kb segment of the human α1(I) procollagen structural gene. Very
high levels of triple helical procollagen are produced via this system (8 mg/mL)
(52). An advantage for transgenic expression of protein polymers in milk is that
the expression levels of proteins can be increased via the use of a species with a
higher volume production of milk (eg, mouse to rabbit to pig or goat).
Although recombinant natural collagen sequences have been of significant
academic and commercial interest (115,122,123), the production of collagen-based
proteins with additional functions has also been explored. For example, in an effort
to design advanced biomaterials, Fertala and co-workers (124) have identified specific regions in collagen that are responsible for particular biological activity. These
researchers have developed a cDNA cassette expression system (125) that allows
systematic removal of specific amino acid sequences in collagen II. By removal of
certain D blocks in collagen II, followed by expression of the modified protein in
mammalian cells (HT-1080 cells), purification, and assessment of human chondrocyte (cartilage cell) spreading and migration, the amino acid region 704-938
has been identified as critical for the spreading of chondrocytes (124). The studies
demonstrate that collagen (II) domains differ in their ability to support attachment and migration of chondrocytes, and suggest that advanced collagen-based
materials could be designed to present multiple copies of the critical domains,
permitting support of a greater number of cells and potential improvements in
162
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
tissue regeneration. In other efforts to produce collagen-based materials with improved biological activity, Hayashi and co-workers have produced a collagen III
epidermal growth factor (EGF) chimeric protein from Sf9 insect cells (126). The
proteins maintain the fibril-forming properties of the collagen domain, and films
of the protein also improve cell growth via the action of the EGF; the chimeric
protein may therefore be useful for cell culturing, wound healing, and tissue engineering applications. Together, these studies point to the utility of protein engineering strategies to identify and to produce materials containing multifunctional
domains that impart desired biological and materials properties.
In addition to protein engineering investigations that have been based on
natural collagen sequences, there is also significant interest in creating artificial collagen-like sequences with novel chemical and physical properties. Accordingly, genetically directed methods have been employed to produce artificial
protein polymers containing the collagen-based GXY repeat, with substitutions in the X and Y positions to impart desired chemical functionality. The
proteins have been expressed in yeast and bacterial systems. For expression from yeast (Saccharomyces cerevisiae) and certain bacteria (Bacillus brevis), fusion of the α-mating factor secretion signal permits secretion of the
collagen-like proteins (CLPs) into the medium (53,56,57), which simplifies purification and points to the feasibility of commercial scale production of the
proteins. Kajino and co-workers (57) have designed DNA sequences encoding human α1 collagen repeats (GESGREGAPGAEGSPGRDGSPGAKGDRGET)6
and (GAPGAPGAPGPVGPAGKSGDRGETGPAGPP)8,10 according to the codon
usage of B. brevis. The proteins are expressed in B. brevis at estimated yields
of 500 mg/L, and they exhibit reversible sol-gel behavior similar to that of native gelatin. Nonhydroxylated gelatins, with sequences based on partial cDNA
sequences from mouse type I and rat type III collagen, have been produced in
higher yields of 14.8 g/L from P. pastoris, using the S. cerevisiae α-mating factor
(53). Completely synthetic, 36.8-kDa collagen sequences that are rich in GXP and
GXQ sequences can also be produced, without degradation, in P. pastoris at high
yields of approximately 3–6 g/L (58).
Expression from E. coli has not been as successful as the yeast and B. brevis
expression systems. In early attempts to express CLPs from bacterial expression
systems (59,60), a 22-kDa CLP with the sequence (Gly-Pro-Pro)32 was produced
in E. coli under control of a thermally inducible promoter. The resulting protein
degraded in this cellular host, although use of an appropriate mutant host minimized this problem. Reducing the proline content of the collagen-like proteins appears to minimize stability problems, as less proline-rich proteins with sequences
such as {GAP(GPP)4 }x and {{GAP(GPP)4 }2 GPAGPVGSP}x have been successfully produced from E. coli by Ferrari and Cappello (61). Recently, the sequence
(GAPGAPGSQGAPGLQ)52 , which has an even lower proline content than previously produced CLPs, has been expressed in E. coli; the protein can be expressed in
soluble form, at 100–200-mg/L yields, in low cell density, small-scale experiments
(62,63).
The tensile strengths and availability of type I collagen render it the most
widely used natural polymer in the medical field, with uses in tissue engineering,
delivery systems, and cosmetic surgery; the denatured form of collagen (gelatin)
finds widespread use as a food additive and photographic emulsion. In addition to
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
163
minimizing risks associated with isolation of collagen from animal sources, genetically directed strategies for the production of recombinant collagen-like protein
polymers are also appealing as a result of the potential for CLPs in the above
applications. The progress toward expression of collagen-like proteins from bacterial and other expression systems may permit the rational design of artificial
CLPs with specialized materials uses; for example, understanding the individual roles of collagen domains in eliciting biological responses may result in the
design of CLP-based scaffolds for directed interactions with cells. The ability to
produce designer collagen and gelatin polymers via genetically directed methods
may therefore drive the growth of these strategies as a large-scale source of recombinant human collagens and artificial collagen-like polymers for a variety of
additional applications in nanotechnology, materials science, and medicine.
Elastin-Like Protein Polymers
Elastin is another protein found in connective tissues, and provides strength,
flexibility, and elasticity to organs and tissue. When expressed in tissue, elastin
is produced as the 750–800 amino acid protein tropoelastin, which then assembles into microfibrils, is modified via hydroxylation of proline, and is cross-linked
via lysyl oxidase oxidation of lysine-rich domains (127). This assembly and insolubilization of tropoelastin results in the mature elastin network, which confers
elastic and biological functionality to tissue. Elastin is composed of large, mobile,
highly hydrated hydrophobic domains and smaller alanine-rich regions. The hydrophobic domains of elastin contain proline, alanine, valine, leucine, isoleucine,
and glycine, with valine and glycine being most abundant. The sequence of the hydrophobic domain is highly repetitive, containing repeats of the general sequences
GX, PX, GGX, or PGX (X = G, A, V, L, or I); the common repeat in mammalian
elastin is VPGVG, with up to 11 consecutive pentapeptide repeats (127,128). In
contrast to silk and collagen, elastin, even at high molecular weights, is soluble in aqueous solution, which, along with the highly repetitive nature of the
hydrophobic regions of this functional protein, has fueled an enormous amount
of interest in the use of elastin-based repeats in genetically engineered protein
polymers. Whereas protein engineering has been employed in the production of
tropoelastins for studies of biological activity, essentially all materials investigations have focused on the biosynthesis of elastin-like proteins (ELPs) comprising
repetitions of the VP(LCST)GVG motif.
One unique feature of the VPGVG sequence is that, although it is generally soluble, it exhibits a lower critical solution temperature (LCST), which
causes aqueous solutions of the protein to separate into solvent-rich and polymerrich phases upon heating. This transition is analogous to that of the well-known
poly(N-isopropylacrylamide) polymers, in which an entropy-driven dehydration of
the polymer chain results in phase separation above a critical temperature. The
ability of elastin-like polymers to undergo an LCST transition is conserved as long
as the glycine and valine residues are present. The LCST transition is observed
even with substitution, in the repeat sequence VPGXG, of any amino acid X (except proline); indeed, choice of the amino acid residue X can be used to control the
LCST of the protein. This unique behavior of elastin confers mechanical properties
164
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
that make it a versatile platform for the engineering of protein-based materials
for a variety of applications such as biomaterials, drug delivery, responsive silica
membranes, and tissue engineering.
Because of this versatility, both chemical and protein engineering methods
have been used widely for the synthesis of elastin-based protein materials based
on the VPGXG sequence. A single repeat of the VPGVG unit is sufficient to observe
the random coil to β-turn transition (129), which has made chemical strategies
accessible for the synthesis of elastin-based peptide materials. However, limitations of solid-phase peptide synthesis plague these materials at higher molecular
weights, since the presence of hard-to-purify deletion products significantly alters
the properties of the materials (64). Protein engineering methods have therefore
been used widely for synthesis, with relatively high yields (generally 50–100 mg/L,
and up to 800 mg/L) facilitating the characterization and use of these materials
for commercial applications. Elastin-like proteins have been produced in E. coli
(64–68), fungi (69), chloroplasts (70), and plants (71), although most materials
investigations with elastin-like proteins utilize bacterial expression.
The broad range of interest in elastin-like protein polymers has fueled the
development of new genetic strategies for the assembly of the genes encoding the
elastin repeats (70,74). Seamless cloning methods (72) permit the assembly of
high molecular weight genes, in a controlled head-to-tail fashion, directly into the
expression vector. Clones of up to 5000 bp (200 kDa) have been prepared via these
strategies, with yields commonly on the order of 200–800 mg/L. Since the multimerization is random in nature, seamless cloning results in the formation of a
library of genes of different lengths, although the exact length of the genes cannot
be controlled purposefully. Recursive strategies have therefore also been applied
for the synthesis of repetitive artificial proteins with specific and predetermined
chain lengths (68) to enable control of elastin-like protein transition temperature
via purposeful control of ELP molecular weight and block architecture. By sequential cloning steps, gene products carrying a specified number of repeats can
be produced. A variety of (VPGXG)n ELPs have been produced via these strategies,
with molecular weights up to 130 kDa (330 pentapeptide repeats).
The elastin-like proteins produced by genetic methods have proven useful
for a number of applications in biotechnology, materials science, and medicine.
In early demonstrations by Urry and co-workers (64), ELPs with the general sequence G–(VPGVG)19 –VPGV have been expressed from E. coli, are easily purified from cellular proteins, and exhibit a sharp LCST at approximately 48◦ C, as
expected for a VPGVG sequence of this molecular weight. By appropriate design of the elastin-like polymer, thermal, chemical, and electrochemical stimuli can all be used to alter and control the LCST of ELPs (64). Urry and coworkers have demonstrated, for example, that substitution of any of the natural
amino acids in the fourth position of the VPGVG pentapeptide provides a range
of LCST transitions. Attachment of redox-active cofactors such as FAD/FADH2
(flavin adenine dinucleotide) or NAD/NADH (nicotinamide adenine dinucleotide)
to glutamic acids in a VPGEG repeat permits electrochemical control over the
hydrophobic collapse of the protein (73). Upon reduction of FAD to FADH2 or of
NAD to NADH, the increased hydrophobicity of the pentapeptide results in the
hydrophobic collapse of the elastin-like polymer at substantially lower temperatures (73). If the two different states of the ELP (oxidized and reduced) have LCSTs
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
165
above and below room temperature, the electrochemical transition can stimulate
an observable transition under ambient conditions.
Following up on the extensive body of work by Urry (74,75,130), others have
also taken advantage of the control of LCST possible for these elastin-like polymers in the design of ELPs for drug delivery applications. In one example, Chilkoti
and co-workers (19,68,76) have produced a family of polymers of the general sequence VPGXG in E. coli, where X = V, A, and/or G. The ratios of V:A:G are
varied in order to tune the LCST, with one library containing no A or G, and
two others containing the residues in ratios of 5:2:3 and 1:8:7. The A and G substitutions in the fourth position of the pentamer are dispersed throughout the
sequence to reduce the repetition within the gene. These polymers have 120 to
330 pentapeptide repeats and molecular weights of 50 kDa to 130 kDa, and the
LCST of the polymers is dependent on both the molecular weight and the guest
residue sequence. Transition temperatures that vary smoothly between approximately 28◦ C and 78◦ C are obtained, demonstrating the potential for genetically
designing an ELP with a desired transition temperature for drug delivery applications. In addition, ELPs designed to exhibit an LCST at 35◦ C (131) exhibit
complex shear moduli similar to those observed for collagen and hyaluronan at
physiological temperature, and are able to support cartilaginous cell growth. The
combination of these results suggests that these materials may also be useful as
injectable scaffolds for cartilaginous tissue repair (131), although chemical crosslinking would likely be required to impart sufficient mechanical integrity upon gel
formation. These investigators have also designed block copolymers based on the
VPGXG sequences, with one block comprising 64 repeats of the V:A:G composition
1:8:7 (LCST > 90◦ ) and the other comprising 60 repeats of VPGVG (LCST = 35◦ C).
Upon heating, these block copolymers form 40–100-nm size nanoparticles, owing
to the hydrophobic collapse and aggregation of the VPGVG block. The mechanical, chemical, and/or biological properties can be tuned to enable self-assembly
with incremental changes in temperature, pH, and ionic strength, making these
block copolymers attractive candidates for controlled delivery, tissue engineering
applications, and stimuli responsive surfaces and membranes (132,133).
Genetically directed synthetic strategies also permit the preparation of other
complex block copolymers with tunable properties. Conticello and co-workers
(77,78) have designed and synthesized a series of elastin-mimetic diblock (AB)
and triblock (BAB) copolymers for assembly on the mesoscopic scale, and have
expressed these polymers in E. coli. The B blocks consist primarily of the hydrophobic elastin repeats {VPAVG(IPAVG)4 }16 , and the A blocks comprise more
hydrophilic repeats such as {VPGEG(IPGAG)4 }14 , {VPGEG(VPGVG)4 }30 , and
{VPGEG(VPGVG)4 }48 (see Table 2). The hydrophobic B blocks exhibit an LCST
below 37◦ C, which is desired so that collapse of the hydrophobic block occurs under physiologically relevant conditions. The substitution of alanine (A) in place of
glycine (G) in position three of the pentamer results in a change of mechanical
properties of the material from elastomeric to plastic, which permits control of
the mechanical properties of the block. The hydrophilic blocks are chosen because of their high LCST values, so that phase separation of the two blocks occurs at physiological conditions to form ordered polymeric structures. Indeed,
BAB block copolymers with an A block comprising {VPGEG(VPGVG)4 }30 undergo a reversible sol–gel transition at 23◦ C. The same group has demonstrated
166
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
that diblock copolymers of the general sequence {VPGEG(IPGAG)4 }14 VPGEG–
{VPGFG(IPGVG)4 }16 VPGFG assemble into spherical particles and beaded filaments in water at 25◦ C (79).
