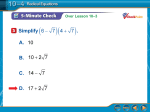
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Woodward–Hoffmann rules wikipedia , lookup
Cracking (chemistry) wikipedia , lookup
George S. Hammond wikipedia , lookup
Fischer–Tropsch process wikipedia , lookup
Discodermolide wikipedia , lookup
Kinetic resolution wikipedia , lookup
Enantioselective synthesis wikipedia , lookup
Ene reaction wikipedia , lookup
Asymmetric hydrogenation wikipedia , lookup
Tiffeneau–Demjanov rearrangement wikipedia , lookup
Asymmetric induction wikipedia , lookup
Polythiophene wikipedia , lookup
Elias James Corey wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Petasis reaction wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
Stille reaction wikipedia , lookup
Hydrogenation wikipedia , lookup
Hydroformylation wikipedia , lookup
Barton Deoxygenation Radical-induced deoxygenation of O-thiocarbonate derivatives of alcohols in the presence of hydrogen-atom donors is a versatile and widely-used method for the preparation of an alkane from the corresponding alcohol. The Barton deoxygenation is a two-step process. In the initial step, the alcohol is acylated to generate an O-thiocarbonate derivative, which is then typically reduced by heating in an aprotic solvent in the presence of a hydrogen-atom donor. The method has been adapted for the deoxygenation of primary, secondary, and tertiary alcohols. In addition, monodeoxygenation of 1,2- and 1,3-diols has been achieved. www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf http://www.organic-chemistry.org/namedreactions/barton-mccombie-reaction.shtm Barton Deoxygenation - Mechanism Mechanism of this reduction proceeds by attack of a tin radical on the thiocarbonyl sulfur atom. Subsequent fragmentation of this intermediate generates an alkyl radical which propagates the chain. Initiation The catalytic cycle, in which low concentration of .SnBu3 effects the reaction: www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf http://www.organic-chemistry.org/namedreactions/barton-mccombie-reaction.shtm Applications In the following example, the radical generated during the deoxygenation reaction undergoes 6-exo-trig radical cyclization. (Baldwin's Rules for Ring Closure) Tin-Free Barton-Type Reduction Employing Water as a Hydrogen Atom Source: Simple concentration of the reaction mixture provides products in high purity. Trialkylborane acts as both the radical initiator and an activator of water prior to hydrogen www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Barton Decarboxylation O-Esters of thiohydroxamic acids are reduced in a radical chain reaction by tin hydride reagents. These are typically prepared by the reaction of commercial N-hydroxypyridine-2thione with activated carboxylic esters. Proceed via free radical pathway Barton, D. H. R.; Circh, D.; Motherwell, W. B. J. Chem. Soc., Chem. Commun. 1983, 939-941. www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Barton Decarboxylation - Mechanism The initiation of the Barton Decarboxylation ( Bu3Sn-H -> Bu3Sn. ) is effected with a radical initiator, and as with the BartonMcCombie Deoxygenation, the driving force for the reaction itself is the formation of the stable S-Sn bond. In addition, Barton esters can also be cleaved photolytically or thermally: If an excess of a suitable radical trapping agent is present in the reaction medium, substitution will occur; otherwise, radical recombination takes place to give the pyridyl sulfide: http://www.organic-chemistry.org/namedreactions/barton-decarboxylation.shtm Diazene-Mediated Deoxygenation Deoxygenation proceeds by Mitsunobu displacement of the alcohol with onitrobenzenesulfonylhydrazine (NBSH) followed by in situ elimination of o-nitrobenzene sulfinic acid. The resulting monoalkyl diazene is proposed to decompose by a freeradical mechanism to form deoxygenated products. The deoxygenation is carried out in a single step without using metal hydride reagents. The method is found to work well for unhindered alcohols, but sterically encumbered andβ- oxygenated alcohols fail to undergo the Mitsunobu displacement and are recovered unchanged from the