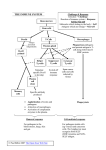
Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Herd immunity wikipedia , lookup
Lymphopoiesis wikipedia , lookup
Vaccination policy wikipedia , lookup
Childhood immunizations in the United States wikipedia , lookup
Hygiene hypothesis wikipedia , lookup
Hepatitis B wikipedia , lookup
Monoclonal antibody wikipedia , lookup
Immune system wikipedia , lookup
Immunosuppressive drug wikipedia , lookup
Duffy antigen system wikipedia , lookup
Adoptive cell transfer wikipedia , lookup
Innate immune system wikipedia , lookup
Molecular mimicry wikipedia , lookup
Adaptive immune system wikipedia , lookup
Cancer immunotherapy wikipedia , lookup
Psychoneuroimmunology wikipedia , lookup
Immunocontraception wikipedia , lookup
Polyclonal B cell response wikipedia , lookup
DOI: 10.2478/nanca-2014-0001 Nanocarriers 2014; volume 1, 10–45 Review article Open Access Priyanka Prabhu and Vandana Patravale* Potential of Nanocarriers in Antigen Delivery: The Path to Successful Vaccine Delivery Abstract: Vaccination has indubitably made noteworthy contribution to global health. Recent years have witnessed the employment of subunit antigens rather than inactivated or live attenuated vaccines, owing to the superior safety of the former. The intrinsic weak immunogenicity of subunit antigens makes it imperative to formulate them with an adjuvant. Presently, the armamentarium of approved vaccine adjuvants is very poor. Nanocarriers hold great promise for successful vaccine delivery owing to their versatility, excellent cellular uptake properties, capacity to protect antigen, amenability to targeting, and ability to offer prolonged antigen presentation. All these attributes ultimately endow nanocarriers with immense potential to achieve needle-free vaccine delivery, reduce the number of vaccinations, attain dose sparing of antigen, and lead to stronger immune response generation. Nanocarriers can be explored in manifold ways to accomplish targeted antigen delivery to antigen presenting cells. They can be formulated to contain both antigen and immunostimulant molecules, and they can be engineered from specific materials to achieve antigen presentation through the desired pathway to stimulate a particular arm of the immune response. This review discusses the basics of immune response generation, mechanisms of adjuvanticity by nanocarriers, parameters influencing their adjuvanticity, and finally describes the incredible opportunities offered by a gamut of nanocarriers for vaccine delivery. Keywords: Vaccine, nanoparticles, antigen, adjuvant, dendritic cells, liposomes, chitosan, polymeric, calcium phosphate, emulsions. *Corresponding author: Vandana Patravale: Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Matunga, Mumbai-400019, India, Telephone no: 91-223361 2217, Fax no: 91-22-3361 1020, E-mail: [email protected] Priyanka Prabhu: Department of Pharmaceutical Sciences and Technology, Institute of Chemical Technology, Matunga, Mumbai-400019, India List of Abbreviations APCs - Antigen Presenting Cells AMVAD- Archaeal lipid Mucosal Vaccine Adjuvant and Delivery BRSV - Bovine Respiratory Synctial Virus BSA - Bovine Serum Albumin BALT - Bronchus Associated Lymphoid Tissue CMIS - Common Mucosal Immune System CFA - Complete Freund’s Adjuvant CALT - Conjunctiva Associated Lymphoid tissue DOTMA - N-[1-(2, 3-dioleyloxy)propyl]-N,N,N-trimethylammonium chloride GIT - Gastrointestinal tract GSK - GlaxoSmithKline GALT - Gut Associated Lymphoid Tissue HSV- Herpes simplex virus HIV - Human Immunodeficiency Virus HPV - Human PapillomaVirus ISCOMs - Immunostimulating complexes ID - Intradermal IM - Intramuscular LPS - Lipopolysaccharide MHC - Major Histocompatibility Complex MVP - Major Vault Protein MPLA - Monophosphoryl Lipid A MALT - Mucosa Associated Lymphoid Tissues MDP - Muramyl dipeptide NALT - Nasal Associated Lymphoid Tissue NOD - Nucleotide-binding oligomerization domain PAMPs - Pathogen Associated Molecular Patterns PRRs - Pattern Recognition Receptors PAMAM - Polyamidoamine PCPP - Poly [di(carboxylatophenoxy)phosphazene] PCEP - Poly [di(sodiumcarboxylatoethylphenoxy) phosphazene] PEI - Polyethyleneimine PLA - Poly Lactic Acid PLGA - Poly Lactic-co-glycolide RSV - Respiratory Syncytial Virus SWCT - Single walled carbon nanotubes © 2014 Priyanka Prabhu, Vandana Patravale licensee De Gruyter Open. This work is licensed under the Creative Commons Attribution-NonCommercialNoDerivs 3.0 License. Unauthenticated Download Date | 6/17/17 12:05 AM SALT SLN SC TMC TLRs TDB TNF VLPs Nanocarriers for vaccine delivery - Skin Associated Lymphoid Tissue - Solid Lipid Nanoparticles - Subcutaneous - N-trimethyl chitosan - Toll-like receptors - Trehalose 6’6-dibehenate - Tumour Necrosis Factor - Virus like particles 1 Introduction The main aim of vaccination is to incite a strong pathogenspecific immune response to confer life-long protection against subsequent exposure to the pathogen [1]. An ideal vaccine must be efficacious, non-toxic, cost-effective, and amenable to large scale manufacture [2]. Since its inception, vaccination has played a pivotal role in reducing the morbidity and mortality of several debilitating infections [3]. Eradication of small pox and significant reduction in the global burden of polio, tetanus, pertussis, mumps, rubella, diphtheria, yellow fever, and measles [4] exemplify the incredible impact of vaccines on global healthcare. Despite these impressive outcomes, there are a few diseases for which effective vaccines are unavailable, 11 including AIDS, malaria, and tuberculosis [3]. As far as vaccine delivery is concerned, there are three major hurdles. Firstly, the majority of vaccines suffer from poor thermal stability, necessitating cold storage, which poses a hurdle for vaccine storage in developing nations [5]. Secondly, most vaccines require multiple administrations resulting in poor patient compliance [6]. Thirdly, due to the low immunogenicity of antigens administered via the noninvasive mucosal route of administration, most vaccines are intended for parenteral administration. This not only results in poor patient compliance but is also unable to elicit mucosal immunity, eliciting only a systemic antibody response, which is often insufficient to tackle pathogens that employ mucosal surfaces for ingress into the host or which result in disease at mucosal surfaces [7]. Additionally, most vaccines only generate a humoral immune response, which only deals with extracellular pathogens, whereas cell-mediated immunity needs to be induced in order to tackle intracellular pathogens [8]. There are three different types of vaccines, shown in Table 1 [4,9-11]. Vaccines may be administered by three routes: parenteral (invasive), mucosal (non-invasive), and topical/transdermal (non-invasive) routes. Figure 1 Table 1: Types of vaccines [4,9,10,11]. Type Key features Killed/ Inactivated Include microbes inactivated by application of heat / chemical treatment [9] Live attenuated Advantages Drawbacks Examples Not able to multiply and enter into host cells [9] Require multiple doses to elicit an adequate immune response [9] Prepared by repeated passage Ability to multiply within Probability of causing disease in of pathogen in culture to the host and closely mimic immunocompromised individuals render microbe non-pathoge- natural infection [9] and their probable reversion to nic [9] Single administration results virulence [9] in development of a strong humoral and cell-mediated immune response [9] Subunit vaccines Include portions of pathogenic microbes (toxins, proteins, polysaccharides, or recombinant proteins) [9,10] Possess superior safety to live vaccines and lower antigen competition due to small number of defined components [11] Amenable to targeting to specific sites where immunity is required [11] Vaccinated animals can be distinguished from infected animals [11] Their cost effective production is possible through recombinant DNA technology [11] Inherent poor immunogenicity [9] Low uptake by antigen presenting cells (APCs) [9] In vivo instability [9] Parenteral Polio vaccine, Influenza, and Hepatitis A vaccines [9] Small pox, oral polio vaccine, measles, mumps, rubella, and Bacillus Calmette Guerin (BCG) [4,9] Tetanus, Diphtheria, and Human Papilloma virus (HPV) [9] Unauthenticated Download Date | 6/17/17 12:05 AM 12 Priyanka Prabhu and Vandana Patravale Figure 1: Comparison of mucosal v/s parenteral immunization [12]. compares the key features of mucosal v/s parenteral immunization [12]. 