There are also significant opportunities for controlling the mechanical properties of ELP materials via covalent cross-linking strategies. Several different
strategies have been employed to cross-link soluble elastin molecules into a crosslinked, elastomeric network. Early work by Urry employed γ irradiation for crosslinking, which yields elastomeric networks with an elastic modulus very similar to native elastin. Although this method does not require a specific amino
acid for cross-linking, it affords little control over the position of cross-linking
and can also result in chain scission. Therefore, in addition to γ irradiation
(134), other radical-based cross-linking methods employing dicumyl peroxide have
been developed, as well as chemical cross-linking strategies such as carbodiimide coupling (80) and chemical reactions with electrophilic reagents. Chemical cross-linking approaches, explored by several groups, offer the advantage
of control of the positions of the chemically reactive cross-linking sites, which
can in turn control cross-linking density, molecular weight between cross-links,
and resultant mechanical properties. Elastin-like proteins with the repetitive sequences {(VPGVG)4 VPGKG}39 have been produced by the Conticello group; the
lysines are reacted with the electrophilic N-hydroxysuccinimide esters of bifunctional carboxylic acids, bis(sulfosuccinimidyl)suberate (in phosphate buffer), or
disuccinimidylsuberate (in DMSO) to produce cross-linked gels (67). The elastinmimetic hydrogels exhibit expected elastomeric behavior, with a reversible contraction (62.4%) and expansion upon cycling between 10 and 45◦ C. Trifunctional
tris-succinimidyl aminotriacetate has also been used by other groups to crosslink elastin-like polymers {VPGKG(VPGVG)6 }n (with n = 56, 112, and 224) and
{VPGKG(VPGVG)16 }n (with n = 51, 102, and 204) (81). The mechanical properties of the cross-linked hydrogels are dependent on molecular weight, the density
of lysine residues, and temperature, and range from 0.24 to 15 KPa. Cross-linked
elastin-like polymer hydrogels exhibit an LCST, although one that is gradual,
as compared to the abrupt LCST transition observed for soluble elastin-like polymers. In additional studies by Urry and co-workers (80), polymers with the general
sequence {(GVGVP)2 (GXGVP)(GVGVP)2 }n , with X = lysine or glutamic acid, have
been mixed and cross-linked via carbodiimide-mediated coupling. The swelling of
the cross-linked materials varies as a function of the cross-linking reaction temperature, and filamentous structures are formed in elastin-like polymers cross-linked
above their LCST.
Methods to produce elastomeric fibers from ELPs have also been investigated. Uncross-linked polymers can be spun into fibrous mats via electrospinning
protocols (135), with long uniform fibers resulting from spinning solutions greater
than 10 wt% in polymer; tensile strengths of 35 MPa and a material modulus of
1.8 GPa are possible via these strategies. Acrylate groups have been incorporated
via reaction with the lysine (K) amines to permit production of cross-linked elastinmimetic fibers (136), which exhibit increased tensile strengths and moduli that
are similar to those reported for native elastin.
Alternate strategies to control the mechanical properties of elastin-like
proteins, via combination with amino acid sequences from different fibrous
proteins, have also been explored. Combining the temperature-responsiveness
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
167
of elastin-based repeats with the excellent mechanical properties of silk-like
proteins (vide supra) yields protein polymers that have properties similar to
those observed in segmented polyurethanes, with the advantage of controlled
sensitivity to stimuli (pH and temperature). Cappello and co-workers have
combined the (GVGVP) repeat from mammalian elastin with the (GAGAGS)
sequence from B. mori silk to create copolymers of over 10 different compositions (82). The relative lengths of the silk-like and elastin-like blocks are
varied, and the ionic sensitivity of the polymers is controlled via the selection of amino acids in the elastin-like block. Representative copolymer compositions include the sequences {(GVGVP)4 (GEGVP)(GVGVP)3 (GAGAGS)}16 and
{(GVGVP)4 (GXGVP)(GVGVP)4 (GAGAGS)}11 , where X is either glutamic acid or
valine (83,84); these silk-elastin-like polymers (SELPs) are easily expressed from
E. coli hosts. Characterization of the physical behavior of these proteins via turbidity measurements indicates that the transition temperature (LCST) of the polymers containing glutamic acid is sensitive to pH, and that the LCST of both the
glutamic acid- and valine-containing polymers can be modulated with changes in
ionic strength, temperature, polymer concentration, and polymer length (83,84).
The simultaneous control over amino acid sequence and molecular weight afforded by genetically directed methods of polymer synthesis therefore provides
opportunities for fine-tuning the stimuli-responsive behavior of these polymers.
The materials have potential applications as injectible urethral bulking agents for
treating incontinence, cell culture coatings, drug delivery systems, or soft-tissue
augmentation or bone repair.
Cappello and co-workers have also investigated polymers in which the number of silk-like domains in the polymer is increased to produce polymers that
spontaneously form hydrogels in aqueous solution. For example, the sequence
{(GVGVP)4 (GKGVP)(GVGVP)3 (GAGAGS)4 }12 undergoes an irreversible sol-gel
transition in aqueous solution of physiological pH and ionic strength, mediated
by hydrogen-bond controlled crystallization of the silk-like domains (85), unlike the polymers above, which contain fewer silk-like repeats and do not form
hydrogels. The hydrogels (12 wt%) exhibit mechanical and swelling properties
that do not change as a function of temperature, as the presence of the silk domains eliminates their temperature sensitivity (86). X-ray diffraction patterns
of SELP polymers are very similar to those of simple silk-like polymers (SLPs),
confirming the presence of similar crystalline arrangements of silk-like blocks
in both SELPs and SLPs. Crystalline silk-fibroin domains are also observed, via
NMR, in the solid state of other SELPs (137). Proteins and DNA can be delivered from the hydrogels without loss of activity (85,87,138,139); bioactive DNA
can be released from the gels for up to 28 days, with the rate of release for
2.6–11-kb plasmids being dependent on the size of the plasmid DNA (87,139).
DNA released from these gels can also be delivered in the active form in a
mouse breast cancer model (139). These results suggest the potential use of these
polymers as in situ gel-forming implants for protein and gene delivery applications
(140,141).
Protein engineering strategies have also allowed the incorporation of biological function into elastin-like protein polymers and SELPs. The incorporation
of cell-binding amino acid sequences, such as RGD (82,142) and REDV (from the
CS5 domain of fibronectin) (66) (Table 2), permits endothelial cell adhesion to
168
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
these materials for their application in tissue engineering. Tirrell and co-workers
have also incorporated cross-linking domains into biologically active elastin-like
polymers (Table 2) to permit the production of biologically active, cross-linked
materials with mechanical properties similar to those of native elastin. The incorporation of lysine residues at the termini or in the VPGXG repeat of elastin-CS5
copolymers permits cross-linking via gluteraldehyde or N-hydroxysuccinimide activated suberic acids (88,89). Molecules of the general sequence RKTMG{LD-CS5G(VPGIG)20 VP}x LEKAAKLE, with x = 1, 3, and 5, have been cross-linked via
reaction with gluteraldehyde. The observed molecular weight between cross-links
(M c ) and mechanical properties of the cross-linked polymers approximate those
of elastin (0.3–0.6 MPa), with elongation-to-break values of 100–220% (88). A
broader range of mechanical properties can be obtained from elastin-based proteins in which lysine residues are incorporated in the VPGXG repeats. Proteins
of the general sequence {LD-CS5-G((VPGIG)2 (VPGKG)(VPGIG)2 )4 VP}3 , when
cross-linked via reaction with NHS-activated suberic acids, yield cross-linked
materials with Young’s moduli ranging from 0.07–0.97 MPa, with Mc values of
3,000–38,000 (89). These values span the range of mechanical properties of natural elastins and vary as expected with measured extents of cross-linking. Films
prepared from these proteins have also been shown to support endothelial cell adhesion under shear stresses that mimic those in blood vessels (143). The polymers
are therefore being considered for applications in engineered artificial grafts for
the surgical reconstruction of small- and medium-diameter blood vessels.
Overall, the applications for elastin-based protein polymers are numerous,
given their useful elastomeric properties, biological behavior, and low immunogenicity. The versatility of these systems includes the ability to engineer the LCST
behavior of (VPGXG)n polymers via choice of the X residue, to produce cross-linked
elastomeric gels, and to synthesize elastin-like block copolymers that assemble
into nanoparticles. The amino acid sequence of these polymers can also be engineered to allow the incorporation of biologically active domains. Their possible
applications as drug and gene delivery vehicles, membranes, elastomeric fibers,
and vascular grafts have continued to motivate interest in their scientific and
technological potential, and a variety of academic and industrial research groups
continue to investigate these polymers.
Artificial Proteins Designed de Novo
In addition to the design of protein polymers based on repetitive amino acid sequences found in naturally occurring proteins, de novo design has also been applied
to the production of artificial proteins with desired conformational properties.
Knowledge of the secondary structure preferences (ie, α-helix, β-sheet, coiled-coil,
reverse turn, etc) of amino acids and amino acid sequences has permitted the
design of completely artificial proteins with prescribed structures, chemical reactivity, and assembly. These short amino acid sequences are encoded into genes and
multimerized into target polymer lengths via the genetic strategies previously described. Proteins are expressed from E. coli, purified by appropriate protocols, and
their structure and assembly is characterized. A variety of novel protein materials, such as crystalline lamellar solids, smectic-like liquid crystals, and reversible
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
169
hydrogels have been produced via these methods, and showcase the control over
macromolecular structure and function that can be achieved by genetically directed polymer synthesis.
The earliest example of this approach involved the design and synthesis of
crystalline lamellar solids by Tirrell and co-workers. Although the folded chain
lamellar crystal is a well-known motif in polymer science, this architecture is
formed in synthetic polymers for largely kinetic reasons. The trapping of the polymer chain into a folded conformation upon cooling makes it essentially impossible to precisely control the thickness and surface chemistry of such chain-folded
lamellar structures. The genetic engineering approach, however, has afforded such
folded structures in which both variables can be controlled, simply by controlling
the sequence of amino acids in a protein polymer. Polymers with the general sequence {(AG)x EG}y (with x ranging from 3 to 6, and y ranging from 5 to 54) have
been designed on the basis of the knowledge that alanyl–glycyl dyads form thermodynamically stable β-sheets in natural silk proteins. When precipitated from
formic acid, this family of proteins forms crystalline solids with primarily β-sheet
structure, as ascertained from infrared and Raman spectroscopic analysis, cross
polarization/magic angle spinning NMR, and wide-angle X-ray diffraction (WAXD)
(90,144). X-ray diffraction investigations indicate that the lamellar thickness is
always shorter than the chain length of the polymers, as expected for the chainfolded architecture, and consistent with this observation, X-ray structure refinement identifies an antiparallel β-sheet arrangement of the chains as the best fit
to experimental X-ray data. The glutamic acid residues of this sequence would be
expected to lie at the lamellar surface, and indeed, deprotonation of the carboxylic
acid groups does not cause any change in the chain conformation or intersheet
packing distance as observed via vibrational spectroscopy and WAXD, respectively (145). Consistent with these results, X-ray diffraction characterization of
crystalline lamellar solids of {(AG)3 XG} sequences (with X = Asn, Phe, Ser, Val,
or Tyr) demonstrates that the interlamellar spacing increases linearly with the
increasing volume of the amino acid side chain in position X (91), but that neither
the turn length nor the intrasheet packing distance is altered. These results suggest that crystal surface functionality can be designed via the choice of the amino
acid in position X of the {(AG)3 XG}y sequences.
The self-assembling properties of certain β-sheet-forming peptides have also
been captured in protein polymers. Sequences of alternating polar and nonpolar
residues comprise a prevalent sequence motif in the antiparallel β-sheet regions
of natural proteins, and accordingly, the peptide (AEAEAKAK)2 has been demonstrated by Zhang and co-workers to self-assemble into an organized supramolecular structure (111). Protein polymers of the sequence (AEAEAKAKAEAEAKAK)9
have been produced by Goeden-Wood and co-workers in an effort to capture the
self-assembling characteristics of the peptides while improving the mechanical
properties of the supramolecular gels (146,147). The proteins (which contain a
decahistidine fusion sequence) can be expressed from E. coli and purified via
metal-chelate affinity chromatography with yields of approximately 5 mg/L. The
protein is shown, via CD and FTIR investigations, to adopt a stable β-sheet structure, and is suggested to form β-sheet fibrils via Congo Red dye binding assays
and characterization by scanning electron microscopy (SEM). Under physiological
conditions, the protein forms hydrogels with a storage modulus of approximately
170
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
100 Pa, suggesting the potential application of these polymers in soft tissue engineering or drug delivery applications.
In addition to the de novo design of β-sheet forming protein polymers in which
self-assembly is controlled by monomer sequence, the design of helical proteins
in which assembly is controlled by molecular weight has also been demonstrated.
Helical rods form well-defined self-assembled structures; one particularly wellstudied example is poly(γ -benzyl-α,L-glutamate) (PBLG), which forms oriented
films and liquid crystalline solutions. The large dipole moment along the helical
axis permits orientation of these rod-like polymers via the use of an electric field
to yield materials with interesting piezoelectric and nonlinear optical properties.
Since the conventional approach for making these molecules utilizes the ringopening polymerization of N-carboxy-α-amino acid anhydrides, molecules made
by this method have broad molecular weight distributions, and therefore form
cholesteric liquid crystalline phases in which there is orientational order but
no longitudinal registry of individual chains. Smectic liquid crystalline phases,
in which there is both orientational order and longitudinal registry, are not observed for PBLG synthesized by the chemical methods. In contrast, genetically directed synthetic strategies permit the synthesis of monodisperse poly(α,L-glutamic
acid) (PLGA), which can be chemically benzylated to form biosynthetically derived PBLGs capable of forming smectic-like liquid crystalline phases in solution
(92,93). Monodisperse polypeptides with the sequence ED(E17 D)x EE (referred to
as PBLG-x after benzylation), where x = 3–6, have been produced in E. coli (93,94).
Aspartic acid (D) is included periodically to provide recognition sites for the enzyme BbsI and to preserve the genetic stability of the repetitive glutamic acid
sequence. The biosynthetically produced PLGAs are benzylated by alkylation of
the PLGA with phenyldiazomethane, and analysis by NMR indicates 94–98%
benzylation. Small-angle X-ray diffraction patterns of films dried from solutions
of PBLG-4 and PBLG-5 (in a 97/3 mixture of ◦chloroform and trifluoroacetic acid)
show well-defined spacings of 114.5 and 140 A, respectively. These values almost
exactly
match the expected lengths of the monodisperse PBLG helices (114.5 and
◦
141 A) (92,93), suggesting alignment of the helical molecules in a smectic-like
liquid crystalline phase. The films are thought to specifically orient in a twisted
grain boundary (TGB)-like phase, owing to the superposition of a smectic-A layering (from the monodisperse PBLG) and the chirality of the helical rod (148).