reaction mixture. www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Applications In the following example, the radical generated from decomposition of the diazene intermediate underwent a rapid 5-exo-trig radical cyclization. This generated a second radical that was trapped with oxygen to provide the cyclic carbinol shown after work-up with methyl sulfide. Myers, A. G.; Movassaghi, M.; Zheng, B. J. Am. Chem. Soc. 1997, 119, 8572-8573. www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Deoxygenation via Alkyl Tosylates p-Toluenesulfonate ester derivatives of alcohols are reduced to the corresponding alkanes with certain powerful metal hydrides. Among hydride sources, lithium triethylborohydride (Super Hydride, LiEt3BH) has been shown to rapidly reduce alkyl tosylates efficiently, even thoes derived from hindered alcohols. Krishnamurthy, S.; Brown, H. C. J. Org. Chem. 1976, 41, 3064-3066 Evans, D. A.; Dow, R. L.; Shih, T. L.; Takacs, J. M.; Zahler, R. J. Am. Chem. Soc. 1990, 112, 5290-5313. www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Advancement In related studies, it was shown that alkyllithium reagents add to N-tert-butyldimethylsilyl aldehyde tosylhydrazones at –78 °C and that the resulting adducts can be made to extrude dinitrogen in a free-radical process. Myers, A. G.; Movassaghi, M. J. Am. Chem. Soc. 1998, 120, 8891-8892. www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Corey–Bakshi–Shibata (CBS) reduction Corey–Bakshi–Shibata (CBS) reduction is a type of reduction in which reaction in which an ketone is enantioselectively reduced to produce the corresponding chiral alcohol, non-racemic alcohol. For this reaction a mixture of borane and a chiral oxazaborolidine as catalyst (CBS catalyst) is used. CBS reagents predictably give one enantiomer as the major product of ketone reduction, as illustrated with acetophenone as the starting material. Using CIP rules if the substituents rank high priority to low priority clockwise then this is the Re –face . If they rank high priority to low priority anti-clockwise then this is the Si-face. • Borane in catalyst is Lewis acid; Nitrogen is Lewis base to coordinate second borane • One B—H bond serves as the source of hydride in this reduction. Borane coordination increases Lewis acidity of catalyst (at B) and activates BH3 as hydride donor • Carbonyl coordination trans to bulky or electron rich group • Hydride transfer via 6-membered TS • Disproportionation between 8 and BH3 + (RO)2BH allows <1 equiv BH3 http://en.wikipedia.org/wiki/Corey%E2%80%93Itsuno_reduction CBS - Mechanism http://en.wikipedia.org/wiki/Corey%E2%80%93Itsuno_reduction CBS – Mechanism – key points First step coordination of BH3 to the nitrogen atom of the oxazaborolidine CBS catalyst Activation the BH3 as a hydride donor which enhance the Lewis acidity of the catalyst’s endocyclic boron Endocyclic boron of the catalyst coordinates to the ketone at the sterically more accessible electron lone pair (i.e. the lone pair closer to the smaller substituent, Rs). Binding in 3 acts to minimize the steric interactions between the ketone and the R’ group of the catalyst aligns the carbonyl and the coordinated borane for a favorable, face-selective hydride transfer through a six-membered transition state 4 Hydride transfer yields chiral boron enolate 5, which upon acidic workup yields the chiral alcohol 6. Last step to regenerate the catalyst may take place by two different pathways (Path 1 or 2). The predominant driving force for this face-selective, intramolecular hydride transfer is the simultaneous activation of the borane reagent by coordination to the Lewis basic nitrogen and the enhancement of the Lewis acidity of the endocyclic catalyst boron for coordination to the ketone. http://en.wikipedia.org/wiki/Corey%E2%80%93Itsuno_reduction Radical Dehalogenation Alkyl bromides and iodides are reduced efficiently to the corresponding alkanes in a free-radical chain mechanism with tri-n-butyltin hydride. The reduction of chlorides usually requires more forcing reaction conditions and alkyl fluorides are practically unreactive. The reactivity of alkyl halides parallels the thermodynamic stability of the radical produced and follows the order: tertiary > secondary > primary. Triethylboron-oxygen is a highly effective free-radical initiator. Reduction of bromides and iodides can occur at –78 °C with this initiator. Neumann, W. P. Synthesis 1987, 665-683. Curran, D. E.; Chen, M.