1.1 Parenteral vaccination Parenteral routes commonly employed for vaccination purposes include the intramuscular (IM), subcutaneous (SC), and intradermal (ID) routes. 1.1.1 Intramuscular route The IM route is a very popular route for routine vaccination. Cellular and humoral immunization achieved through this route is comparable to that achieved after SC and ID administration. Myoblasts are antigen-presenting cells (APCs) found in the muscle tissue responsible for immune response generation after IM administration. However, due to the limited number of dendritic cells in the muscle in comparison to the skin, in order to elicit an effective immune response it is necessary to administer higher doses of antigen by the IM route in comparison to ID immunization [2]. Myocytes do not express major histocompatibility complex (MHC) class II molecules and are devoid of costimulatory compounds, rendering them incapable of directly activating T cells [13]. IM immunization results in a predominant Th2 response unless boosters are given subsequently [2]. Examples of vaccines which are currently administered via the IM route include hepatitis A, hepatitis B, diphtheria toxoid, inactivated polio vaccine, pertussis, human papilloma virus (HPV), rabies vaccine, Streptococcus pneumonia, and tetanus toxoid [10]. 1.1.2 Subcutaneous route The SC route is beneficial for antigen delivery owing to the drainage of antigen from the injection site to lymph nodes which house the immunocompetent cells [14]. Particles in the micrometer as well as nanometer size range have been explored by the subcutaneous route for vaccine adjuvant purposes [15,16]. The delivery of carriers administered via the SC route to the lymph nodes is governed by their size. Particles greater than 1000 nm stay at the site of injection until they are broken down into particles of a smaller size. They are neither able to gain entry into the lymphatics directly nor are they phagocytosed easily. Particles less than 100 nm in size are able to enter the lymphatic capillaries via the gaps between lymphatic endothelial cells. Particles between 100-1000 nm undergo phagocytosis by APCs, such as dendritic cells, followed by passage into lymphatic capillaries [14]. Benefits associated with the SC route include lower clearance and longer persistence at the site of administration resulting in prolonged antigen presentation to the immune system [17]. Examples of vaccines which are currently administered via the SC route include the anthrax vaccine, Haemophilus influenza type b vaccine, inactivated polio vaccine, Japanese encephalitis, measles, mumps, rubella, chicken pox, S. pneumonia, and yellow fever [10]. Unauthenticated Download Date | 6/17/17 12:05 AM Nanocarriers for vaccine delivery 1.1.3 Intradermal route 13 Mucosal routes explored for vaccination include nasal, oral, pulmonary, sublingual, ocular, rectal, and the vaginal route. The presence of mucus, poor antigen uptake through the mucosa into immunocompetent tissue, antigen degradation, and inability of the mucosal tissue to respond to non-replicating antigens leads to failure of mucosal immunization to generate a potent immune response [19]. in soluble form is known to elicit a systemic immune response because it can traverse the nasal epithelium and gain access to the cervical lymph nodes, resulting in IgG production. On the other hand, antigen administered in particulate form is uptaken by M cells to produce IgA antibody [12]. Based on the physiological environment in the nasal cavity, an ideal intranasal vaccine delivery system should have a number of attributes, as outlined in Table 2 [21]. A major drawback to nasal vaccination is the poor retention of vaccine formulation in the nasal cavity due to nasal mucociliary clearance, which results in removal of the vaccine formulation within 15 minutes of administration, thus limiting vaccine uptake into the nasal mucosa.Mucoadhesive substances, both in soluble and particulate form, nanogels, and in situ gelling systems have been utilized to augment the nasal retention time of antigens and enhance the immune response [22]. Other drawbacks associated with the nasal route are the probable delivery into the brain via the olfactory region, and problems due to asthama, respiratory syndromes, and allergy [21]. 1.2.1 Nasal route 1.2.2 Oral route The nasal route is well suited for the induction of immunity against antigens which enter the body through inhalation (e.g. influenza). The leaky characteristic of the nasal epithelium can result in entry of the antigen into the underlying blood vessels, cervical lymph nodes and lymphocytes to elicit a potent immune response [20]. In case of nasal vaccination, immune response generation occurs through nasal-associated lymphoid tissue (NALT). NALT includes M cells, lymphoid follicles, dendritic cells, and goblet cells. Antigen administered The oral route is the easiest and the most preferred route for vaccination from a patient compliance perspective. Gut-associated lymphoid tissue (GALT) is involved in the generation of an immune response after oral vaccination. GALT is comprised of the Peyer’s patches, where M cells process the antigen and carry it to APCs such as dendritic cells and macrophages. Oral vaccination induces an immune response in the small intestine, ascending colon, and distant tissues such as mammary glands and salivary glands owing to the common mucosal immune This route offers immense potential for successful immunization owing to the localization of the most potent APCs (dendritic cells) and other immune cells in the dermis. The US Center for Disease Control and Prevention has approved two vaccines for ID vaccination, namely the Mycobacterium bovis tuberculosis vaccine, better known as the Bacille Calmette Guerin (BCG), and the Vaccinia smallpox vaccination [18]. 1.2 Mucosal vaccination Table 2: Attributes of an ideal intranasal vaccine delivery system [21]. Attribute Benefit Nanoparticulate system (less than 250 nm) Enhanced cellular uptake by APC Hydrophilic surface Avoid aggregation and allow transport of discrete particles Preferably contain targeting moiety Receptor binding to M cells of nasal mucosa or APC Afford protection to antigen against degradation Dose sparing of antigen and enhanced vaccine efficacy Exhibit prolonged antigen release into APC in nasal tissue Reduce number of vaccinations Comprise of immunostimulant molecule and antigen Non-toxic and non-irritant to nasal tissue Simultaneous delivery of immunostimulant molecule and antigen to same APC for enhanced immune response generation Safety Preferably mucoadhesive Prolonged nasal retention Simple to fabricate Easy public availability Amenable to large scale production Unauthenticated Download Date | 6/17/17 12:05 AM 14 Priyanka Prabhu and Vandana Patravale system (CMIS). But oral vaccination lacks the ability to elicit an immune response in the female genital tract and the tonsils. Administration of antigen in particulate form enhances uptake by the Peyer’s patches [12]. To date, theoral polio vaccine is the most efficient oral vaccine [22]. Other orally administered vaccines include rotavirus, typhoid fever, and cholera vaccine [10]. A major snag associated with oral vaccination is the degradation of antigen in the harsh environment of the GI tract comprising low pH and enzymes [12]. Oral immunization is also associated with antigen tolerance. The immune system has evolved to dampen systemic immune responses against many substances which pass through the gastrointestinal tract (GIT) in huge quantities. As a result, larger doses of antigen have to be administered via the oral route than the parenteral route [8]. The nasal route has some benefits over the oral route [12]. These include the superior permeability of the nasal mucosa in comparison to the gastro-intestinal (GI) mucosa, presence of dendritic cells in the NALT which are reputedly the most potent APCs, rapid and stronger immune response generation than the oral route, ability of the NALT to retain immunological memory resulting in a faster immune response on second exposure, ability to elicit cytotoxic T lymphocyte stimulation to deal with intracellular pathogens, lower dose of antigen required due to absence of dilution of antigen in nasal fluids, low enzymatic action, no exposure to low pH, and potent immune response in the respiratory and genital tracts due to the CMIS [21]. 