Additional helical polymers in which assembly is controlled by monomer sequence have also been designed by Tirrell and co-workers and by Kaplan and
McGrath, and have proven fruitful for assembly of protein-based materials. Helical coiled-coil motifs (comprising leucine zipper peptides) are found in transcription factors and play an important role in controlling protein dimerization and
DNA binding; related helical motifs are also found in structural proteins such as
keratin (the main structural protein in hair and nails). The primary structural
feature of the leucine zipper peptides is the heptad repeat sequence abcdefg, in
which hydrophobic amino acids occupy positions a and d (d is primarily leucine),
while charged residues generally occupy positions e and g. Under appropriate pH
and temperatures, these amino acid sequences adopt helical conformations that
array the hydrophobic residues a and d along a single face of the helix. Assembly of these helices, most often in the form of dimeric coiled-coils, is controlled
by aggregation of the hydrophobic face, with the stability of specific associations
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
171
modulated by the charged residues e and g. The coiled-coil stability, aggregation
number, and aggregate specificity can be manipulated though control of the heptad amino acid sequence and the chain length. Thus, these helical motifs serve as
versatile structures for the assembly of novel protein-based materials.
Earliest work in this area by Kaplan and McGrath focused on the synthesis
of a variety of isolated leucine zipper peptides with controlled assembly properties (149,150). Expanding the approach, Tirrell and co-workers have designed
hydrogel-forming, triblock copolymers carrying short leucine zipper end blocks
flanking a water-soluble polyelectrolyte domain (95). In this triblock copolymer,
there are three main considerations in the design of the protein sequence: (1)
the hydrophobic face of the leucine zipper block is modeled after the a/d residue
pattern of the Jun oncogene product; (2) the b, c, and f positions are chosen on
the basis of the residues that most commonly reside in those positions in naturally occurring coiled-coil proteins; and (3) nine of the 12 e and g positions in the
sequence are occupied by Glu residues in order to facilitate pH control of gelation and viscoelastic behavior. The triblock protein polymer comprises two leucine
zipper end blocks of six heptad repeats, with a central polyelectrolyte block of
the sequence {(AG)3 PEG}10 . It has been expressed in E. coli, and CD characterization of the purified protein confirms the presence of both helical and random
coil structures. At low pH and ambient temperature, these materials form elastic
gels; increases in pH or temperature produce a viscous solution of nonassociated
protein (95). Solid-state NMR investigations of the protein hydrogel show that
the central polyelectrolyte domain is isotropically mobile on the microsecond time
scale, while the leucine zipper domains are rigid, which is consistent with the
association of the leucine zippers as the basis for hydrogel formation (151). On
the millisecond time scale, the leucine zipper domains exhibit rigid body motion,
which supports the hypothesis that the domains act as continuously exchanging
physical cross-links.
Alteration of the composition of these proteins via genetic methods may prove
to be a successful strategy for modulating the physical behavior of protein-based
hydrogels. For example, alteration of the acid/base balance in the helical domains
(positions e and g) can alter the stability of the coiled-coil association, which will
in turn control the stability of the hydrogel network and alter the pH and temperature at which these macromolecular structures assemble into hydrogels. Inclusion of cross-linking or lysis domains and/or cellular or molecular recognition
sequences will permit the design of hydrogels with predetermined physical and
biological properties and may expand their uses as reagents for encapsulation and
controlled delivery.
Genetically directed synthetic methods are also being exploited for the
structure-based design of protein polymers that display desired functional groups.
Kiick and co-workers have designed a series of functionalized, alanine-rich helical proteins with the general sequence {(AAAQ)x (AAAE)(AAAQ)x }h , in which the
position and number of the glutamic acid residues (E) can be varied systematically.
Members of this family of proteins can be produced in E. coli and are easily purified
via metal chelate affinity chromatography at yields of 10–20 mg/L. The proteins
are shown, via CD spectroscopy, to be highly helical under ambient conditions
(96), and, along with related random coil sequences, have intended applications
for purposeful display of biologically active groups and/or organic moieties.
172
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
In all of the cases described above, genetically directed methods have been
used to produce protein polymers with predesigned secondary structures that
control the folding, assembly, and/or the mechanical and biological properties of
the resulting polymer. However, genetically directed methods of polymer synthesis
have also been applied to the production of protein polymers in which a lack
of regular secondary structure is desired. Recursive multimerization strategies
have been used by Won and Barron (97) to produce genes that encode random coil
protein polymers of the general sequence (GKGSAQA)x , with molecular weights up
to 46.25 kDa. Protein polymers of this repetitive, nonnatural sequence have been
produced in E. coli at yields of approximately 15 mg/L, and CD studies confirm the
random coil nature of the protein. The ultimate goal of this line of investigation
is the creation of long, repetitive water-soluble proteins for diverse biomaterials
and tissue engineering applications. Inclusion of the lysine residues (K) at regular
intervals in this random-coil, water-soluble protein is intended for the attachment
of bioactive factors or for cross-linking.
Multisite Incorporation of Nonnatural Amino Acids
A very important recent development in these synthetic methods is the expansion of their chemical versatility via the incorporation of nonnatural amino acids.
Standard methods of in vivo protein synthesis employ the 20 naturally occurring
amino acids normally encoded by the mRNA templates. This repertoire of amino
acids includes carboxylic acids, amines, and thiols as the primary chemically reactive sites, which limits the chemical versatility of protein polymers. Accordingly,
there has been a great deal of interest in incorporating functional groups such
as alkenes, alkynes, halogens, or azides into proteins. Such incorporation would
permit chemical modification of protein polymers via reactions that are orthogonal to those of the natural amino acids and would also afford opportunities to
alter surface properties, impart photosensitivity, and control association of protein
polymers. However, investigations of this kind have been challenging owing to the
strict controls over the fidelity of protein biosynthesis exerted by the aminoacyltRNA synthetases (aaRS), the class of enzymes that controls the incorporation of
amino acids into proteins in vivo.
There are several strategies for incorporating nonnatural amino acids into
proteins that circumvent the discriminatory power of the aaRS; for example, nonnatural amino acids can be incorporated directly into polypeptides via chemical
synthesis strategies. Whereas the number of nonnatural amino acids that can
be incorporated in this fashion is essentially limitless, there are several disadvantages. In solution-based N-carboxyanhydride polymerizations (13), sequence
control is lost, while in solid-phase peptide synthetic methods (152), the less
than perfect efficiency of the amino acid coupling step limits peptide length to
approximately 50–75 amino acids, which is generally insufficient for materials
applications.
A second strategy that avoids the synthetases combines both chemical and
biological synthesis. Chemical aminoacylation methods were introduced by Hecht
and co-workers in the late 1970s (153) and have since been exploited by a variety
of other investigators (154–157). In these strategies, the nonnatural amino acid is
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
173
chemically attached to a suppressor tRNA, which decodes a single stop codon that
is engineered into the mRNA sequence of a target protein. The nonnatural amino
acid is then incorporated into the protein by suppression (decoding) of the stop
codon by the suppressor rRNA during in vitro translation. This method permits
the site-specific incorporation of nonnatural amino acids into proteins, and the
chemical aminoacylation step permits the incorporation of essentially any nonnatural amino acid. The in vitro translation protocols, however, yield only microgram quantities of protein. To address these low protein yields, in vivo strategies
employing heterologous synthetases have been developed for the site-specific incorporation of nonnatural amino acids into proteins produced from E. coli and
from yeast (158–163); these methods have proven a versatile strategy for the sitespecific incorporation of electrophilic, acetylenic, azido-functionalized, and redoxactive amino acids into proteins (164–166). Such strategies require the inclusion,
in the expression host, of both a suppressor tRNA that decodes the stop codon
during protein synthesis (and is not recognized by any of the E. coli aaRS) and
a synthetase that will aminoacylate only the suppressor tRNA (and none of the
endogenous E. coli tRNAs) with only the nonnatural amino acid. A recent advance
in this line of investigation is the generation of a bacterial expression host that
carries the heterologous synthetases necessary for site-specific, in vivo incorporation of a nonnatural amino acid and that can also synthesize the nonnatural
amino acid from basic carbon sources (167).
Although suppression-based methods permit the incorporation of a variety
of nonnatural amino acids, modest suppression efficiencies limit the incorporation of the nonnatural amino acid to a single site in the protein. Whereas in many
instances it may be desirable to limit incorporation to a single site (eg, studies of
protein folding or enzymatic activity), multisite incorporation has many distinct
advantages in the synthesis of protein polymer materials, in which alteration of
bulk material properties is desirable. In multisite incorporation strategies, nonnatural amino acids are incorporated into protein polymers at some or all of the
positions normally occupied by the natural amino acid, generally via the utilization of the nonnatural amino acid by the protein biosynthetic apparatus of the
expression host. These strategies therefore also afford the opportunity to control
the placement of the nonnatural amino acids in the protein polymer, which offers
additional advantages in macromolecular synthesis.
Perhaps surprisingly, given the extremely high fidelity of amino acid incorporation during protein biosynthesis, it has been known for decades that a number
of nonnatural amino acids are able to replace natural amino acids in bacterial
proteins. For example, selenomethionine is incorporated in place of methionine in
E. coli, can be used in all steps of protein biosynthesis, and supports cell growth
(168–170). Amino acids that differ in structure and functionality from the natural amino acids, such as p-fluorophenylalanine, norleucine, and trifluoroleucine,
have also been known for decades to replace the natural amino acids in bacterially
synthesized proteins (170–172). Over the past several years, a variety of additional
nonnatural amino acids bearing novel chemical functionality have also been incorporated into protein polymers in vivo (Fig. 4). Unsaturated, azido-functionalized,
and ketone-decorated analogues have been incorporated and subsequently modified via chemical strategies that are orthogonal to the chemistries of natural
amino acid side chains (173–175). Fluorinated amino acids offer opportunities for
174
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
Fig. 4. Nonnatural amino acids and the strategies used for their multisite incorporation
into proteins.
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
175
controlling the surface properties of protein films (98), and also afford strategies for
increasing the thermodynamic stability of protein assemblies (176). Incorporation
of aryl halide function permits chemical modification of proteins via palladiumcatalyzed reactions, and also offers new opportunities for protein structure determination (177,178).
The incorporation of nonnatural amino acids into proteins in vivo requires
that the nonnatural amino acid meet several criteria. First the analogue must
be transported across the cell membrane into the cell, either by amino acid specific or general transport mechanisms, and it must not be degraded once inside
the cell. Second, the nonnatural amino acid must be recognized by the aaRS during translation, and must also be able to form a stable aminoacyl-tRNA that is
not subject to the aaRS editing mechanisms that normally prevent misacylation
of tRNA. Finally, the nonnatural amino acid must be an efficient substrate for
the elongation factor Tu (EF-Tu) and must be accepted at the ribosomal A site.
Transport into the cell does not appear to be limiting, based on the number of
nonnatural amino acids that have been demonstrated to replace natural amino
acids in biosynthetically derived proteins. Recognition of the misacylated-tRNA
by EF-Tu and acceptance at the ribosomal A site are also not suggested to be
limiting factors, based on the enormous number of nonnatural amino acids that
can be incorporated into proteins by in vitro translation protocols. Many different investigations have indicated that the key determinant to the success of the
in vivo approach, therefore, is the ability of the nonnatural amino acid to be
charged to a tRNA by an aminoacyl-tRNA synthetase.
The high specificity and efficiency that the aaRS exhibit for the natural amino
acids requires that the concentration of natural amino acid in the culture medium
during protein expression must be strictly limited relative to the concentration of
the nonnatural amino acid, so that incorporation of the nonnatural amino acid will
be favored. This requires that the natural amino acid that will be replaced must
be chosen prior to protein expression, and the appropriate bacterial auxotroph (a
strain that is unable to synthesize the chosen natural amino acid) must be obtained/produced. The general experimental procedure for nonnatural amino acid
incorporation involves several steps. The auxotroph is grown first in medium that
contains the natural amino acid (to maximize the number of protein-producing
cells), and then at a desired cell density the cells are centrifuged and washed
quickly in order to remove the natural amino acid, and are then resuspended in a
medium that contains the nonnatural amino acid but not the natural amino acid.
Protein expression is then induced, and in cases where the nonnatural amino acid
supports protein biosynthesis, the protein is isolated and purified. The extent of
amino acid replacement (ie, nonnatural amino acid incorporation) can be determined via amino acid analysis, Edman degradation analysis, mass spectrometry,
tryptic digest/mass spectrometry, and/or NMR, depending on the identity of the
nonnatural amino acid.
One of the more recent developments in the in vivo incorporation of nonnatural amino acids has been the correlation of the activation of a nonnatural amino
acid by an aaRS with the ability of the nonnatural amino acid to support protein
biosynthesis (ie, to be incorporated into proteins in vivo). The covalent attachment of an amino acid to its cognate tRNA proceeds in two steps, activation and
176
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
aminoacylation:
aaRS + aa + ATP [aaRS : aa∼AMP] + PPi
[aaRS : aa∼AMP] + tRNAaa aaRS + AMP + aa∼tRNAaa
where aaRS is the aminoacyl-tRNA synthetase, aa is the amino acid, ATP is
adenosine triphosphate, AMP is adenosine monophosphate, PPi is pyrophosphate, aaRS:aa∼AMP is the aminoacyladenylate complexed with the enzyme, and
aa∼tRNAaa is the aminoacyl-tRNA. The rate of activation of the amino acid by the
enzyme can be measured in assays that monitor the rate at which radiolabeled
pyrophosphate is incorporated into ATP (the reverse of the top reaction). The rate
of ATP–PPi exchange is measured as a function of analogue concentration, and the
data are fit to a standard Michaelis–Menten model to determine kcat /K m , which
can be used to compare the relative rates of activation of different amino acids by
a given aaRS. The measured rates of activation of a variety of nonnatural amino
acids by the appropriate aaRS have been shown by Tirrell and co-workers to correlate well with the ability of the bacterial host to utilize the amino acids during
protein biosynthesis.
The quantitative establishment of the direct role of the aaRS in the multisite incorporation of nonnatural amino acids has resulted in the development of
several strategic approaches that can be taken to control incorporation. The first
relies on the ability of the wild-type biosynthetic apparatus to utilize nonnatural
amino acids, and uses a standard auxotrophic expression host for protein synthesis. The second approach relies on increasing the level of activity of the wild-type
aaRS in the bacterial host by engineering the host to produce extra copies of
the desired enzyme. In the third approach, the aaRS is also engineered; the active site can be altered to permit activation of a broader set of nonnatural amino
acids, and/or the editing activity of the aaRS can be abolished so that misacylated
tRNAs are no longer destroyed by the editing pathway of the enzyme. The mutant
aaRS is then overexpressed in the bacterial host. An additional strategy, which
permits simultaneous incorporation of both a nonnatural amino acid and its natural counterpart in specified positions, expands the genetic code by the expression
of a heterologous aaRS/tRNA pair. The aaRS with desired new activities can be
identified via evolutionary-based mutagenesis strategies and/or by rational computational approaches. The application of these methods, as described in more
detail below, has resulted in a large set of nonnatural amino acids that can be
incorporated into proteins in vivo (Fig. 4).