-H. Tetrahedron Lett. 1985, 26, 4991-4994. In the above reaction, the radical generated during the dehalogenation reaction undergoes a tandem radical cyclization. www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Radical Cyclization The radical can react with multiple bonds in an intramolecular fashion to yield cyclized radical intermediates. The two ends of the multiple bond constitute two possible sites of reaction. If the radical in the resulting intermediate ends up outside of the ring, the attack is termed "exo"; if it ends up inside the newly formed ring, the attack is called "endo." In many cases, exo cyclization is favored over endo cyclization (macrocyclizations constitute the major exception to this rule). 5-hexenyl radicals are the most synthetically useful intermediates for radical cyclizations, because cyclization is extremely rapid and exo selective. Although the exo radical is less thermodynamically stable than the endo radical, the more rapid exo cyclization is rationalized by better orbital overlap in the chair-like exo transition state. http://en.wikipedia.org/wiki/Radical_cyclization Reduction of Acid Halides The Rosemund reduction is a classic method for the preparation of aldehydes from carboxylic acids by the selective hydrogenation of the corresponding acid chloride. Over-reduction and decarbonylation of the aldehyde product can limit the usefulness of the Rosemund protocol. The reduction is carried out by bubbling hydrogen through a hot solution of the acid chloride in which the catalyst, usually palladium on barium sulfate, is suspended. Sodium tri-tert-butoxyaluminohydride (STBA), generated by the reaction of sodium aluminum hydride with 3 equivalents of tert-butyl alcohol, reduces aliphatic and aromatic acid chlorides to the corresponding aldehydes in high yields. www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Hydrogenation of Olefines Thereductionofalkenesbyhydrogeninthepresenceofametalcatalyst(“catalytichydrogenation”) a reaction developed by Paul Sabatier and got Nobel Prize for Chemistry in 1912 along with Victor Grignard. The products of this reaction are a part of our daily lives – modern margarine is produced from hydrogenation of vegetable oils. Heterogeneous catalysts means the form of catalysis where the phase of the catalyst differs from that of the reactants; like finely divided insoluble platinum, palladium or nickel catalysts. Homogeneous catalysts means that involve a catalyst in the same phase as the reactants; catalyst (typically rhodium or ruthenium based) is soluble in the reaction medium – Wilkinson’scatalystis[RhCl(PPh3)3] This process is called a reduction or hydrogenation An unsaturated compound becomes a saturated (with hydrogen) compound http://en.wikipedia.org/wiki/Hydrogenation Mechanism Heterogeneous catalysts Homogeneous catalysts http://en.wikipedia.org/wiki/Asymmetric_hydrogenation Hydrogenation of Alkynes Hydrogenation: syn addition – Synthesis of cis-alkene It involve the treatment of the alkyne with hydrogen gas and a metal catalyst (Ni, Pd. Pt etc). To modify the behavior of the catalyst such that it is powerful enough to reduce the first pibondbutnotreactiveenoughtoaffectthesecond.Inotherwords,“poisoning”its reactivity. In practice, this is done by combining palladium on carbon with lead carbonate (PbCO3) and quinoline (an aromatic amine). The resulting mixture, known as “Lindlar’scatalyst”afteritsinventor,iseffectiveforthepartialreductionofalkynes. Other Similar conitions; Ni–B (nickel boride), Pd-CaCO3, palladium on barium sulfate, Pd-CaCO3-quinoline Lindlar, H.; Dubuis, R. (1973), "Palladium Catalyst for Partial Reduction of Acetylenes", Org. Synth.; Coll. Vol. 5: 880 Hydrogenation of Alkynes Hydrogenation: anti addition – Synthesis of trans-alkenes A dissolving metal reaction which uses lithium or