1.2.3 Pulmonary route The respiratory tract is well equipped with immunocompetent cells to tackle the pathogens which commonly enter the body through the respiratory tract. Human lungs are endowed with a huge surface area, are extensively vascularised, and contain APCs such as dendritic cells and alveolar macrophages [22]. Pulmonary vaccines are administered to the alveoli using an aerosol [12]. Bronchus-associated lymphoid tissue (BALT) is present in the lungs, and is able to generate not only a systemic immune response but also a mucosal immune response in both the respiratory tract and the distal mucosal tissues. An example of a vaccine delivered by the pulmonary route is the live Newcastle disease vaccine for poultry. To date, there is no commercially available vaccine for pulmonary administration in humans [22]. 1.2.4 Sublingual route Sublingual immunotherapy is utilized to treat type I allergy, which involves desensitization of the patient by sublingual allergen delivery [12]. The route offers numerous benefits over the oral route due to the absence of low pH and comparatively lower enzymatic activity. Both live and inactivated influenza virus, when administered through the sublingual route, have been demonstrated to elicit an immune response and protect mice against influenza infection. Also, sublingual administration of inactivated viruses has been shown to produce secretory IgA antibody and cytotoxic T lymphocyte response in areas. The sublingual route could prove to be useful in children and infants owing to its simple administration [22]. 1.2.5 Ocular route This route could be utilized to achieve successful immunization against microbes infecting the ocular tissue. Both and systemic immune responses are induced due to the conjunctiva-associated lymphoid tissue (CALT). The route does not suffer from the drawbacks of the GI environment, such as low pH, or of the nasal route, such as antigen entry into the brain [12]. 1.2.6 Rectal route This route could be used as a substitute to oral immunization owing to its ability to generate a potent immune response in the intestine and other distant parts without subjecting the antigen to hostile conditions of low pH and proteolytic enzymes. The chief shortcomings of the rectal route are poor patient acceptability and removal of the vaccine after administration [12]. 1.2.7 Vaginal route This route would be ideal for conferring mucosal immunity in the female genital tract against sexually transmitted diseases such as the human immunodeficiency virus (HIV), HPV, and Herpes simplex virus (HSV). A major disadvantage associated with this route for immunization is the dependence of the immune response on hormonal control and the variation according to the stages of the menstrual cycle. Reports suggest that nasal immunization results in a more effective immune response in the vaginal mucosa compared to vaginal immunization due to the CMIS [12]. Unauthenticated Download Date | 6/17/17 12:05 AM 1.3 Topical/transdermal vaccination This route offers several beneficial attributes for vaccination. Firstly, it is non-invasive. Secondly, it prevents exposure of the antigen to low pH or proteolytic enzyme activity [23]. Thirdly, it is inherently endowed with more immunocompetent cells than the muscles or SC tissues because of its close contact with the outer environment [22]. Also, it drains the antigen into the lymph nodes for effective immune response generation [24]. Topical immunization reduces the variation associated with the IM and SC route depending on the site of injection, depth of injection, local blood supply, and movement of the body [23]. Skin-associated lymphoid tissue (SALT) is involved in the induction of immunity after topical immunization. SALT comprises the Langerhans cells and keratinocytes present in the epidermis, dendritic cells and mast cells present in the dermis, and B and T lymphocytes present in the draining lymph nodes [25]. Langerhans cells are potent APCs which take up antigen, process it, and present it to naive T cells in the draining lymph nodes. Keratinocytes in the epidermis produce pro-inflammatory cytokines, such as IL-1 and tumour necrosis factor-α (TNF-α), on encounter with danger signals resulting in the activation, maturation and movement of Langerhans cells and drawing of effector cells at the point of inflammation [26]. A major hindrance to topical vaccine delivery is the barrier property of the skin due to the stratum corneum. Only antigens with high lipophilicity and a molecular mass of less than 500 Da can passively traverse the stratum corneum [25]. Hence, it is imperative to employ permeation enhancement approaches for effective topical delivery of vaccines. These may include physical approaches or formulation of suitable delivery vehicles to deliver antigen across the formidable skin barrier. 2 Basics of immune response generation Complete discussion of the human immune system in all its complexity is too vast and beyond the scope of this review. However, the following section will describe in a nutshell, the functioning of the immune system, and introduce the reader to the commonly used terminologies and phenomena in immunology. This will afford a better understanding of the immune response modulation shown by vaccine adjuvants and eventually guide researchers to design efficient vaccine adjuvants to elicit the desired immune response. Nanocarriers for vaccine delivery 15 2.1 Defence mechanisms of the human immune system The human body is endowed with the ability to defend itself against pathogens encountered within the environment. The human immune system consists of primary lymphoid organs and secondary lymphoid organs. Primary lymphoid organs consist of the bone marrow and the thymus. These are the sites of generation of B lymphocytes and T lymphocytes respectively. Secondary lymphoid organs include the spleen, lymph nodes, skin, adenoids, tonsils, and themucosa-associated lymphoid tissues (MALT), and GALT (Peyer’s patches). Initiation of an adaptive immune response takes place in the secondary lymphoid organs [18]. The human immune system comprises three different types of defence mechanisms. 1. Skin, ciliated epithelial cells and other mucous membranes of the body which form an external barrier and serve as the first line of defence against invading pathogens; and enzymes and other chemical secretions such as stomach acid [27]. 2. Innate immune system. The innate immune system constitutes the first line of protection against pathogens which have entered the human body, and comprises macrophages and dendritic cells [27]. Migratory dendritic cells exist in the periphery and on activation by antigen migration to the lymphoid tissues. Such migratory dendritic cells are present in the skin, lungs, liver, kidneys, and intestinal tract [2]. The innate immune system also has mobile cells such as neutrophils, monocytes, and eosinophils which tour throughout the body via blood and lymph screening for pathogens [27,28]. A rapid immune response can be induced against the pathogen in question owing to the ability of the cells of the innate immune system to be immediately recruited to the site of infection. Cells of the innate immune system are endowed with a set of receptors called “pattern recognition receptors (PRRs)” which identify a number of molecular structures unique to pathogens of a particular class. These structures are called pathogenassociated molecular patterns (PAMPs) [27]. There are different types of PRRs, including toll-like receptors (TLRs), C-type lectin receptors, nucleotide-binding oligomerization domain (NOD) receptors, NOD-like receptors, and retinoic acid-inducible gene 1(RIG-1-like helicases) [29]. TLRs are the most extensively explored PRRs for immunomodulation. TLRs can be categorized according to their localization and the PAMPs they recognize. Different TLRs recognize an array of Unauthenticated Download Date | 6/17/17 12:05 AM 16 Priyanka Prabhu and Vandana Patravale Table 3: List of endogenous and exogenous ligands for various TLRs [2, 27, 30]. TLR Endogenous ligand Exogenous ligand Effect TLR1 Triacyl lipoproteins Microbial peptidoglycans Stimulate inflammatory cytokine secretion TLR2 Unknown TLR3 mRNA Gram positive peptidoglycan, lipoproteins, viral glyco- Stimulate inflammatory cytokine secretion proteins, glycolipids dsRNA, siRNA Production of Type I interferons TLR4 TLR5 Fibrinogen, hyaluronic