Strategies for Nonnatural Amino Acid Incorporation that Employ
the Wild-Type Biosynthetic Apparatus
Methionine. Of the investigations on the incorporation of nonnatural
amino acids into proteins, the incorporation of methionine analogues is among
the earliest studied. Methionine has been an interesting target for replacement by
nonnatural amino acids, as the promiscuity of the biosynthetic apparatus toward
methionine has been known for decades, and a variety of methionine analogues
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
177
are able to replace methionine during protein biosynthesis. There are no editing
mechanisms for hydrolysis of misacylated tRNAMet , which suggests that methionine analogues of varying side-chain structures and chemical function may be readily incorporated into proteins. In addition, methionine-rich interfaces are known
to mediate a variety of protein–protein assembly processes, and the replacement
of methionine may therefore provide opportunities for purposeful manipulation of
protein–protein interactions.
The first report of multisite inclusion of nonnatural amino acids into genetically engineered protein polymers was reported by Tirrell and co-workers in
the early 1990s, with the inclusion of selenomethionine (in place of methionine)
into the repetitive β-sheet-forming polypeptide {(GA)3 GM}9 (99). A methionine
auxotroph was transformed with an expression plasmid that contained the DNA
sequence encoding the polypeptide. The ratio of selenomethionine to methionine
in the product protein, as determined via radiolabelling assays, correlated linearly with the ratio in the growth medium, indicating that selenomethionine
is capable of essentially perfect replacement of methionine. Standard auxotrophic expression hosts were used to produce the selenomethioninecontaining proteins, with the wild-type protein biosynthetic machinery accepting selenomethionine in place of methionine in every stage of protein
biosynthesis.
In addition to the selenomethionine example above, telluromethionine, norleucine, trifluoromethionine, and ethionine have all demonstrated translational
activity in wild-type bacterial hosts as well (179–181). The translational activity
(ie, ability to support protein biosynthesis) of several additional chemically novel
methionine analogues (1-10) has therefore been investigated (Figure 4). In all of
these studies, the incorporation of methionine analogues into the target protein
murine dihydrofolate reductase (mDHFR) has been determined; mDHFR carries
methionine in 8 positions, and therefore serves as a reasonably stringent test
of incorporation of methionine analogues. The mDHFR is encoded in a commercially available pQE15 expression plasmid, which is transformed into an E. coli
methionine auxotroph.
Methionine analogues 1, 2, 3, and 4 have been shown to replace methionine in vivo in these conventional bacterial expression hosts (174,182,183); the
remaining methionine analogues, 5-10, do not support protein biosynthesis under similar assay conditions. Analogues 1 (azidohomoalanine, Aha) and 2 (homopropargylglycine, Hpg) replace methionine quantitatively, as assessed via amino
acid analysis, Edman degradation, and mass spectrometry. Furthermore, cultures
supplemented with these analogues support the production of mDHFR in the
same yields as cultures supplemented with methionine (approximately 40 mg/L)
(174,183). Analogue 4 (homoallylglycine, Hag) replaces methionine at levels of
approximately 92%, although yields of mDHFR from cultures supplemented with
Hag are approximately 28% of those from cultures supplemented with methionine.
Further assessment of the activation of the methionine analogues by MetRS has
also been conducted and has suggested additional strategies for the incorporation
of analogues 5–10 (vide infra).
These results suggest that the replacement of methionine by chemically reactive analogues can be used in the synthesis of chemically and physically novel
protein polymers. Indeed, mDHFR decorated with the azido-functionalized Aha
178
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
(1) can be selectively modified by Staudinger-type reactions with triarylphosphine
reagents (174), in both its purified form as well as in whole cell lysates. Cellsurface proteins equipped with Aha can also be efficiently modified with alkynefunctionalized biotinylating reagents via copper(I)-catalyzed triazole formation
(184). Upon incubation with an avidin-functionalized fluorescent agent after biotinylation, cells decorated with Aha can be differentiated from cells decorated
with methionine via flow cytometry methods (184). These results suggest opportunities for chemical modification of protein polymers and for labeling of proteins
in vivo.
Fluorinated Amino Acids. Synthetic fluoropolymers exhibit many useful surface properties, such as low surface energy, low friction coefficient, excellent
solvent resistance and hydrolytic stability, and chemical and biological inertness,
and this has enabled these polymers to find use in many disparate applications,
such as Teflon-coated pans, self-lubricating parts, pipe liners, membranes, and
vascular grafts. The possibility of imparting similar properties to well-defined
protein architectures has been of long-term research interest. Fluorinated leucine
analogues have been of particular focus owing to leucine’s abundance in natural
proteins, and substitution by leucine analogues may provide a general strategy
for controlling protein folding and function. Furthermore, 5,5,5-Trifluoroleucine
(Tfl, 11) has been known for over 30 years to support bacterial cell growth and to be
incorporated into proteins in the absence of leucine. Tfl has been incorporated into
β-sheet forming polypeptides with the general sequence {(GA)3 GL}12 , via protein
expression in a standard leucine auxotroph, in an attempt to produce protein materials with similar surface characteristics as fluoropolymers. The incorporation
of Tfl significantly lowers the surface energy of resulting protein films; contact
angles of hexadecane on fluorinated protein films (70◦ ) are much higher than
those on the unfluorinated, leucine-containing protein (17◦ ) (98). The fluorinated
analogue p-fluorophenylalanine (Pff, 21) can also be incorporated into the similar
sequences {(GA)3 GF}13 (98). FTIR and WAXD studies of protein solids containing
Tfl or Pff confirm the antiparallel β-sheet architecture, suggesting that the fluorinated surface may be formed by presentation of the fluorinated amino acids in
the fold regions of the β-sheet proteins.
The incorporation of fluorinated amino acids into proteins also confers other
potential materials property advantages. Since many organofluorine compounds
exhibit lower solubility in water than their hydrocarbon equivalents, fluorinated
amino acids may act as hyper-hydrophobic analogues of the hydrophobic amino
acids, which may impart increased stability to assembled protein structures stabilized by hydrophobic interactions. The fact that the fluorinated amino acids
are nearly isosteric to their natural counterparts also suggests that fluorinated
amino acids could be incorporated into proteins without disrupting protein structure. These factors have motivated the investigation of the effect of fluorinated
analogues of leucine on the assembly and stability of coiled-coil proteins produced in vivo. In experiments to test Tfl incorporation, the pQE9-derived expression plasmid encoding the leucine zipper protein (vide supra) is transformed
into a leucine auxotroph, and the cells are grown on cultures supplemented with
Tfl (11). As might be expected on the basis of many previous reports of the translational activity of Tfl, the analogue supports the synthesis of the leucine zipper
protein (176), with a maximum level of incorporation of 92%, as indicated by
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
179
amino acid analysis and MALDI-MS. Varying the ratio of leucine to Tfl in the
culture medium permits various amounts of each amino acid to be incorporated
into the protein, although the yields of protein decrease steadily to 20 mg/L (from
40 mg/L) as the percentage of Tfl in the culture medium is increased to 100%.
Analysis of the purified proteins via CD indicates that the protein outfitted with
Tfl exhibits essentially identical secondary structures as the leucine-containing
protein, with greater than 90% helicity. Additionally, the proteins show increasing
stability to thermal and chemical denaturation with increasing degrees of fluorination, with “melting” temperatures increased to a maximum of 67◦ C as compared
to 54◦ C for the leucine-containing protein (176). The increased stability of these assembled structures with respect to thermal and chemical denaturation may have
important consequences on controlling the stability and stimuli-responsiveness
of not only leucine zipper based, but other types of protein-based materials as
well.
Similar strategies have also been employed to incorporate trifluoroisoleucine
(Tfi, 15) into proteins in vivo (185). The analogue Tfi was used to supplement
cultures of an E. coli isoleucine auxotroph equipped with a pQE-15-derived expression plasmid encoding mDHFR (which contains 14 Ile residues). In this bacterial expression host, Tfi was shown to support protein synthesis, with levels
of replacement of 93% (185). In an effort to deduce the impact of the incorporation of fluorinated amino acids on protein function, Tfi was also incorporated
into murine interleukin-2 (mIL-2), which contains five isoleucine residues in the
helical core of the protein. Fluorinated mIL-2 elicits an equivalent maximal proliferative response as the wild-type mIL-2, which indicates that the fluorinated
protein still folds into an active, native structure. The combination of the results for the isoleucine and leucine analogues suggests that at least some protein domains will tolerate side-chain fluorination without loss of function. They
also indicate the potential for maintaining desired secondary structures and biological activity in repetitive artificial proteins equipped with nonnatural amino
acids.
Unsaturated and Structural Amino Acids. The earliest investigations
of the incorporation of unsaturated analogues into genetically engineered proteins were conducted in the 1990s. Unsaturated proline analogues were of initial
interest for their potential ability to modify protein folding behavior and present
reactive chemical functionality at fold surfaces; the potential for substitution of
proline had been indicated by earlier studies indicating incorporation of proline
analogues into cellular proteins (186). Polymers of the repeating unit {(AG)3 PEG}x
had been successfully produced in E. coli hosts (100) but were difficult to crystallize, presumably because the conformationally restricted proline residue prevents alignment and hydrogen bonding of the alanylglycyl diads in the repetitive polymer. The incorporation of the analogues dehydroproline (Dhp, 18) and
azetidine-2-carboxylic acid (Aze, 19) has therefore been assessed via expression of
{(AG)3 PEG}16 in cultures of a proline auxotroph grown on media supplemented
with Dhp or Aze (173,187). The Dhp is most readily incorporated (nearly 100%),
while the Aze is incorporated at levels of approximately 40%, as determined by
1
H NMR spectroscopy and amino acid analysis.
The physical properties of the Dhp variant are qualitatively very similar
to those of the proline-containing variant; both form optically clear, amorphous
180
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
solids and are water-soluble, indicating a lack of crystallization. The chemical
properties of the Dhp variant, however, are strikingly different, as reaction of the
protein with H2 O2 or Br2 produces hydroxylated and brominated forms of Dhp
in quantitative reactions, as determined by the loss of the alkene resonance in
the NMR spectrum of the modified protein (173). Whereas the Aze variant is not
chemically reactive, the physical structure of this protein is very different than
the proline-containing protein, with FTIR spectroscopy indicating β-sheet structure (187). The smaller size of the Aze appears to increase the chain flexibility
sufficiently to permit folding; the fact that the addition of only 4 to 6 Aze residues
out of 148 amino acids can cause such a pronounced change in physical properties
may also have important consequences for purposeful control of polymer physical and chemical behavior. Incorporation of the unsaturated leucine analogue
12 and isoleucine analogues 16 and 17 into the test protein mDHFR has also
been reported and may have similar potential for modification of protein folding
and reactivity (188,189). The isoleucine analogues can support protein synthesis
at levels of approximately 50% of that observed in cultures supplemented with
isoleucine, and can replace isoleucine with extents of replacement of 80% (16) and
70% (17).
The modification of protein structural properties via incorporation of nonnatural amino acids has also recently been realized for human type I collagen expressed in E. coli. Proline residues in collagen are hydroxylated to permit folding
and to improve the stability of the collagen triple helix (vide supra). In mammalian
systems, this enzymatic post-translational modification permits the accumulation
of correctly folded protein. In prokaryotic systems, however, these enzymes are not
present, and thus the modification of the proline has been accomplished via the
introduction of the enzyme prolyl 4-hydroxylase in the expression host. The direct
incorporation of trans-4-hydroxyproline (Hyp, 20) in place of proline would obviate the need for the introduction of such additional enzymatic pathways. Buechter
and co-workers have demonstrated the feasibility of this approach by the expression, in E. coli, of a protein with a sequence derived from the α 1 fragment of human
type I collagen; the protein contains 64 Gly-X-Y repeating units and 52 proline
residues in both positions X and Y (190). The protein has been expressed in an
E. coli proline auxotroph, under conditions in which the auxotroph is grown in
proline-depleted cultures that contain hydroxyproline and hyperosmotic concentrations of sodium chloride (500 mM). Mass spectrometry and amino acid analysis of the protein indicate quantitative replacement of Pro by Hyp in proteins
isolated from these cultures. Hyp is activated 5 orders of magnitude less efficiently than Pro by ProRS in vitro (190), and therefore does not support protein
biosynthesis under normal culture conditions. However, subjecting E. coli to hyperosmotic conditions increases the intracellular Hyp concentration sufficiently
to recover its ability to support protein biosynthesis; it may be accumulated via
mechanisms similar to those that cause active accumulation of Pro under hyperosmotic conditions (191). The Hyp-containing collagen fragments fold into the
correct triple-helical structure, as assessed via CD spectroscopy (190), indicating that the global substitution of Hyp for Pro does not appear to destabilize the
triple helix, even though Hyp is normally only found in the Y position of GlyX-Y repeats in vertebrate collagens. Full-length fragments of α 1 (I) chain, with
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
181
338 G-X-Y repeats and 255 proline residues, have also been expressed in prolinedepleted cultures supplemented with Hyp under hyperosmotic conditions, and
although the extent of Hyp substitution has not been quantitatively determined,
these results suggest that Hyp can replace Pro in even very large numbers of
Propositions.
Electroactive Amino Acids. Conducting polymers have been actively investigated for decades because of their potential applications in lightweight batteries, electrodes, nanowires, and nerve regeneration. The production of conducting
polymers, however, is often plagued by the insolubility of the highly conjugated,
rigid polymer. The incorporation of electroactive amino acids into genetically engineered protein polymers has been explored as a strategy for circumventing these
solubility issues, as well as for generating biomedically relevant materials with
combined biological and electrical function useful for nerve regeneration. Early
investigations explored the incorporation of 3-thienylalanine (3TA, 22) into the
repetitive sequence {(GA)3 GF}13 ; protein polymers were expressed from a pETderived plasmid in a phenylalanine auxotroph grown on media supplemented
with 3TA. The 3TA replaces Phe at levels of approximately 85%, as determined
by 1 H NMR spectroscopy, amino acid analysis, and UV spectroscopy (192). The
3TA analogue alone can be oxidatively polymerized to produce conjugated systems (193), which could be used for the formation of electroactive films or the
direct electropolymerization and attachment of biologically active motifs to electrode surfaces.