sodium metal in low temperature ammonia or amine solvent produces trans-alkenes. This dissolving metal reduction process is different than other catalytic hydrogenation process. In this reaction, electrons from Na metal sequentially add to the alkyne, resulting in an anion that is protonated by the NH3 solvent. An interesting aspect of this reaction is the stereochemistry: due to electronic repulsion, the geometry of the resulting alkene is trans. The dissolving metal reductions always form the more stable trans product preferentially. Hydrogenation of Alkynes - Mechanism The trans alkene is formed because the vinyl carbanion intermediate that is formed is more stable when the larger R groups are further away from each other to avoid steric interactions. Protonation of this anion leads to the more stable trans adduct. Sodium metal has an extremely low ionization energy and will readily give up its electron. http://www.masterorganicchemistry.com/ Reduction of α,β-Unsaturated Carbonyl to carbonyl The carbon-carbondoublebondofα,β-unsaturated carbonyl compounds can be reduced selectively by catalytic hydrogenation, affording the corresponding carbonyl compounds. This method is not compatible with olefins, alkynes, and halides. α,β-Unsaturated carbonyl compounds undergo selective 1,4-reduction with [(Ph3P)CuH]6. [(Ph3P)CuH]6 is stable indefinitely, provided that the reagent is stored under an inert atmosphere. The reagent can be weighed quickly in the air, but the reaction solutions must be deoxygenated. The reaction is unaffected by the presence of water (in fact, deoxygenated water is often added as a proton source). The reduction is highly steroselective, with addition occurring to the less hindered face of the olefin: http://www.masterorganicchemistry.com/ Reduction of miscellaneous species Reduction deselenation Beilstein J. Org. Chem. 2013, 9, 2669–2674 Reduction of miscellaneous species Reduction silylation JOC, 1979, 44, 421 Reduction of miscellaneous species Reduction desilylation Org. Lett. 2006, 8, 179–182 Reduction of miscellaneous species Corey–Winter olefin synthesis Mechanism The reaction mechanism involves the formation of a cyclic thio-carbonate from the diol and thiophosgene. The second step involves treatment with trimethyl phosphite, which attacks the sulfur atom, producing S=P(OMe)3 (driven by the formation of a strong P=S double bond) and leaving a carbene. This carbene collapses with loss of carbon dioxide to give the olefin. http://en.wikipedia.org/wiki/Corey%E2%80%93Winter_olefin_synthesis Reduction of miscellaneous species Eastwood Deoxygenation: A vicinal diol is treated with ethyl orthoformate at high temperature (140-180 °C), followed by pyrolysis of the resulting cyclic orthoformate (160-220 °C) in the presence of a carboxylic acid (typically acetic acid) --- The elimination is stereospecific --- Not suitable for functionalized substrates. Crank, G.; Eastwood, E. W. Aust. J. Chem. 1964, 17, 1385 Base Induced Decomposition of Benzylidene Acetals Key points; The elimination is stereospecific. Long reaction times and high temperature under extremely basic conditions make this an unsuitable method for functionalized substrates. J. Chem. Soc. Chem. Commun. 1964, 1593 www.chem.harvard.edu/groups/myers/handouts/1_Reduction.pdf Reduction of miscellaneous species Desulfurization Beilstein J. Org. Chem. 2010, 6, No. 17. Reduction of miscellaneous species Desulfonylation http://en.wikipedia.org/wiki/Desulfonylation_reactions Reduction of Alkyl halides Addition of Hydride Addition of metals R3C-X + 2Li ——> R3C-Li + LiX R3C-X + Mg ——> R3C-MgX An Alkyl Lithium Reagent A Grignard Reagent Reduction of Alkyl halides Mechanism J. Org. Chem., 2010, 75 (8), pp 2645–2650 Reduction of epoxides Reduction cynation Selective Cleavage of C-O and/or Si-O Bonds Komiya, S. et al. Organometallics, 1985, 4, 1504-1508 Mechanism – cleavage of C-O bond Komiya, S. et al. Organometallics, 1985, 4, 1504-1508 Mechanism – cleavage of Si-O bond Komiya, S. et al. Organometallics, 1985, 4, 1504-1508 Mechanism – cleavage of Si-O bond Komiya, S. et al. Organometallics, 1985, 4, 1504-1508 Overview - Reduction