acid, defensin 2 Unknown TLR6 Unknown Lipopolysaccharide, RSV fusion protein, virus envelop Production of Type I interferons protein Flagellin Present in intestinal and lung epithelia, plays a key role in mucosal immune response generation Lipopeptides Stimulate inflammatory cytokine secretion TLR7/8 Unknown ssRNA, imiquimod, resiquimod, imidazoquinoline TLR9 Chromatin complex CpG DNA TLR10 Unknown Unknown molecular motifs unique to pathogens. Table 3 shows different TLRs with their ligands and the effect seen on their activation [2, 27, 30]. TLRs 1, 2, 4, 5 and 6 are chiefly expressed on the surface of the cell membrane and recognize products of bacterial origin, whereas TLRs 3, 7, 8, and 9 are found inside the intracellular compartments and identify virus-derived products and nucleic acids. The innate immune system is not only capable of sensing infection through direct PRRmediated pathogen identification, but also the effects of an infection via recognition of the danger signals or stress signals released by infected cells. These signals include uric acid or ATP, which are released during cell lysis owing to infection. The innate immune system exerts its activity through phagocytosis. All the cells of the innate immune system, irrespective of whether they are resident or moving, show efficient phagocytosing ability. Once the innate immune cell encounters a pathogen, it engulfs it and traps it within an intracellular vesicular structure, followed by its destruction by digestive enzymes or reactive oxygen species [27]. 3. Adaptive immune system. Pathogens manifesting a high mutagenic rate may eventually evade the innate immune system owing to the narrow diversity of PRRs. Intracellular multiplication of microbes such as viruses and parasites further makes their elimination challenging. The adaptive immune system is a highly advanced system developed by the body to tackle highly mutagenic and intracellularly replicating pathogens [27]. The innate immune response differs from the adaptive immune response in that the former responds immediately (minutes to a few hours) to Anti-viral action invading pathogens, whereas the adaptive immune system takes longer (days to weeks) to respond. The innate immune response is short-lived owing to the numerous feedback regulation mechanisms which intervene to curb any harm to tissues from the potent non-specific effects of the innate immune system. Innate immune responses are also devoid of memory effects, and as a result do not show a faster and stronger immune response on second exposure to the same antigen [31]. Adaptive immune responses are of two types: humoral and cellular. The humoral arm consists of B cells which produce antibodies leading to humoral immunity. The cellular arm consists of CD4+ T lymphocytes and CD8+ T lymphocytes [32]. 2.2 Stages in immune response generation There are three key steps involved in the generation of an immune response: antigen uptake by APCs, antigen presentation by APCs, and antigenic elimination by the effector cells of the adaptive immune system. 2.2.1 Antigen uptake by APCs Antigen may take three different pathways postvaccination. Firstly, the antigen may enter the blood where it will be exposed to plasmacytoid dendritic cells, blood resident monocytes, B lymphocytes, splenic macrophages or splenic dendritic cells. Secondly, the soluble antigen may enter the lymphatic capillaries and vessels and be carried through lymphatic fluid to the lymph nodes where it would encounter lymph node-resident dendritic Unauthenticated Download Date | 6/17/17 12:05 AM cells. Lastly, antigens which neither enter the blood nor lymph may interact with the APCs at the site of administration/injection. These include Langerhans cells, dermal dendritic cells, and immune cells lining the gut and respiratory mucosa, depending on the route of administration [18]. Both macrophages and dendritic cells are capable of phagocytosis, however, macrophages phagocytose larger particles, 500-2000 nm in size, such as bacteria, whereas dendritic cells phagocytose smaller particles, 20-100 nm in size, such as viruses [33]. Only dendritic cells are capable of activation of naive helper T cells. Dendritic cells act as sentinels in peripheral tissues and monitor the environment for pathogens. They exist in an immature state characterized by potent endocytic potential. These immature dendritic cells take up antigen, process it, and present it on their surface in association with MHC class II molecules followed by their migration to lymphoid organs where they activate naive helper T cells through a combination of antigenic, cytokine, and co-stimulatory molecules. [27]. Uptake of virus sized nanocarriers (20-200 nm) occurs via receptor-mediated endocytosis, whereas particles > 0.5 µm in size are taken up by phagocytosis [29]. Macropinocytosis is involved in the uptake of small nanoparticles (50 nm) [34]. Antigen uptake by dendritic cells results in their transformation from an immature state adept at capturing antigen to a mature state. Mature dendritic cells show increased surface expression of co-stimulatory molecules such as CD40, CD80, and CD86, which induce activation of T cells. These dendritic cells then migrate to the T cells to co-deliver the antigenic stimulus and co-stimulatory signals to T cells resulting in their activation to eventually mount a strong antigen-specific immune response [27]. A single mature dendritic cell has the ability to activate 1001000 T cells [35]. 2.2.2 Antigen presentation by APCs and elimination of antigen by the effector cells of the adaptive immune system Dendritic cells serve as a valuable link between the innate and adaptive arms of the human immune system. After uptake of antigen, they process the antigen and present it to the adaptive immune system comprising T cells and B cells. Once antigens are internalized by APCs through phagocytosis or endocytosis, they are degraded within the endosome to shorter peptides which are then presented in the context of MHC class II molecules. There are two types of MHC molecules: MHC class I and MHC class II molecules. MHC class II molecules are expressed only Nanocarriers for vaccine delivery 17 by the cells of the immune system, unlike MHC class I molecules, which all nucleated cells of the human body display on their surface. When the intracellular proteins undergo degradation as a part of the normal cell quality control process, some are not completely degraded, resulting in the formation of short chain peptides. These peptides are delivered from the cytoplasm into the endoplasmic reticulum where they are bound by transmembrane presenting molecules encoded by the major histocompatibility complex (MHC) gene. These molecules consist of two chains which undergo folding to form a cleft in which the peptide rests. These peptidebound MHC-encoded molecules are then transported to the cell surface for peptide (antigen) presentation. Lymphocytes are the key players in the adaptive immune response, and are present in the lymph, blood, and spleen and lymph nodes. B cells and T cells differ with respect to their origin, their ability to recognize pathogens, and their manner of destruction of pathogen. B cells originate in the bone marrow, while T cells originate in the thymus. B cells are capable of recognizing the pathogen directly with the help of B cell surface receptors. Each B cell expresses a number of copies of a unique antibody specific for a particular antigen as a cell surface receptor. This results in the production of B cells which are able to react with a single antigen. Once the B cell recognizes a specific antigen along with the presence of auxiliary cells and signals, it undergoes activation to divide into plasma cells and memory B cells. The majority of plasma cells return to the bone marrow and generate huge quantities of soluble antibody which are released in the blood and lymph. Antibodies produced by B lymphocytes take care of the pathogen within body fluids, however, due to their large molecular size, antibodies are unable to cross the plasma membrane and hence are ineffective against intracellularly multiplicating pathogens. T lymphocytes recognize antigens through MHC class I/II molecule presentation. Unlike B lymphocytes, T cell receptors are unable to directly recognize pathogens; they require the pathogen/antigen to be presented in association with MHC molecules. There are two distinct types of T lymphocytes: CD4+ and CD8+ T lymphocytes. CD4+ T lymphocytes express CD4+ markers on their surface and recognize antigen loaded onto MHC class II molecules. CD8+ T lymphocytes express CD8+ markers on their surface and recognize antigen loaded onto MHC class I molecules. Peptides of endogenous origin are generally presented in the context of MHC class I molecules, whereas peptides of exogenous origin are presented in the context of MHC class II molecules. However, certain APCs such as dendritic cells, macrophages, and B lymphocytes are capable Unauthenticated Download Date | 6/17/17 12:05 AM 18 Priyanka Prabhu and Vandana Patravale of “cross-presentation”. Cross-presentation is defined as the ability of the endocytosed antigenic material to escape from the endosome and enter the cytoplasm for presentation in the context of MHC class I molecules. T cell surface receptors exhibit some peculiar features as regards to their mode of antigen recognition. Firstly, they are unable to react with soluble antigens, they can only destroy the antigen presented to them in association with MHC class I/II molecules. Secondly, they can mainly only react to proteins. CD8+ T lymphocytes, after their recognition of antigen/MHC class I complex, proliferate and differentiate into CD8+ cytotoxic T lymphocytes that secrete a pore forming protein called perforin. This in turn causes the release of proteases into the cytoplasm of the infected cells. These proteases initiate apoptotic responses leading to the deathof the antigen expressing cell. Antigen-specific T lymphocytes also secrete cytokines such as interferon-γ (IFN-γ) and TNF-α, which interfere with multiplication of viruses and inhibit their growth in the cell. CD4+ T lymphocytes on activation by mature dendritic cells differentiate into antigen-specific CD4+ T cells and CD4+ helper T cells; the latter play a pivotal role in the initiation and regulation of an effective immune response. Regulation of immune responses by CD4+ helper T cells occurs through the activity of soluble mediators called cytokines secreted by activated T cells. There are different subsets of helper T cells. Th1 cells secrete mainly IFN-γ, and TNF-α. Th 2 cells secrete IL-4, IL-5, IL-10, and IL-13. Th1 cells thus mount an effective immune response against viruses and other intracellular pathogens. Th2 cells activate eosinophils and mastocytes and help to tackle the extracellular pathogens [27]. Commonly, Th2 cells result in an antibody-mediated immune response and the Th1 cells lead to a strong cellmediated immune response [32]. Cytokines produced by Th1 cells lead to the production of IgG2a antibody whereas cytokines produced by Th2 cells enhance production of IgG1 antibody [36]. The third subset is follicular helper T cells which exist in close association with B lymphocytes in follicles of lymphoid organs. Follicular helper T cells produce IL-21 and augment increased production of antigen-specific antibodies by B cells. The fourth subset is Th17 cells which serve to regulate the local immune responses against gut and lung pathogens. Th17 cells secrete IL-17 and IL-22. Dendritic cells themselves are able to produce certain cytokines and thereby influence the differentiation of naive CD4+ T cells into a particular subset of helper/ effector cells. For example, IL-12 produced by dendritic cells results in the differentiation of naive CD4+ T cells into Th1 cells. IL-6 is involved in the differentiation into follicular helper T cells and Th17 cells. Th1 and Th2 cells possess the ability to inhibit each other’s function [27]. IL-10 is reported to hamper the development of Th1 cells, and IFN-γ is known to avert the activation of Th2 cells [35]. Antibody production by B lymphocytes and production of cytotoxic CD8+ T lymphocytes can occur even in the absence of CD4+ helper T cells. However, this results in the generation of low affinity antibodies. Also, the antibody response is short-lived and does not elicit a memory response on subsequent exposure to the same antigen. A stronger antigen-specific secondary immune response is only elicited when B cells are activated in a T cell dependent manner. T cell dependent humoral responses require simultaneous activation of both B and T cells. Once an antigenic protein is injected into the human body, the primary immune response is slow and involves low affinity IgM antibodies. Subsequent exposure to the same antigen results in the induction of a faster and a stronger immune response involving higher affinity IgG antibodies. Antigen-specific helper T cells play a pivotal role in providing B cells with the necessary stimuli to give rise to IgG antibodies of high affinity [27]. Memory B cell responses exhibit different features from the primary B cell responses with the former being generated more rapidly, resulting in higher antibody levels predominantly of the IgG, IgA, and IgE isotypes, and generating antibodies of higher affinity [37]. T cell activation results in their proliferation and differentiation into effector T cells. 90% of effector T cells are not long lived, and after destroying antigen they are removed from the body by apoptosis. The remaining 10%, however, become memory T cells and are retained for several years or even for a lifetime. There are two types of memory T cells: central memory T cells and effector memory T cells. Central memory T cells reside in the lymphoid organs and require time for activation to produce large levels of IL-2 and differentiate into effector memory T cells, whereas effector memory T cells are present at the peripheral sites and exhibit rapid activation on subsequent exposure to antigen [32]. Figure 2 shows aschematic representation of the steps involved in the generation of an immune response [29]. 3 What are Vaccine Adjuvants? A vaccine adjuvant can be defined as any substance or a mixture of two or more substances or approaches to enhance the nature, magnitude, and longevity of the antigen/vaccine specific immune response stimulated by the vaccine as compared to the antigen/vaccine alone without causing any significant immunomodulation or Unauthenticated Download Date | 6/17/17 12:05 AM Nanocarriers for vaccine delivery 19 Figure 2: Schematic representation of steps involved in the generation of immune response ([29], modified). toxicity by its own [36, 38]. The selection of an appropriate adjuvant for a vaccine is governed by a number of factors including type of antigen, whether it is being co-administered with adjuvant or co-formulated with adjuvant, the proposed route of vaccination, vaccination schedule, and the nature of the desired immune response [38]. There are two types of vaccine adjuvants based on the mechanism of adjuvanticity: immunostimulants/ immunopotentiators, and particulate vaccine delivery systems. Immunostimulants/immunopotentiators directly activate the immune system through their interaction with specific receptors of the innate immune system. These include TLR agonists, NOD like receptor agonists, etc. [39]. Figure 3 depicts the different immunostimulants [40]. Particulate vaccine delivery systems, on the other hand, act by a number of mechanisms to enhance the immunogenicity of the antigen. Several mechanisms through which they act as adjuvants include: 1. Enhanced APC uptake. Particulate delivery systems have similar dimensions to pathogenic microbes, which our immune system has evolved to battle. This results in their enhanced uptake by APCs which are sentinels for pathogens [38]. 2. Co-formulation of antigen, immunostimulant, and targeting ligand. Particulate delivery systems offer an opportunity to co-deliver antigen, immunostimulant, and targeting ligand together in one system to the same APC, which has dual benefits. Firstly, it results in 3. 4. 5. 6. 