Nonnatural Amino Acid Incorporation via Overexpression
of Wild-Type Aminoacyl-tRNA Synthetases
The examples above demonstrate the flexibility of the wild-type translational apparatus for the incorporation of nonnatural amino acids. Nevertheless, the range
of chemical functionality available via utilization of a wild-type bacterial host is
substantially more limited than that available via chemical polymerization methods, which has prompted further investigation of the promiscuity of the aaRS in
the incorporation of nonnatural amino acids. Such investigations have relied on
developing an understanding of the relationship between the rates of activation
of a nonnatural amino acid by its cognate aaRS and the ability of the nonnatural
amino acid to be incorporated into proteins in vivo.
Studies by Kiick and Tirrell have demonstrated the correlation between the
kinetics of MetRS-catalyzed analogue activation in vitro and methionine analogue
incorporation into proteins in vivo. The results of ATP-PPi activation assays indicate that the translationally active analogues 1, 2, 3, and 4 are activated by E. coli
MetRS, although they are 390-, 500-, 1050- and 1850-fold poorer substrates for the
enzyme than methionine, respectively (174,194). In the case of analogues 1 and
2, the lower rates of activation do not impede the rate of protein synthesis or the
extent of incorporation of the analogue (vide supra). At the sufficiently lower rates
of activation of analogues 3 and 4, however, the yield of protein recovered from
cultures supplemented with the analogue correlates with the rate of activation of
182
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
the analogue (57% and 28% yields, respectively). Analogues 5-10 are increasingly
poor substrates for the MetRS, consistent with their lack of translational activity
in a conventional bacterial host (195).
Overexpression of MetRS in a bacterial expression host has been successfully employed as a strategy for incorporation of methionine analogues 5-10. In
this approach, the E. coli metG gene and its promoter are ligated into the expression plasmid pQE-15; bacterial hosts equipped with this plasmid (pQE15-MRS)
exhibit approximately 50-fold increased rates of methionine activation. When this
modified bacterial expression host is grown in cultures supplemented with analogue 5, the rate of activation of analogue 5 is increased sufficiently to permit its
incorporation into proteins at levels of replacement of approximately 92% (with
protein yields of approximately 12 mg/L) (196). The analogues 6-10, which are activated up to 340,000 more slowly than methionine, can also be incorporated into
proteins via similar strategies (195), although the engineered expression host
must be grown in medium supplemented with large amounts of the analogue
(500 mg/L). In protein produced under these conditions, the level of methionine
replacement by analogues 6-10 is 60–98%. Overexpression of the MetRS has also
been shown to increase protein yields from cultures supplemented with poor substrates of MetRS.
Overexpression of wild-type aaRS has also been used to expand the number
of fluorinated amino acid analogues that can be incorporated into proteins (197).
The successful modification of protein properties via incorporation of Tfl, 11, suggests fluorination as a strategy for modifying surface properties and assembly, and
substitution by Hfl, 13, which carries six fluorine atoms, may have a correspondingly greater impact on protein stabilization. ATP-PPi exchange assays indicate
that Tfl is only 240-fold poorer a substrate for LeuRS relative to Leu, consistent
with its ability to replace Leu in proteins produced by a conventional bacterial
host. These assays, however, show that Hfl is approximately a 4000-fold poorer
substrate for LeuRS versus leucine. Overexpression of the wild-type LeuRS in an
engineered expression host has therefore been employed as a strategy to permit
incorporation of Hfl (197). The E. coli leuS gene and its promoter were ligated
into the expression vector pQEA1 (which encodes the leucine zipper motif), to
yield plasmid pA1EL. Bacterial hosts equipped with the pA1EL plasmid exhibit
LeuRS activity that is elevated approximately 8 times that of unmodified bacterial hosts. Leucine zipper proteins produced from Hfl-supplemented cultures of
this bacterial host show a maximum level of Hfl incorporation of 74%, as assessed
via amino acid analysis and MALDI-MS. CD measurements confirm that the Hflcontaining protein is >90% helical, and that the melting transition temperature
for this fluorinated protein is elevated to 76◦ C [which is 22◦ C greater than that
of the leucine-containing protein and 9◦ C greater than that of the Tfl containing
protein (vide supra)]. The Hfl-containing protein remains 100% folded in urea
concentrations up to 4 M (197). Overexpression of the LeuRS has also permitted
the incorporation of cyclobutenylglycine, 14, into leucine zipper proteins (198);
the side-chain functionality of this analogue may have uses for chemical modification or graft polymerization via ring-opening metathesis polymerization methods.
Combined, these results suggest that overexpression of a wild-type aaRS is a general strategy for expanding the set of nonnatural amino acid analogues that can
be incorporated into proteins in vivo.
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
183
Nonnatural Amino Acid Incorporation via Overexpression
of Mutant Aminoacyl-tRNA Synthetases
Active Site Mutations. Alteration of the amino acid specificity of the
aminoacyl-tRNA synthetases offers another strategy for incorporating into proteins nonnatural amino acids that are not utilized by the wild-type translational
apparatus. Such alteration was initially demonstrated in investigations focused
primarily on altering the specificity of an aaRS for different natural amino acids
(199,200). More recently, investigations by Tirrell and co-workers have expanded
the number of nonnatural amino acids that can be incorporated into proteins via
mutagenesis of the active site of PheRS (175,177,178). Wild-type PheRS is active toward phenylalanine and p-fluorophenylalanine (Pff, 21), but not toward
p-chlorophenylalanine (Pcf, 23), presumably because the additional steric bulk of
the chlorine substituent precludes acceptance of Pcf at the active site of PheRS. In
1991, an engineered mutant form of this enzyme that charges tRNAPhe with Pcf
was reported (201); the enzyme carries the mutation A294G, in which the alanine
residue at position 294 has been mutated to glycine. This mutation effectively
enlarges the active site and permits the activation of phenylalanine analogues
that carry larger substituents in the para position; indeed, investigations indicated that the mutant PheRS permitted incorporation of Pcf into proteins in vivo
(202). Overexpression of this mutant PheRS in E. coli has also permitted the multisite incorporation of a larger set of chemically diverse phenylalanine analogues
into target proteins; analogues incorporated include p-bromo (24) (177), p-iodo
(25), p-cyano (26), p-ethynyl- (27), and p-azido-phenylalanine (28), and 2-, 3-, and
4-pyridylalanine (29-31) (178).
To achieve these results, the mutant α subunit of PheRS is encoded on an
expression plasmid that also encodes the target protein mDHFR (which contains
nine Phe residues). The resulting plasmid is transformed into an E. coli phenylalanine auxotroph (177), and the auxotroph is grown in media depleted of Phe
and supplemented with one of the phenylalanine analogues (250–500 mg/L). Target protein isolated from these cultures is then characterized to determine the
level of analogue incorporation. The analogue p-Br-Phe, 24, is incorporated into
mDHFR at levels of replacement of 88% (as determined via amino acid analysis),
with protein yields of 20–25 mg/L (approximately 70% of the expression levels
of cultures supplemented with Phe) (177). The other phenylalanine analogues,
25-31, can be incorporated into mDHFR with levels of replacement ranging from
45 to 90%, with protein yields of 6–18 mg/L (178). The UV spectra of proteins
outfitted with analogues 27, 28, 29, 30, or 31 show peaks whose positions and
intensities are consistent with those of the free amino acid analogues, indicating
that the functional groups are not modified by the bacterial host during protein
biosynthesis or modified by photodegradation.
The introduction of these functional groups provides numerous opportunities
to engineer protein polymer properties. Introduction of aryl halide functionality
into engineered proteins, for example, opens possibilities for a variety of controlled
chemical modifications of proteins via chemistries that are orthogonal to existing methods of protein modification, such as transition metal-catalyzed coupling,
amidation, and cyanation. The aryl azide side chain provides the capacity for
intramolecular photoactivated cross-linking via the liberation of an amine group
184
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
upon exposure to UV light. The use of such modification strategies would allow new
approaches to side-chain modification, immobilization on surfaces, cross-linking,
and synthesis of graft copolymers. Several of the phenylalanine analogues also
show distinct photophysical behavior in the X-ray, UV, and IR regions that may
facilitate crystal structure determination via X-ray diffraction and biophysical
studies of protein folding and assembly. Bromination, for example, has been explored as a new tool for use in X-ray diffraction studies of protein structure via
multiwavelength anomalous diffraction methods.
The potential for incorporating additional Phe analogues via further mutagenesis of the PheRS has also been investigated by the Tirrell group. The crystal
structure of the Thermus thermophilus wild-type PheRS complexed with Phe (203)
has served as a starting point for the design for additional mutant E. coli PheRS.
Computational approaches have been used to identify important cavity-forming
mutations in the T. thermophilus enzyme (175), and show that mutation of Ala314
(Ala294 in E. coli) to Gly, and Val261 (Thr251 in E. coli) to Gly, enlarges the active site. Overexpression of the double mutant PheRS (A294G and T251G) in a
bacterial expression host (via similar strategies as those above) permits incorporation of p-acetophenylalanine (Paf, 32) into mDHFR, at levels of replacement of
approximately 80% and yields of 20 mg/L (60% of those obtained from cultures
supplemented with Phe) (175). The Paf-containing mDHFR has been shown to be
active toward hydrazide reagents, without chain cleavage. These studies demonstrate the power of computational approaches for the design of aaRS that permit
incorporation of new nonnatural amino acids. They also demonstrate the versatility of the biosynthetic apparatus for production of proteins containing chemically
novel nonnatural amino acids.
In addition to these investigations of PheRS, other investigations have suggested the potential for altering the amino acid specificity of TyrRS (199,204)
to permit incorporation of tyrosine analogues. An active site mutant of TyrRS,
F130S, shows an increased specificity for azatyrosine, 34, via in vitro assays of
aminoacylation. The mutant TyrRS is overexpressed in E. coli expression hosts,
and when cultures of the host are grown on media supplemented with radiolabelled 3 H-azatyrosine, azatyrosine is incorporated into E. coli cellular proteins,
as assessed via SDS-PAGE with detection via film exposure (204). The results for
cellular protein uptake are likely to be generally applicable to the incorporation
of similar analogues into artificial repetitive protein polymers.
Editing Site Mutations. Mutations of aaRS editing sites have also been
a target for increasing the number of nonnatural amino acids that can be incorporated into proteins. IleRS, LeuRS, and ValRS all exhibit an editing mechanism
in which a separate editing domain of the aaRS controls hydrolysis of noncognate aminoacyladenylates and misaminoacylated tRNAs on the basis of the size
and hydrophilicity of the noncognate amino acid side chain. LeuRS, in particular, employs this editing mechanism to prevent the incorporation of methionine
and isoleucine in place of leucine during protein biosynthesis. Tang and Tirrell
have reported the generation of a T252Y editing site mutant of E. coli LeuRS
with impaired editing activity, as assessed via in vitro aminoacylation assays
(205). A leucine zipper target protein (vide supra) has been produced in cultures
of a leucine auxotroph that overexpresses the mutant LeuRS; the cultures are
supplemented with various nonnatural amino acids but are depleted of leucine,
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
185
methionine, isoleucine, and valine. Analysis of the target protein via amino acid
analysis and tryptic digest mass spectrometry indicates that the methionine analogues norleucine (3) and norvaline (9) are incorporated at the leucine sites in
the leucine zipper protein at levels of 91 and 79%, respectively (205), consistent
with the attenuation of the editing activity of LeuRS against methionine. The analogues also replace methionine at levels of 92 and 68%, respectively (205), as is
expected on the basis of the reported ability of these analogues to replace methionine in vivo (vide supra). Overexpression of the mutant LeuRS also permits the
incorporation of the unsaturated methionine analogues homopropargylglycine (2),
homoallylglycine (4), 2-butynylglycine (7), and allylglycine (8), at levels of 74, 37,
37, and 76%, respectively, as assessed via mass spectrometric analysis. In other
investigations, mutations at the editing site of valyl-tRNA synthetase have also
been demonstrated to permit the incorporation of aminobutyrate in place of valine
in cellular proteins produced by E. coli (206). Modulation of the editing activities
of select aaRS therefore provides an additional strategy for incorporation of novel
amino acids into proteins in vivo.
Nonnatural Amino Acid Incorporation via Introduction
of Heterologous aaRS/tRNA Pairs
The multisite incorporation of nonnatural amino acids in protein polymers has
relied on the replacement of a given natural amino acid by a desired nonnatural amino acid. Protein polymers that contain both a natural amino acid and its
analogue can be produced via methods in which the ratio of the amino acids in
the culture medium is controlled (vide supra). These strategies, however, do not
permit precise control of the placement of the natural and nonnatural amino acids
in the protein chain, since the nonnatural amino acid must “share” codons with
the natural amino acid it replaces. The degeneracy of the genetic code, which
arises from the use by the protein biosynthetic apparatus of 61 mRNA sense
codons to direct the templated polymerization of 20 amino acid monomers, can be
used to overcome this limitation. The ability to reassign degenerate triplet codons
to nonnatural amino acids offers possibilities for expanding the combinations of
building blocks (natural and nonnatural) that can be used to construct artificial
proteins. Tirrell and co-workers have recently demonstrated the feasibility of this
strategy for the simultaneous incorporation of phenylalanine and its analogue
3-(2-naphthyl)alanine (Nal, 33) at specified positions in a target protein (207).
Phenylalanine is encoded by two codons, UUC and UUU, and both codons
are read by a single tRNAPhe , which carries the anticodon GAA. Introduction
of a tRNAPhe with an AAA anticodon, therefore, offers a strategy to incorporate
nonnatural amino acids only at phenylalanine positions encoded by UUU, while
maintaining the incorporation of Phe at positions encoded by UUC. To achieve this
experimentally, Kwon and co-workers have introduced a heterologous PheRS and
its cognate tRNAPhe into an E. coli expression host (207). Specifically, the investigations have employed a yeast tRNAPhe (with an AAA anticodon, ytRNAPhe AAA ),
which is not charged by E. coli PheRS, and a mutant yeast PheRS (T415G) that
charges only the yeast tRNAPhe and preferentially activates Nal over Phe (Fig. 5).
186
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
Fig. 5. Strategy for specific multisite incorporation of both natural and non-natural amino
acids. Adapted, with permission, from Ref. 208.