7. effective activation of the APCs for a stronger adaptive immune response generation. Secondly, it avoids the toxic effects of the immunostimulant by restricting its systemic uptake [38]. Multivalent/multimeric presentation of antigen. Particulate systems like dendrimers and liposomes can also be designed so as to display multivalent copies of the antigen on their surface which closely mimics natural pathogens thereby generating a strong immune response [41]. Stability enhancement of vaccine/antigen. Particulate delivery systems, due to encapsulation or covalent association with antigen/vaccine, serve to protect the antigen/vaccine from pH or enzyme based degradation [9]. Allow mucosal vaccine delivery. Particulate delivery systems serve to enhance the transport of antigens through the mucosal tissue which otherwise show poor uptake through mucosa [9]. Targeting to APC. Delivery systems for vaccine delivery can be designed in such a way so as to achieve targeted delivery of antigen to APCs for maximum uptake using low doses of antigen [42]. Ability to trigger the cell-mediated arm of the immune response. It is generally seen that administration of antigen in soluble form leads to the induction of a humoral response whereas administration of antigen in a particulate carrier mimics a natural infection and is able to induce a cell-mediated immune response [9]. Unauthenticated Download Date | 6/17/17 12:05 AM 20 Priyanka Prabhu and Vandana Patravale Figure 3: Summary of immunostimulant molecules [40]. 8. Dose sparing effect. Vaccine delivery vehicles may reduce the dose of antigen required to elicit an immune response due to protection of antigen against degradation, due to enhanced mucosal transport, or due to targeted delivery to APCs [9]. 9. Enhance patient compliance. Vaccine delivery systems possess the ability to provide controlled release of the antigen resulting in prolonged presentation to the immune system thus mimicking natural infection. This reduces the number of vaccinations required, thereby increasing patient compliance [9]. Particulate vaccine delivery systems include micronsized (> 1000 nm) and nanosized systems (1 to 1000 nm). Nanoparticles offer an advantage over microparticles as vaccine delivery systems owing to their better uptake through APCs due to their similar size to invading pathogens, and the capacity of some nanocarriers such as liposomes, archaeosomes, niosomes, and dendrimers to offer multivalent immunostimulant and antigen presentation resulting in simulation of viral invasion [12]. 3.1 Need for vaccine adjuvants The need for vaccine adjuvants stems from the fact that the recent years have witnessed a paradigm shift from using inactivated/live attenuated vaccines to subunit antigens. These subunit antigens show poor immunogenicity owing to their inability to activate APCs and their limited half life in the body [9]. Secondly, although the clinical use of vaccine adjuvants can be traced back about 90 years, only a handful has been approved. In addition, the approved adjuvants are all for parenteral administration and licensed for a particular antigen by a single specified route only. Multiple reasons are responsible for this phenomenon. Firstly, prophylactic vaccines are administered to huge populations of healthy persons emphasizing the paramount requirement of safety. Secondly, approval of vaccine adjuvants by regulatory agencies entails separate registration of a given antigen/adjuvant combination for a particular route of administration [22]. Thirdly, in vitro APC uptake and activation studies, and immune response generation in rodent animal models in vivo, which are utilized to gauge the efficacy and safety of novel proposed vaccine adjuvants do not completely predict or translate into effective immune responses in humans for a number of reasons. Expression patterns of PRRs differ between humans and mice [31]. TLR9 expression occurs in different subsets of dendritic cells in mice and in humans. Also, TLR4 expression is substantial in mouse B cells whereas TLR4 expression is absent in human B cells during normal conditions [43]. Non-human primates offer a better option to study the mechanism of adjuvanticity and safety since they show greater resemblance to humans than mice as Unauthenticated Download Date | 6/17/17 12:05 AM regards to PRR expression and dendritic cell subsets. A major drawback associated with the use of non-human primates is their availability and huge maintenanceassociated expenses [31]. Alum has enjoyed unparalleled monopoly as the only FDA approved vaccine adjuvant for almost eight decades [1]. However, there are some serious drawbacks associated with alum. It induces a biased humoral immune response, rendering it useless for vaccination against intracellular infections where there is an urgent need for cellular immunity [4]. Additionally, alum cannot be freeze dried since it results in destruction of the colloidal structure, and it is ineffective for mucosal vaccination [29]. Alum is also ineffective in inducing immunity against small peptides and vaccines such as influenza and typhoid fever vaccines [19], and is known to cause local adverse effects such as granuloma at the site of injection [42]. However, the last twenty years have witnessed the approval of a few novel vaccine adjuvants. MF59 is a vaccine adjuvant approved in Europe in the Fluad® influenza vaccine by Novartis. It is an oil-inwater emulsion (160 nm) containing 5% squalene, 0.5% polysorbate 80, and 0.5% sorbitan trioleate. AS03™ by GlaxoSmithKline (GSK) is another approved emulsion based adjuvant (150 nm) employed in the Pandemrix™ influenza vaccine. It contains squalene, tocopherol, and Tween 80 [39,44]. AS04 by GSK is another approved adjuvant containing alum and monophosphoryl lipid A (MPLA). It is used in two licensed vaccines made by GSK, namely, Fendrix® for hepatitis B and Cervarix® for cervical cancer caused by HPV [45]. Nanocarriers for vaccine delivery 21 Salient features of an ideal vaccine adjuvant [18,42] are summarized in Figure 4. 3.2 Parameters impacting immune response of nanocarriers The various factors influencing the immune response elicited by nanocarriers are discussed below. 3.2.1 Particle size Particle size of the antigen delivery system plays a pivotal role in its uptake by the APCs at the site of action or by the lymphatic vessels to reach the lymph nodes. Particles with a size of less than 1000 nm are apt for uptake by macrophages and dendritic cells. The impact of antigen delivery system size on the resultant immune response generated also depends on the route employed. Microparticles elicit effective immunity after oral and nasal administration. Their larger size probably facilitates their uptake into the GALT and NALT for greater mucosal immunity. Particles in the size range of 20-50 nm are suitable for transport through lymphatic vessels and lymph nodes [8]. Particles of 500 nm or smaller are ideal for dentritic cell or macrophage uptake, 20-200 nm particles are mostly uptaken via endocytosis resulting in a Th1 type immune response, whereas particles greater than 500 nm in size are uptaken via phagocytosis or macropinocytosis resulting in a humoral immune response [46]. Figure 4: Salient features of an ideal vaccine adjuvant [18,42]. Unauthenticated Download Date | 6/17/17 12:05 AM 22 Priyanka Prabhu and Vandana Patravale However, study results comparing the immune responses elicited by nanoparticles and microparticles are contradictory. Some reports support the use of nanoparticles to elicit a stronger immune response whereas some studies report the opposite. Kanchan et al. demonstrated that hepatitis B surface antigen loaded poly lactic acid (PLA) microparticles (2-8 µm) showed higher antibody levels than hepatitis B surface antigen-loaded PLA nanoparticles (200-600 nm). The microparticles elicited a Th2 type response whereas the nanoparticles elicited a Th1 type immune response [47]. Jung et al. compared the immune response of tetanus toxoid adsorbed onto sulfobutylated poly(vinyl alcohol)-graftpoly lactic-co-glycolide (PLGA) particles and reported that particles of 100 and 500 nm resulted in greater antibody levels as compared to the particles greater than 1000 nm by the oral and intranasal route [48]. Caputo et al. demonstrated using HIV Tat protein that adsorption onto cationic polymeric nanoparticles (220 or 630 nm) elicited a strong cell-mediated immune response and a poor antibody based immune response than the protein adsorbed onto microparticles (2-8 µm) [49]. 3.2.2 Surface characteristics Surface characteristics such as shape, hydrophobicity, and surface charge are reported to influence phagocytic uptake by APCs. Positive charge and cylindrical or spherical shape enhance more efficient phagocytic uptake than negatively charged and disk shaped particles. Also, hydrophobic surfaces are readily opsonised by endogenous serum proteins and in the interstitial fluid at the site of injection resulting in augmented uptake by APCs [38]. 3.2.3 Route of administration Administration of the same vaccine delivery system of a particular particle size via different administration routes results in different types of immune responses which can be attributed to the different types of APCs and different numbers present at various sites in the body. Intraperitoneal immunization results in uptake by macrophages present in the peritoneal cavity whereas ID immunization leads to greater uptake by the dendritic cells. Different APCs vary in their ability to process antigen and produce immunity [50]. 3.2.4 Type of antigen Bovine serum albumin (BSA), ovalbumin, tetanus toxoid, hepatitis B surface antigen, and HIV Tat protein are the most widely used antigens to evaluate potential vaccine adjuvants. Different antigens vary with respect to their endotoxin content, inherent immunogenicity, and purity as a result of which a given adjuvant may elicit a different immune response depending on the antigen [50]. A good example of this is a report highlighting the difference in immune response obtained with ovalbumin and Bacillus anthracis protective antigen. Conjugation of ovalbumin onto lecithin solid lipid nanoparticles (SLN) showed greater antibody levels than those obtained with ovalbumin and aluminium hydroxide. Conjugation of Bacillus anthracis protective antigen onto the lecithin SLN showed similar immune response to that of alum adjuvanted Bacillus anthracis protective antigen [46]. 3.2.5 Antigen release Release of antigen is governed by the manner in which it is loaded onto or associated with the nanocarrier. Antigens can be covalently attached to nanocarriers or physically adsorbed onto the surface via electrostatic interactions or entrapped/encapsulated within the core of the nanocarrier. Adsorption onto the nanocarrier surface results in burst release of antigen or premature release of antigen before uptake by the APCs. Covalent coupling requires enzymatic activity to break the linkage and release the free antigen. Encapsulation or entrapment involves dependence on the degradation of the matrix material in order to achieve antigen release. This leads to prolonged or controlled release of antigen [50]. However, the ideal method of loading would be to have some antigen on the surface which would be released immediately to initiate the immune response, in addition to sustained release of the entrapped antigen over a period of time to help to boost the immune response, thus eliminating the need for multiple booster administrations and improving patient compliance. Constant antigen delivery commonly results in the generation of an efficient immune response as a persistent encounter with the antigen permits sufficient affinity maturation and antibody isotype switching, both of which play a key role in generation of a strong antigenspecific immune response [51]. Unauthenticated Download Date | 6/17/17 12:05 AM 3.2.6 Presence of immunostimulants Inclusion of immunostimulants along with nanocarriers further enhances the immune response compared to nanocarriers alone [39]. 3.3 Upcoming strategies for successful vaccine delivery This section highlights some upcoming approaches which are employed to further enhance adjuvanticity of nanocarriers. 3.3.1 Targeted delivery to APCs Various ligands may be employed for the targeted delivery of antigen-loaded nanoparticles to the dendritic cells or other APCs. Nanocarriers decorated or functionalized with mannose, fucose, or N-acetylglucosamine can be used to interact with the lectin-like receptors (e.g. mannose receptors, DC-SIGN, and DEC-205) present on dendritic cell surfaces. Phosphatidyl serine may be included while fabricating liposomes to promote interaction with phosphatidyl serine receptors on monocytes. Monoclonal antibodies against DC-SIGN or CD11c receptors may also be employed to target dendritic cells [29, 52]. DC-SIGN is mainly expressed on dendritic cells at mucosal areas, skin, and lymph nodes [53]. Receptors employed for dendritic cell targeting are chosen based on the following criteria. Firstly, the receptor must show preferential expression on dendritic cells compared to other cells. Secondly, antigen delivered to the dendritic cell via the receptor should be internalized, processed and presented in association with both MHC class I and II molecules to elicit a balanced humoral and cellular immune response. Thirdly, the receptor binding should induce immunostimulatory signals essential for dendritic cell maturation and migration in order to avoid tolerance and to efficiently activate naive T cells [52]. 3.3.2 Use of intelligent particulate carriers to achieve cytosolic delivery of antigen Strategies which may be employed in order to achieve cytosolic antigen delivery include use of endosome disrupting agents in order to allow antigen delivery to the cytosol where MHC class I presentation commences [54]. There are six different types of intelligent carriers, which disrupt the endosome in different ways. The first Nanocarriers for vaccine delivery 23 type of carriers is pH responsive: when they reach the acidic endosome (pH 5.9-6), they release the antigen and disturb the endocytic vacuole resulting in antigen delivery to cytosol. The second type includes use of fusogenic components in the liposomal membrane. Sendai virus proteins, when included in liposomes, confer a fusogenic property to the carrier resulting in fusion with endosomes to deliver antigen into the cytosol. The third type involves use of cell penetrating peptides for delivery to cytosol [55]. Octaarginine, when included in liposomes containing ovalbumin, led to more efficient cytotoxic T cell responses than cationic liposomes and pH responsive liposomes [56]. The fourth type involves use of pore forming substances which directly perforate the endosomal membrane and reach the cytoplasm. Pore forming proteins which have been used to deliver antigen into the cytosol of dendritic cells are porins from Shigella dysenteriae and Listeriolysin O from Listeria monocytogenes [55]. The fifth type includes bubble liposomes. Perfluoropropane gas is included inside the liposomes which, on exposure to ultrasound, are able to disrupt the cell membrane and deliver antigen into cytoplasm [55]. Exposure of dendritic cells to bubble liposomes and ultrasound resulted in 3 fold higher IL-2 production compared to ovalbumin solution alone or ovalbumin solution plus ultrasound [57]. The sixth type includes use of virosomes and virus-like particles (VLPs) [55]. Virosomes are liposomes containing glycoproteins extracted from isolated viruses and inserted into the lipidic membrane bilayer [58]. Glycoproteins include hemagglutinin and neuraminidase [39]. The first subunit of hemagglutinin helps in anchoring to the sialic acid residues present on the APC surface, and the second subunit assists fusion of the endocytosed virosome with the endosome leading to delivery of the antigen into the cytoplasm [59]. Virosomes are unilamellar and spherical, with a size of 150 nm [60]. Virosome based vaccines approved in Europe include Epaxal ® (hepatitis A) and Inflexal® V (influenza) [58]. The vaccine adjuvant activity of virosomes may be attributed to number of mechanisms. Their virus particle mimicking structure results in repetitive antigen presentation to B cells. They also provide a depot effect for sustained antigen presentation [60]. Additionally, virosomes also promote crosspresentation of antigen. They undergo uptake by APCs via receptor-mediated endocytosis and subsequently fuse with the acidified endosomes to release their antigen load into the cytosol [29]. VLPs are composed of viral structural proteins which self-assemble to form VLPs. They act as PAMPs and show potent B and T cell stimulation owing to the repetitive presentation of antigenic epitopes on their surface. Commercially available VLP based vaccines Unauthenticated Download Date | 6/17/17 12:05 AM 24 Priyanka Prabhu and Vandana Patravale include Gardasil® and Cervarix ® [58]. VLPs show antigen cross-presentation owing to their fusogenic capability resulting in antigen delivery to cytosol for effective MHC class I presentation leading to CD8+ T cell stimulation [58]. 