The mutant yeast PheRS gene is encoded on an expression plasmid that also encodes the target protein mDHFR, which contains four Phe residues encoded by
UUC and five by UUU; the yeast tRNAPhe AAA is encoded on the repressor plasmid pREP4. Protein expression is induced in cultures of a Phe auxotroph that is
equipped with these plasmids; the culture medium is supplemented with Nal and
depleted of Phe. Amino acid analysis of the isolated target protein indicates that
4.4 of the 9 Phe residues are replaced by Nal, and mass spectral analysis shows replacement of 5 phenylalanine residues by Nal, which corresponds exactly with the
number of UUU codons present in the mDHFR gene. Analysis, via MALDI-MS,
of tryptic digest fragments of mDHFR indicates that Nal is indeed incorporated
at positions encoded by UUU, while Phe is incorporated at positions encoded by
UUC (207). This approach will have important consequences for the engineering
of protein polymers, particularly upon the identification of additional heterologous
aaRS that can charge specific tRNAs with desired nonnatural amino acids.
The versatility of nonnatural amino acid incorporation afforded by overexpression of mutagenized aaRS suggests the potential of evolutionary-based and
rational strategies for the design of aaRS with engineered activity toward specific
nonnatural analogues. Directed evolution strategies developed by Arnold (209) can
be applied to the random mutagenesis of aaRS, followed by appropriate screening
to identify aaRS that can charge its tRNA with a desired nonnatural amino acid.
Such evolutionary strategies have been applied by Schultz and co-workers to the
production of heterologous aaRS that can aminoacylate suppressor tRNAs with
nonnatural amino acids in vivo (158,162–167); similar strategies should be applicable to the design of aaRS/tRNA pairs for multisite incorporation as well. Rational
methods are also potentially powerful approaches to aaRS design. Goddard and
co-workers have recently applied computational methods for the determination
of Phe analogue/PheRS binding energies, and have demonstrated the correlation
between computationally determined binding energies and the incorporation of
the Phe analogues into proteins in vivo (210). Given the expanding kinetic data
now available for a variety of nonnatural amino acid/aaRS pairs, these computational methods can be further developed to allow the rational design of new sets of
amino acid/aaRS pairs that will find application in protein polymer engineering.
The prospects are therefore promising for utilizing both evolutionary and rational
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
187
approaches to engineer the biosynthetic apparatus for the purposeful production
of chemically and physically novel protein polymers.
Prospects and Commercial Viability
The expansion in the design and synthesis of de novo designed protein polymers over the last decade has had important consequences in polymer science.
The genetically directed synthesis of protein polymers has allowed elucidation of
structure-function relationships in protein polymers and has experienced enormous growth as an approach to the design of new polymeric materials, with both
interesting academic and commercial applications. Protein polymers modeled after natural fibrous proteins have shown novel mechanical, assembly, and biological
properties. Artificial protein polymers that exhibit controlled chain folded architectures, liquid crystalline phases, and self-assembled hydrogels on the basis of
sequence and molecular weight control have been produced. The development
of additional artificial protein polymers will offer continued opportunities for the
production of novel materials that display functional groups in a manner that controls materials assembly, mediates interactions with biological targets, and yields
polymers with interesting mechanical, biological, and/or surface properties. The
expansion of the synthetic methods to include the incorporation of a wide array of
nonnatural amino acids also suggests a variety of new strategies for macromolecular synthesis.
In addition to the functional advantages offered by protein-based polymers,
continued interest in exploring their commercial use is motivated by the fact that
the polymers are produced from renewable resources and are environmentally
friendly materials from production through disposal. Practically, the relative cost
and amount of time required to design and synthesize a protein polymer with
desired physical and chemical characteristics can be much higher than the costs
of designing and producing a chemically derived polymer. However, once the protein is designed and methods for its reliable production are defined, the costs
of producing the protein polymer via fermentation is generally independent of
the complexity of the polymer produced, and can therefore be much lower than
the costs of chemical synthesis of complex macromolecules, since chemical synthesis costs increase quickly with increasing polymer complexity and control of
monomer sequence. The costs associated with the biosynthesis of protein-based
polymers depend on the costs of the materials used in synthesis and the scale of
the synthesis; the raw materials are often as simple as glucose or methanol, ammonia, and salts. Larger-scale synthesis is achieved via the use of high cell density
fermentation procedures, in which oxygen, glucose, and pH levels are controlled
to allow high cell density growth in large quantities of culture medium; in the
laboratory, multigram quantities (>25 g) of protein can be produced from E. coli
via such strategies, and yields as high as 15g/L are possible from yeast. Given that
commercial fermentations can be scaled to thousands of liters of culture, yields
of proteins suitable for technological and market applications are possible. Hundreds of tons of protein per year can be produced via technologies currently in
place for the production of amino acids, antibiotics, and protein therapeutics.
188
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
The costs of purification of protein polymers can be high if purification requires multiple chromatographic separations; however, most repetitive protein
polymers have physicochemical properties very different from cellular proteins,
and can be separated from these proteins by selective precipitation methods involving changes in temperature and pH. Similarly, protein polymers whose sequences alone may not permit such simple purification strategies can be expressed
with fusion tags that facilitate purification. Secretion into the culture medium can
also simplify purification and provide opportunities for less complicated largescale syntheses, as expression hosts could be immobilized on a solid support, and
protein removed in a continuous fashion without the requirement to turn over the
culture entirely.
Recombinant enzymes have been produced and purified via these methods
at market costs as low as $23/lb (110); these costs are likely acceptable for protein
polymers with a primary commercial use as a high value biomedical material.
For large commercial materials applications, however, lower cost production and
purification will be required. Additional advances in fermentation, such as the use
of secretion, may help drive costs down, although currently the yields of protein
obtained by protein secretion systems have generally tended to be much lower
than those of the standard intracellular accumulation systems.
Nevertheless, production of protein-based materials continues to progress in
the areas of biomedically related polymers and fibers, and both prokaryotic and eukaryotic expression systems are used commercially. Since 1988, Protein Polymer
Technologies, Inc (PPTI) has been a pioneer in protein polymer design and synthesis, and has extensive patent literature in the area of silk-based and elastin-based
polymeric materials. The high molecular weight, genetically engineered biomaterials are processed into products with properties tailored to specific clinical needs;
over 50 protein polymer sequences have been designed by PPTI for commercial
application, and some can be produced in kilogram quantities. Targeted products
include urethral bulking agents for the treatment of stress urinary incontinence,
dermal augmentation products for cosmetic and reconstructive surgery, tissue
adhesives and sealants, scaffolds for wound healing and tissue engineering, and
depots for local drug delivery. The first commercial product of PPTI combined the
B. mori silk motif with a cell adhesion sequence of fibronectin to yield protein polymers that form autoclavable, stable coatings on plastic and glass via adsorption
of the β-sheet blocks of the polymer; these polymers are now sold as cell-culture
products by Sanyo Chemical Industries. Silk-elastin-like proteins are used for injectable hydrogels; after injection, stable, water-insoluble hydrogels are formed
via β-sheet hydrogen bonding and can be used for both surgical and drug delivery
applications. In addition to medical applications, industrial applications are also of
interest for the protein polymers, and Genencor International, Inc has obtained a
worldwide exclusive license to develop PPTI’s protein polymers in both industrial
and personal care applications. Nexia Biotechnologies Inc manufactures complex
recombinant proteins with industrial and medical applications; its lead product,
BioSteel, is based on recombinant spider-silk proteins, and production of these
proteins from transgenic goats is under continued development. Targeted applications for these materials include medical sutures, surgical meshes, and artificial
ligaments, as well as materials applications such as technical sporting gear (eg,
biodegradable fishing lines), soft body armor, and composites.
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
189
The challenges facing commercial implementation of protein polymers has
not quenched motivation for research and technological progress in this area. In
fact, many important contributions have been published first as patent applications because of their potential commercial significance. Numerous patents have
been awarded, with topics focused on natural protein sequences, construction and
expression of artificial genes/proteins, specific application needs addressed by the
unique properties of protein materials, and the incorporation of nonnatural amino
acids.
BIBLIOGRAPHY
1.
2.
3.
4.
5.
6.
7.
8.
9.
10.
11.
12.
13.
14.
15.
16.
17.
18.
19.
20.
21.
22.
23.
G. Moretti and P. Pino, Polymer 28, 683–692 (1987).
P. Corradini, Macromol. Symp. 89, 1–11 (1995).
M. Szwarc, Nature 178, 1168–1169 (1956).
R. Waack, A. Rembaum, J. D. Coombes, and M. Szwarc, J. Am. Chem. Soc. 79,
2026–2027 (1957).
M. Szwarc and M. VanBeylen, Ionic Polymerization and Living Polymers, Chapman
and Hall, New York, 1993.
R. Faust and J. P. Kennedy, Polym. Bull. 15, 317–323 (1986).
R. R. Schrock, Acc. Chem. Res. 23, 158–165 (1990).
H. H. Brintzinger, D. Fischer, R. Mulhaupt, B. Rieger, and R. M. Waymouth, Angew.
Chem., Int. Ed. Engl. 34, 1143–1170 (1995).
K. Matyjaszewski, K. Davis, T. E. Patten, and M. Wei, Tetrahedron 53, 15321–15329
(1997).
E. L. Dias, T. N. SonBinh, and R. H. Grubbs, J. Am. Chem. Soc. 119, 3887–3897
(1997).
J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne,
G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo, and S. H. Thang, Macromolecules 31,
5559–5562 (1998).
D. M. Lynn, S. Kanaoka, and R. H. Grubbs, J. Am. Chem. Soc. 118, 784–790 (1998).
T. J. Deming, Nature 390, 386–389 (1997).
M. Cheng, A. B. Attygalle, E. B. Lobkovsky, and G. W. Coates, J. Am. Chem. Soc. 121,
11583–11584 (1999).
J. Louie, C. W. Bielawski, and R. H. Grubbs, J. Am. Chem. Soc. 123, 11312–11313
(2001).
J. Parker, Microbiol. Rev. 53, 273–298 (1989).
G. H. Altman, F. Diaz, C. Jakuba, T. Calabro, R. L. Horan, J. Chen, H. Lu, J. Richmond,
and D. L. Kaplan, Biomaterials 24, 401–416 (2003).
J. Gosline, M. Lillie, E. Carrington, P. Guerette, C. Ortlepp, and K. Savage, Philos.
Trans. R. Soc. London, B 357, 121–132 (2002).
A. Chilkoti, M. R. Dreher, and D. E. Meyer, Adv. Drug Deliv. Rev. 54, 1093–1111
(2002).
U.S. Pat. 5,149,657 (Sept. 22, 1992), K. J. Maugh and D. M. Anderson (to Enzon Labs,
Inc.).
J. P. O’Brien, R. H. Hoess, K. H. Gardner, R. L. Lock, Z. R. Wasserman, P. C. Wever,
and F. R. Salemme, in D. Kaplan, W. W. Adams, B. Farmer, and C. Viney, eds., Silk
Polymers, American Chemical Society, Washington, D.C., 1994.
C. Wang, R. J. Stewart, and J. Kopecek, Nature 397, 417–420 (1999).
L. Chen, J. Kopecek, and R. J. Stewart, Bioconjugate Chem. 11, 734–740
(2000).
190
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
24. S. Halstenberg, A. Panitch, S. Rizzi, H. Hall, and J. A. Hubbell, Biomacromolecules 3,
710–723 (2002).
25. S. Arcidiacono, C. Mello, D. L. Kaplan, S. Cheley, and H. Bayley, Appl. Microbiol.
Biotechnol. 49, 31–38 (1998).
26. J. Cappello, J. Crissman, M. Dorman, M. Mikolajczak, G. Textor, M. Marquet, and
F. Ferrari, Biotechnol. Prog. 6, 198–202 (1990).
27. J. P. Anderson, J. Cappello, and D. C. Martin, Biopolymers 34, 1049–1058 (1994).
28. T. Asakura, H. Kato, J. Yao, R. Kishore, and M. Shirai, Polymer J. 34, 936–943 (2002).
29. World Patent 9429450 (Dec. 22, 1994), S. R. Fahnestock, (to E.I. du Pont de Nemours
& Co.).
30. S. R. Fahnestock and S. L. Irwin, Appl. Microbiol. Biotechnol. 47, 23–32 (1997).
31. U.S. Pat. 6,268,169 (July 31, 2001), S. R. Fahnestock (to E. I. du Pont de Nemours &
Co.).
32. S. R. Fahnestock, Z. Yao, and L. A. Bedzyk, Rev. Mol. Biotechnol. 74, 105–119 (2000).
33. S. R. Fahnestock, and L. A. Bedzyk, Appl. Microbiol. Biotechnol. 47, 33–39 (1997).
34. J. T. Prince, K. P. McGrath, C. M. DiGirolamo, and D. L. Kaplan, Biochemistry 34,
10879–10884 (1995).
35. Y. Fukushima, Biopolymers 45, 269–279 (1998).
36. E. E. Hood and J. M. Jilka, Curr. Opin. Biotechnol. 10, 382–386 (2000).
37. J. Scheller, K.-H. Guhrs, F. Grosse, and U. Conrad, Nature Biotechnol. 19, 573–577
(2001).
38. A. Lazaris, S. Arcidiacono, Y. Huang, J.-F. Zhou F. Duguay, N. Chretien, E. A. Welsh,
J. W. Soares, and C. N. Karatzas, Science 295, 472–476 (2002).
39. U.S. Pat. 2001042255 (Nov. 15, 2001), A. L. Karatzas, C. N. Karatzas, and J. D. Turner
(to Nexia Biotechnologies, Inc.).
40. S. Arcidiacono, C. M. Mello, M. Butler, E. Welsh, J. W. Soares, A. Allen, D. Ziegler,
T. Laue, and S. Chase, Macromolecules 35, 1262–1266 (2002).
41. S. Winkler, S. Szela, P. Avtges, R. Valluzzi, D. A. Kirschner, and D. Kaplan, Int. J.
Biol. Macromol. 24, 265–270 (1999).
42. R. Valluzzi, S. Szela, P. Avtges, D. Kirschner, and D. Kaplan, J. Phys. Chem. B. 103,
11382–11392 (1999).
43. S. Szela, P. Avtges, R. Valluzzi, S. Winkler, D. Wilson, D. Kirschner, and D. L. Kaplan,
Biomacromolecules 1, 534–542 (2000).
44. S. Winkler, D. Wilson, and D. L. Kaplan, Biochemistry 39, 12739–12746 (2000).
45. T. Asakura, K. Nitta, M. Yang, J. Yao, Y. Nakazawa, and D. L. Kaplan, Biomacromolecules 4, 815–820 (2003).
46. Y. T. Zhou, S. X. Wu, and V. P. Conticello, Biomacromolecules 2, 111–125 (2001).
47. Y. Qu, S. C. Payne, R. P. Apkarian, and V. P. Conticello, J. Am. Chem. Soc. 122, 5014–
5015 (2000).
48. A. Lamberg, T. Helaakoski, J. Myllyharju, S. Peltonen, H. Notbohm, T. Pihlajaniemi,
and K. I. Kivirikko, J. Biol. Chem. 271, 11988–11995 (1996).
49. M. Nokelainen, T. Helaakoski, J. Myllyharju, H. Notbohm, T. Pihlajaniemi, P. P. Fietzek, and K. I. Kivirikko, Matrix Biol. 16, 329–338 (1998).
50. A. Vuorela, J. Myllyharju, R. Nissi, T. Pihlajaniemi, and K. I. Kivirikko, EMBO J. 16,
6702–6712 (1997).
51. D. C. A. John, R. Watson, A. J. Kind, A. R. Scott, K. E. Kadler, and N. J. Bulleid, Nature
Biotechnol. 17, 385–389 (1999).
52. P. D. Toman, F. Pieper, N. Sakai, C. Karatzas, E. Platenburg, I. de Wit, C. Samuel,
A. Dekker, G. A. Daniels, R. A. Berg, and G. J. Platenburg, Transgenic Res. 8, 415–427
(1999).
53. M. W. T. Werten, T. J. van den Bosch, R. D. Wind, H. Mooibroek, and F. A. de Wolf,
Yeast 15, 1087–1096 (1999).
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
191
54. E. C. de Bruin, M. W. T. Werten, C. Laane, and F. A. de Wolf, FEMS Yeast Res. 1,
291–298 (2002).
55. J. Myllyharju, A. Lamberg, H. Notbohm, P. P. Fietzek, T. Pihlajaniemi, and K. I.
Kivirikko, J. Biol. Chem. 272, 21824–21830 (1997).
56. U.S. Pat. 5,670,616 (Sept. 23, 1997), S. C. Weber and J. A. McElver (to Eastman Kodak
Co.).
57. T. Kajino, H. Takahashi, M. Hirai, and Y. Yamada, Appl. Environ. Microbiol.
66, 304–309 (2000).
58. M. W. T. Werten, W. H. Wisselink, T. J. Jansen-van den Bosch, E. C. de Bruin, and
F. A. de Wolf, Protein Eng. 14, 447–454 (2001).
59. I. Goldberg, A. J. Salerno, T. Patterson, and J. I. Williams, Gene 80, 305–314 (1989).
60. I. Goldberg and A. J. Salerno, in P. C. Rieke, P. D. Calvert, and M. Alper, eds., Materials
Synthesis Utilizing Biological Processes, Materials Research Society, Pittsburgh, Pa.,
1990, pp. 229–236.
61. F. A. Ferrari and J. Cappello, in K. McGrath and D. Kaplan, eds., Protein Based
Materials, Birkhauser, Boston, 1997, pp. 37–60.
62. U.S. Pat. 5,773,249 (June 30, 1998), J. Cappello and F. A. Ferrari (to Protein Polymer
Technologies, Inc.).
63. J. Yin, J. Lin, W. Li, and D. I. C. Wang, J. Biotechnol. 100, 181–191 (2003).
64. D. T. McPherson, C. Morrow, D. S. Minehan, J. Wu, E. Hunter, and D. W. Urry, Biotechnol. Prog. 8, 347–352 (1992).
65. C. Guda, X. Shang, D. T. McPherson, J. H. Cherry, D. W. Urry, and H. Daniell, Biotechnol. Lett. 17, 745–750 (1995).
66. A. Panitch, T. Yamaoka, M. J. Fournier, T. L. Mason, and D. A. Tirrell, Macromolecules
32, 1701–1703 (1999).
67. R. A. McMillan and V. P. Conticello, Macromolecules 33, 4809–4821 (2000).
68. D. E. Meyer and A. Chilkoti, Biomacromolecules 3, 357–367 (2002).
69. R. W. Herzog, N. K. Singh, D. W. Urry, and H. Daniell, Appl. Microbiol. Biotechnol.
47, 368–372 (1997).
70. C. Guda, S. B. Lee, and H. Daniell, Plant Cell Rep. 19, 257–262 (2000).
71. X. Zhang, D. W. Urry, and H. Daniell, Plant Cell Rep. 16, 174–179 (1996).
72. R. A. McMillan, T. A. T. Lee, and V. P. Conticello, Macromolecules 32, 3643–3648
(1999).
73. D. W. Urry, A. Q. Peng, L. C. Hayes, D. McPherson, J. Xu, T. C. Woods, D. C. Gowda,
and A. Pattanaik, Biotech. Bioeng. 58, 175–190 (1998).
74. D. W. Urry, Prog. Biophys. Mol. Biol. 57, 23–57 (1992).
75. D. W. Urry, A. Pattanaik, M. A. Accavitti, C.-X. Luan, D. T. McPherson, J. Xu, D. C.
Gowda, T. M. Parker, C. M. Harris, and J. Naijie, in A. J. Domb, J. Kost, and D. M.
Wiseman, eds., Drug Targeting and Delivery, Handbook of Biodegradable Polymers,
Vol. 7, Harwood Academic Publishers, Amsterdam, 1997, pp. 367–386.
76. D. E. Meyer, B. C. Shin, G. A. Kong, M. W. Dewhirst, and A. Chilkoti, J. Controlled
Release 74, 213–224 (2001).
77. E. R. Wright and V. P. Conticello, Adv. Drug Delivery Rev. 54, 1057–1073 (2002).
78. E. R. Wright, R. A. McMillan, A. Cooper, R. P. Apkarian, and V. P. Conticello, Adv.
Funct. Mater. 12, 149–154 (2002).
79. T. A. T. Lee, A. Cooper, R. P. Apkarian, and V. P. Conticello, Adv. Mater. 12, 1105–1110
(2000).
80. J. Lee, C. W. Macosko, and D. W. Urry, Biomacromolecules 2, 170–179 (2001).
81. K. Trabbic-Carlson, L. A. Setton, and A. Chilkoti, Biomacromolecules 4, 572–580
(2003).
82. Z. Megeed, J. Cappello, and H. Ghandehari, Adv. Drug Delivery Rev. 54, 1075–1091
(2002).
192
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
83. A. Nagarsekar, J. Crissman, M. Crissman, F. Ferrari, J. Cappello, and H. Ghandehari,
J. Biomed. Mater. Res. 62, 195–203 (2002).
84. A. Nagarsekar, J. Crissman, M. Crissman, F. Ferrari, J. Cappello, and H. Ghandehari,
Biomacromolecules 4, 602–607 (2003).
85. J. Cappello, J. W. Crissman, M. Crissman, F. A. Ferrari, G. Textor, O. Wallis, J. R.
Whitledge, X. Zhou, D. Burman, L. Aukerman, and E. R. Stedronsky, J. Controlled
Release 53, 105–117 (1998).
86. A. A. Dinerman, J. Cappello, H. Ghandehari, and S. W. Hoag, Biomaterials 23,
4203–4210 (2002).
87. Z. Megeed, J. Cappello, and H. Ghandehari, Pharm. Res. 19, 954–959 (2002).
88. E. R. Welsh and D. A. Tirrell, Biomacromolecules 1, 23–30 (2000).
89. K. Di Zio and D. A. Tirrell, Macromolecules 36, 1553–1558 (2003).
90. M. T. Krejchi, E. D. T. Atkins, A. J. Waddon, M. J. Fournier, T. L. Mason, and D. A.
Tirrell, Science 265, 1427–1432 (1994).
91. E. J. Cantor, E. D. Atkins, S. J. Cooper, M. J. Fournier, T. L. Mason, and D. A. Tirrell,
J. Biochem. 122, 217–225 (1997).
92. S. M. Yu, V. Conticello, G. Zhang, C. Kayser, M. J. Fournier, T. L. Mason, and D. A.
Tirrell, Nature 389, 187–190 (1997).
93. S. M. Yu, C. M. Soto, and D. A. Tirrell, J. Am. Chem. Soc. 122, 6552–6559
(2000).
94. G. Zhang, M. J. Fournier, T. L. Mason, and D. A. Tirrell, Macromolecules 25, 3601–3603
(1992).
95. W. A. Petka, J. L. Hardin, K. P. McGrath, D. Wirtz, and D. A. Tirrell, Science 281,
389–392 (1998).
96. R. S. Farmer and K. L. Kiick, manuscript in preparation.
97. J.-I. Won and A. E. Barron, Macromolecules 35, 8281–8287 (2002).
98. E. Yoshikawa, M. J. Fournier, T. L. Mason, and D. A. Tirrell, Macromolecules 27,
5471–5475 (1994).
99. M. J. Dougherty, S. Kothakota, T. L. Mason, D. A. Tirrell, and M. J. Fournier, Macromolecules 26, 1779–1781 (1993).
100. K. P. McGrath, M. J. Fournier, T. L. Mason, and D. A. Tirrell, J. Am. Chem. Soc. 114,
727–733 (1992).
101. Y. Tsujimoto and Y. Suzuki, Cell 18, 591–600 (1979).
102. M. Xu and R. V. Lewis, Proc. Natl. Acad. Sci. USA 87, 7120–7124 (1990).
103. J. D. van Beek, S. Hess, F. Vollrath, and B. H. Meier, Proc. Natl. Acad. Sci. U.S.A. 99,
10266–10271 (2002).
104. F. Vollrath and D. P. Knight, Nature 410, 541–548 (2001).
105. U.S. Pat. 6,380,154 (Apr. 30, 2002), J. Cappello and E. R. Stedronsky (to Protein Polymer Technologies, Inc.).
106. R. V. Lewis, M. Hinman, S. Kothakota, and M. J. Fournier, Protein Expr. Purif. 7,
400–406 (1996).
107. World Pat. 03057720 (July 17, 2003), C. N. Karatzas, Y. Huang, and C. Turcotte (to
Nexia Biotechnologies, Inc.).
108. World Pat. 03057727 (July 17, 2003), C. N. Karatzas and C. Turcotte (to Nexia Biotechnologies, Inc.).
109. World Pat. 03060099 (July 24, 2003), C. N. Karatzas, A. Rodenheiser, A. Alwattari,
and S. Islam (to Nexia Biotechnologies, Inc.).
110. S. Fahnestock, in S. Fahnestock and A. Steinbuchel, eds., Biopolymers, Vol. 8:
Polyamides and Complex Proteinaceous Materials II, Wiley-VCH, Weinheim, 2003,
pp. 47–79.
111. S. Zhang, T. Holmes, C. Lockshin, and A. Rich, Proc. Natl. Acad. Sci. U.S.A. 90,
3334–3338 (1993).
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
193
112. K. E. Kadler, D. F. Holmes, J. A. Trotter, and J. A. Chapman, Biochem. J. 316, 1–11
(1996).
113. World Pat. 9307889 (Apr. 29, 1993), D. J. Prockop, L. Ala-Kokko, A. Fertala, A. Sieron,
K. I. Kivirikko, and A. Geddis (to Thomas Jefferson University).
114. A. Fichard, E. Tillet, F. Delacoux, R. Garrone, and F. Ruggiero, J. Biol. Chem. 272,
30083–30087 (1997).
115. World Pat. 9818918 (May 7, 1998), P. R. Vaughan, M. Galanis, J. A. M. Ramshaw, and J.
A. Werkmeister (to Commonwealth Scientific and Industrial Research Organisation).
116. P. D. Toman, G. Chisholm, H. McMullin, L. M. Giere, D. R. Olsen, R. J. Kovach, S. D.
Leigh, B. E. Fong, R. Chang, G. A. Daniels, R. A. Berg, and R. A. Hitzemann, J. Biol.
Chem. 275, 23303–23309 (2000).
117. D. Olsen, S. D. Leigh, R. Chang, H. McMullin, W. Ong, E. Tai, G. Chisholm, D. E. Birk,
R. A. Berg, R. A. Hitzeman, and P. D. Toman, J. Biol. Chem. 276, 24038–24043 (2001).
118. U.S. Pat. 5,895,833 (Apr. 20, 1999), R. A. Berg (to Cohesion Technologies, Inc.).
119. F. Ruggiero, J.-Y. Exposito, P. Bournat, V. Gruber, S. Perret, J. Comte, B. Olagnier,
R. Garrone, and M. Theisen, FEBS Lett. 469, 132–136 (2000).
120. S. Perret, C. Merle, S. Bernocco, P. Berland, R. Garrone, D. J. S. Hulmes, M. Theisen,
and F. Ruggiero, J. Biol. Chem. 276, 43693–43698 (2001).
121. M. Nokelainen, H. Tu, A. Vuorela, H. Notbohm, K. I. Kivirikko, and J. Myllyharju,
Yeast 18, 797–806 (2001).
122. World Pat. 9714431 (Apr. 24, 1997), P. D. Toman, R. A. Berg, G. A. Daniels, R. A.
Hitzeman, and G. E. Chisholm (to Collagen Corporation and Genotypes, Inc.).
123. World Pat. 9738710 (Oct. 23, 1997), K. I. Kivirikko and T. Pihlajaniemi (to Fibrogen,
Inc.; Academy of Finland).
124. A. Fertala, W. B. Han, and F. K. Ko, J. Biomed. Mater. Res. 57, 48–58 (2001).
125. W. V. Arnold, A. L. Sieron, A. Fertala, H. P. Gachinger, D. Mechling, and D. J. Prockop,
Matrix Biol. 16, 105–116 (1997).
126. M. Hayashi, M. Tomita, and K. Yoshizato, Biochim. Biophys. Acta 1528, 187–195
(2001).
127. L. Debelle and A. M. Tamburro, Int. J. Biochem. Cell. Biol. 31, 261–272 (1999).
128. D. W. Urry, T. M. Parker, M. C. Reid, and D. C. Gowda, J. Bioact. Compat. Polym. 6,
263–282 (1991).
129. H. Reiersen, A. R. Clarke, and A. R. Rees, J. Mol. Biol. 283, 255–264 (1998).
130. D. W. Urry, J. Phys. Chem. B. 101, 11007–11028 (1997).
131. H. Betre, L. A. Setton, D. E. Meyer, and A. Chilkoti, Biomacromolecules 3, 910–916
(2002).
132. G. V. R. Rao, S. Balamurugan, D. E. Meyer, A. Chilkoti, and G. P. Lopez, Langmuir
18, 1819–1824 (2002).
133. N. Nath and A. Chilkoti, Anal. Chem. 75, 709–715 (2003).
134. J. Lee, C. W. Macosko, and D. W. Urry, Macromolecules 34, 5968–5974 (2001).
135. L. Huang, R. A. McMillan, R. P. Apkarian, B. Pourdeyhimi, V. P. Conticello, and E. L.
Chaikof, Macromolecules 33, 2989–2997 (2000).
136. K. Nagapudi, W. T. Brinkman, J. E. Leisen, L. Huang, R. A. McMillan, R. P. Apkarian,
V. P. Conticello, and E. L. Chaikof, Macromolecules 35, 1730–1737 (2002).
137. J. M. Yao and T. Asakura, J. Biochem. 133, 147–154 (2003).
138. A. Dinerman, J. Cappello, H. Ghandehari, and S. Hoag, J. Controlled Release 82,
277–287 (2002).
139. A. Megeed, M. Haider, D. Li, J. B. W. O’Malley, J. Cappello, and H. Ghandehari,
J. Controlled Release 94, 433–445 (2004).
140. M. Haider, Z. Megeed, and H. Ghandehari, J. Controlled Release 95, 1–26 (2004).
141. Z. Megeed, H. Ghandehari, in M. Amiji, ed., Polymeric Gene Delivery: Principles and
Applications, CRC Press, Boca Raton, Fla., in press.
194
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
142. A. Nicol, D. C. Gowda, T. M. Parker, and D. W. Urry, in C. Gebelein and
C. Carraher, eds., Biotechnology and Bioactive Polymers, Plenum, New York, 1994,
p. 95.
143. S. C. Heilshorn, K. A. DiZio, E. R. Welsh, and D. A. Tirrell, Biomaterials 24, 4245–4252
(2003).
144. M. T. Krejchi, S. J. Cooper, Y. Deguchi, E. D. T. Atkins, M. J. Fournier, T. L. Mason,
and D. A. Tirrell, Macromolecules 30, 5012–5024 (1997).
145. C. C. Chen, M. T. Krejchi, D. A. Tirrell, and S. L. Hsu, Macromolecules 28, 1464–1469
(1995).
146. N. L. Goeden-Wood, V. P. Conticello, S. L. Muller, and J. D. Keasling, Biomacromolecules 3, 874–879 (2002).
147. N. L. Goeden-Wood, J. D. Keasling, and S. J. Muller, Macromolecules 36, 2932–2938
(2003).
148. S.-J. He, C. Lee, S. P. Gido, S. M. Yu, and D. A. Tirrell, Macromolecules 31, 9387–9389
(1998).
149. K. P. McGrath and D. L. Kaplan, Macromol. Symp. 77, 183–189 (1994).
150. K. P. McGrath, M. M. Butler, C. M. DiGirolamo, D. L. Kaplan, W. A. Petka, and T. M.
Laue, J. Bioact. Compat. Polym. 15, 334–356 (2000).
151. S. B. Kennedy, E. R. deAzevedo, W. A. Pekta, T. P. Russell, D. A. Tirrell, and M. Hong,
Macromolecules 34, 8675–8685 (2001).
152. R. B. Merrifield, Pure Appl. Chem. 50, 643–653 (1978).
153. S. M. Hecht, B. L. Alford, Y. Kuroda, and S. Kitano, J. Biol. Chem. 253, 4517–4520
(1978).
154. C. J. Noren, S. J. Anthony-Cahill, M. C. Griffith, and P. G. Schultz, Science 244,
182–188 (1989).
155. J. D. Bain, C. G. Glabe, T. A. Dix, and A. R. Chamberlin, J. Am. Chem. Soc. 111,
8013–8014 (1989).
156. V. W. Cornish, D. Mendel, and P. Schultz, Angew. Chem., Int. Ed. Engl. 34, 621–633
(1995).
157. T. Hohsaka, Y. Ashizuka, H. Sasaki, H. Murakami, and M. Sisido, J. Am. Chem. Soc.
121, 12194–12195 (1999).
158. D. R. Liu, T. J. Magliery, M. Pastrnak, and P. G. Schultz, Proc. Natl. Acad. Sci. U.S.A.
94, 10092–10097 (1997).
159. S. Ohno, T. Yokogawa, I. Fujii, H. Asahara, H. Inokuchi, and K. Nishikawa, J. Biochem.
124, 1065–1068 (1998).
160. R. Furter, Protein Sci. 7, 419–426 (1998).
161. A. K. Kowal, C. Kohrer, and U. L. RajBhandary, Proc. Natl. Acad. Sci. U.S.A. 98,
2268–2273 (2001).
162. A. Zhang, L. Wang, A. Brock, and P. G. Schultz, Angew. Chem., Int. Ed. Engl. 41,
2840–2842 (2002).
163. J. W. Chin, T. A. Cropp, J. C. Anderson, M. Mukherji, Z. Zhang, and P. G. Schultz,
Science 301, 964–967 (2003).
164. L. Wang, Z. Zhang, A. Brock, and P. G. Schultz, Proc. Natl. Acad. Sci. U.S.A. 100,
56–61 (2003).
165. A. Dieters, T. A. Cropp, M. Mukherji, J. W. Chin, J. C. Anderson, and P. G. Schultz,
J. Am. Chem. Soc. 125, 11782–11783 (2003).
166. L. Alfonta, Z. Zhang, S. Uryu, J. A. Loo, and P. G. Schultz, J. Am. Chem. Soc. 125,
14662–14663 (2003).
167. R. A. Mehl, J. C. Anderson, S. W. Santoro, L. Wang, A. B. Martin, D. S. King, D. M.
Horn, and P. G. Schultz, J. Am. Chem. Soc. 125, 935–939 (2003).
168. T. Tuve and H. Williams, J. Am. Chem. Soc. 79, 5830–5831 (1957).
169. D. B. Cowie and G. N. Cohen, Biochim. Biophys. Acta 26, 252–261 (1957).
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
195
170. D. B. Cowie, G. N. Cohen, E. T. Bolton, and H. D. Robichon-Szulmajster, Biochim.
Biophys. Acta 34, 39–46 (1959).
171. E. D. Fenster and H. S. Anker, Biochemistry 8, 269–274 (1969).
172. M. H. Richmond, J. Mol. Biol. 6, 284–294 (1963).
173. T. J. Deming, M. J. Fournier, T. L. Mason, and D. A. Tirrell, J. Macromol. Sci., Pure
Appl. Chem. A34, 2143–2150 (1997).
174. K. L. Kiick, E. Saxon, D. A. Tirrell, and C. R. Bertozzi, Proc. Natl. Acad. Sci. U.S.A.
99, 19–24 (2002).
175. D. Datta, P. Wang, I. S. Carrico, S. L. Mayo, and D. A. Tirrell, J. Am. Chem. Soc. 124,
5652–5653 (2002).
176. Y. Tang, G. Ghirlanda, W. A. Petka, T. Nakajima, W. F. DeGrado, and D. A. Tirrell,
Angew. Chem., Int. Ed. Engl. 40, 1494–1496 (2001).
177. N. Sharma, R. Furter, P. Kast, and D. A. Tirrell, FEBS Lett. 467, 37–40 (2000).
178. K. Kirshenbaum, I. S. Carrico, and D. A. Tirrell, Chem. BioChem. 2–3, 235–237
(2002).
179. W. A. Hendrickson, J. R. Horton, and D. M. LeMaster, EMBO J. 9, 1665–1672 (1990).
180. N. Budisa, B. Steipe, P. Demange, C. Eckerskorn, J. Kellermann, and R. Huber, Eur.
J. Biochem. 230, 788–796 (1995).
181. H. Duewel, E. Daub, V. Robinson, and J. F. Honek, Biochemistry 36, 3404–3416 (1997).
182. J. C. M. van Hest and D. A. Tirrell, FEBS Lett. 428, 68–70 (1998).
183. J. C. M. van Hest, K. L. Kiick, and D. A. Tirrell, J. Am. Chem. Soc. 122, 1282–1288
(2000).
184. A. J. Link and D. A. Tirrell, J. Am. Chem. Soc. 125, 11164–11165 (2003).
185. P. Wang, Y. Tang, and D. A. Tirrell, J. Am. Chem. Soc. 125, 6900–6906 (2003).
186. A. B. Mauger and B. Witkop, Chem. Rev. 66, 47–86 (1966).
187. T. J. Deming, M. J. Fournier, T. L. Mason, and D. A. Tirrell, Macromolecules
29, 1442–1444 (1996).
188. T. Michon, F. Barbot, and D. Tirrell, Royal Soc. Chem. 276, 63–72 (2002).
189. M. Mock, T. Michon, and D. A. Tirrell, Polym. Prep. 44, 1065–1066 (2003).
190. D. D. Buechter, D. N. Paolella, G. S. Leslie, M. S. Brown, K. A. Mehos, and E. A.
Gruskin, J. Biol. Chem. 278, 645–650 (2003).
191. J. L. Milner, S. Grothe, and J. M. Wood, J. Biol. Chem. 263, 14900–14905 (1988).
192. S. Kothakota, T. L. Mason, D. A. Tirrell, and M. J. Fournier, J. Am. Chem. Soc. 117,
536–537 (1995).
193. D. W. Flanagan and D. A. Tirrell, Polym. Prep. 44, 892–893 (2003).
194. K. L. Kiick and D. A. Tirrell, Tetrahedron 56, 9487–9493 (2000).
195. K. L. Kiick, R. Weberskirch, and D. A. Tirrell, FEBS Lett. 502, 25–30 (2001).
196. K. L. Kiick, J. C. M. van Hest, and D. A. Tirrell, Angew. Chem., Int. Ed. Engl. 39,
2148–2152 (2000).
197. Y. Tang and D. A. Tirrell, J. Am. Chem. Soc. 123, 11089–11090 (2001).
198. D. A. Tirrell, Y. Tang, and I. S. Carrico, Abstr. Pap. Am. Chem. Soc. 222, U45–U45
(2001).
199. G. D. Gay, H. W. Duckworth, and A. R. Fersht, FEBS Lett. 318, 167–171 (1993).
200. F. Agou, S. Quevillon, P. Kerjan, and M. Mirande, Biochemistry 37, 11309–11314
(1998).
201. P. Kast and H. Hennecke, J. Mol. Biol. 222, 99–124 (1991).
202. M. Ibba and H. Hennecke, FEBS Lett. 364, 273–275 (1995).
203. L. Reshetnikova, N. Moor, O. Lavrik, and D. G. Vassylyev, J. Mol. Biol. 287, 555–568
(1999).
204. F. Hamano-Takaku, T. Iwama, S. Saito-Yano, K. Takaku, Y. Monden, M. Kitabatake,
D. Soll, and S. N., J. Biol. Chem. 275, 40324–40328 (2000).
205. Y. Tang and D. A. Tirrell, Biochemistry 41, 10635–10645 (2002).
196
GENETIC METHODS OF POLYMER SYNTHESIS
Vol. 10
206. V. Doring, H. D. Mootz, L. A. Nangle, T. L. Hendrickson, V. de Crecy-Lagard, P. Schimmel, and P. Marliere, Science 292, 501–504 (2001).
207. I. Kwon, K. Kirshenbaum, and D. A. Tirrell, J. Am. Chem. Soc. 125, 7512–7513
(2003).
208. Chem. Eng. News 40–43 (June 23, 2003).
209. F. H. Arnold, Chem. Eng. Sci. 51, 5091–5102 (1996).
210. P. Wang, N. Vaidehi, D. A. Tirrell, and W. A. I. Goddard, J. Am. Chem. Soc. 124,
14442–14449 (2002).
READING LIST
L. Debelle and A. M. Tamburro, Int. J. Biochem. Cell Biol. 31, 261–272 (1999).
S. Fahnestock, in S. Fahnestock and A. Steinbüchel, eds., Biopolymers, Vol. 8: Polyamides
and Complex Proteinaceous Materials II, Wiley-VCH, Weinheim, 2003, pp. 47–79.
C. W. P. Foo and D. L. Kaplan, Adv. Drug Delivery Rev. 54, 1131–1143 (2002).
R. W. Old and S. B. Primrose, Principles of Gene Manipulation, 5th ed., Blackwell Science,
Cambridge, Mass., 1994.
K. McGrath and D. Kaplan, eds., Protein Based Materials, Birkhauser, Boston, 1997.
J. Sambrook and D. W. Russell, Molecular Cloning: A Laboratory Manual, 3rd ed., Cold
Spring Harbor Press, Cold Spring Harbor, N.Y., 2001.
R. K. Scopes, Protein Purification: Principles and Practice, 3rd ed., Springer, New York,
1994.
J. C. M. van Hest and D. A. Tirrell, Chem. Commum. 1897–1904 (2001).
KRISTI L. KIICK
University of Delaware
List of Abbreviations and Symbols
Natural amino acids, three-letter and one-letter abbreviations
Ala, A
alanine
Arg, R
arginine
Asn, N
asparagine
Asp, D
aspartic acid
Cys, C
cysteine
Gln, Q
glutamine
Glu, E
glutamic acid
Gly, G
glycine
His, H
histidine
Ile, I
isoleucine
Leu, L
leucine
Lys, K
lysine
Met, M
methionine
Phe, F
phenylalanine
Pro, P
proline
Ser, S
serine
Thr, T
threonine
Trp, W
tryptophan
Vol. 10
GENETIC METHODS OF POLYMER SYNTHESIS
Tyr, Y
tyrosine
Val, V
valine
Other abbreviations and symbols
AAA
adenine adenine adenine
CCU
cytosine cytosine uracil
cDNA
complementary deoxyribonucleic acid
CGA
cytosine guanine adenine
DNA
deoxyribonucleic acid
GAA
guanine adenine adenine
GCU
guanine cytosine uracil
GGA
guanine guanine adenine
kcat
turnover number
Michaelis constant
Km
mRNA
messenger ribonucleic acid
RNA
ribonucleic acid
SDS–PAGE sodium dodecyl sulfate–polyacrylamide gel electrophoresis
tRNA
transfer ribonucleic acid
UUC
uracil uracil cytosine
UUU
uracil uracil uracil
GLASS TRANSITION.
GRAFT COPOLYMERS.
GREEN PLASTICS.
See Volume 2.
See Volume 6.
See ENVIRONMENTALLY DEGRADABLE POLYMER.
197