3.3.3 Combination of immunostimulants and particulate delivery systems Another promising strategy is to formulate well known imunostimulants within particulate carriers to achieve a potentiated immune response. This technique also helps to eliminate the untoward effects seen with administration of free immunostimulant [43]. Also, one can reduce the amount of immunostimulant needed thus reducing its toxic effects. GSK has developed a technology called Adjuvant Systems based on the combination of traditional particulate adjuvants such as aluminium salts, liposomes, oil-in-water emulsions with immunostimulants such as MPLA and QS-21. MPLA, a strong TLR4 agonist, is obtained from the lipopolysaccharide present in the cell wall of Salmonella minnesota R595 strain and is detoxified by mild hydrolysis. QS-21 is obtained from the bark of the tree Quillaja saponaria, and induces cytotoxic CD8+ immune responses. A drawback associated with QS-21 is the haemolytic activity of the molecule, which formulation in particulate system helps to overcome. AS04 is composed of MPLA adsorbed onto aluminium hydroxide or aluminium phosphate. AS02 is composed of MPLA and QS-21 formulated in an oil-in-water emulsion to induce both humoral and cellular immunity. The RTS, S/ AS02 vaccine for malaria is currently undergoing clinical trials. AS01 is composed of QS-21 and MPLA formulated in liposomes to induce a strong CD8+ T cell response. The RTS,S/AS01 malaria vaccine is currently undergoing Phase III clinical trials [61]. The following sections will summarize the various nanocarriers explored for vaccine delivery. Figure 5 shows the various nanocarriers used to date for vaccine delivery. Figure 5: List of nanocarriers explored for vaccine delivery. Unauthenticated Download Date | 6/17/17 12:05 AM 4 Polymeric Nanoparticles Polymeric nanocarriers have garnered considerable attention for vaccine delivery over the years owing to their potential for providing continuous as well as pulsatile antigen release by modifying the polymer composition and nanoparticulate structure. This feature may enable single shot vaccination, however, this hypothesis is yet to be established clinically [58]. Polymers explored for vaccine delivery can be biodegradable or non-biodegradable. Polymers from both natural (starch, gelatin, alginate) and synthetic (PLGA, Polyanhydride) sources have been used. The most extensively investigated biodegradable polymers for vaccine delivery include polyesters (PLA, PLGA and polyanhydrides) [62]. Natural polymers offer the beneficial attributes of biocompatibility, low cost, and high aqueous solubility compared to their synthetic counterparts. Drawbacks associated with natural polymers include their poor hydrophobicity, batch to batch inconsistency, and occurrence of unwanted impurities. Synthetic polymers, on the other hand, are amenable to reproducible production and can be designed to achieve the desired rate of degradation, molecular weight, and co-polymer composition. But one major shortcoming of synthetic polymers is their solubility, commonly being soluble only in organic solvents, hampering the antigenicity of the antigen [63]. This section summarizes a number of polymers which have been explored for both parenteral and mucosal vaccination. 4.1 Chitosan nanoparticles Chitosan is a co-polymer of glucosamine and N-acetylglucosamine prepared by the deacetylation of chitin derived from insect skeleton [64]. Chitosan is one of the most intensively explored biomaterials in the field of vaccine delivery, both as an adjuvant and as a delivery platform [65]. The vaccine adjuvant activity of chitosan-based nanoparticles has been evaluated by several different routes such as nasal, SC, oral, IM, transcutaneous, and ID routes. This can be attributed to its peculiar properties which make it a promising material for vaccine delivery.It is non-toxic, biocompatible, and cost effective [66]. It is an FDA approved biomaterial [67]. Chitosan easily forms nanoparticles possessing high loading capacity for several proteins and other antigens [66]. Chitosan nanoparticles can be fabricated without exposure of the antigen to hostile conditions of heat or use of organic solvents [65]. They are spontaneously prepared using a precipitation/coacervation technique with the aid Nanocarriers for vaccine delivery 25 of tripolyphosphate as a precipitating agent [67]. Chitosan has been found to augment both cell-mediated and humoral immune responses. It is mucoadhesive thereby rendering it useful for mucosal vaccination. It also acts as a penetration enhancer by opening the tight junctions of epithelial cells present in the oral and nasal mucosae; thereby increasing the paracellular absorption of antigen [65]. Chitosan has been shown to possess macrophage activation ability and shows cytokine induction [66]. It also activates dendritic cells [65]. Activation of dendritic cells occurs through binding to TLR4 and mannose receptors [68]. Its positive charge confers it with superior uptake by APCs due to ionic interactions with the negatively charged cell membrane. The positive charge also allows electrostatic binding with negatively charged antigens and DNA for vaccine delivery [65]. Chitosan based nanoparticles possess higher stability than other vaccine delivery platforms such as liposomes and ISCOMs [7]. A chief downside associated with the use of chitosan is its poor solubility at physiological pH, as only the protonated form of chitosan in which it is present at acidic pH is able to act as a permeation enhancer. In order to surmount this problem, a partially quaternized derivative of chitosan known as N-trimethyl chitosan (TMC) chloride is synthesized which possesses excellent solubility over a broad pH range [69]. The immune response obtained with chitosan depends on the degree of deacetylation and molecular weight of chitosan [7]. Dzung et al. used chitosan of 30 kDa and 300 kDa to formulate chitosan nanoparticles of around 80 nm and 106 nm respectively loaded with A/H1N1 influenza antigen for SC vaccination. High molecular weight chitosan nanoparticles showed 3-fold higher antibody levels than the alum-adsorbed antigen, 2-fold higher antibody levels than the low molecular weight chitosan (30 kDa), and 100-fold higher antibody levels than plain antigen administered alone [70]. Prego et al. developed chitosan nanoparticles (160-200 nm) using an ionic gelation method. The nanoparticles showed susitained release of antigen and IM immunization in mice showed antibody levels which were 9-fold greater than those obtained by alum adjuvant [71]. Slutter et al. developed ovalbumin-TMC nanoconjugates using thiol chemistry for nasal immunization. The aim was to improve transport across the nasal mucosa using smaller nanoconjugates (28 nm) in comparison to the larger TMC nanoparticles (300 nm). The nanoconjugates demonstrated superior penetration through a lung carcinoma cell monolayer and also elicited higher ovalbumin-specific IgG and nasal IgA levels in mice compared to ovalbumin-loaded N-trimethyl chitosan Unauthenticated Download Date | 6/17/17 12:05 AM 26 Priyanka Prabhu and Vandana Patravale nanoparticles. The nanoconjugates elicited a mixed Th1/ Th2 response (rather than a biased Th1 or biased Th2 response) than ovalbumin-loaded N-trimethyl chitosan nanoparticles [72]. Khatri et al. have investigated the use of chitosan nanoparticles (337 nm) for nasal DNA vaccination using DNA encoding hepatitis B surface antigen. The nanoparticles induced serum antibody levels which were greater than the clinically protective levels. Nasal administration of nanoparticles resulted in generation of mucosal IgA antibody generation in nasal, salivary, and vaginal secretions which was not seen with IM administration of naked DNA and alum adsorbed hepatitis B surface antigen [73]. Sayin et al. have developed nanocomplexes of TMC and mono-N-carboxymethyl chitosan (283 nm) by simple electrostatic interactions for nasal delivery of tetanus toxoid and compared their efficacy with mono-N-carboxymethyl chitosan nanoparticles and TMC nanoparticles. Intranasal immunization of the nanocomplexes resulted in higher levels of both IgG1 and IgG2a compared to the plain antigen solution given nasally or subcutaneously. Also, intranasal immunization of the nanocomplexes led to a stronger immune response compared to nanoparticles fabricated from chitosan, mono-N-carboxymethyl chitosan, and TMC alone [74]. Bal et al. have also demonstrated the ability of ovalbumin- TMC nanoconjugates to elicit higher antibody levels and a greater number of ovalbumin positive dendritic cells in the lymph nodes after transcutaneous immunization compared to ovalbumin-loaded TMC nanoparticles. After ID immunization, there was no significant difference between the immune responses generated by ovalbumin-TMC nanoconjugates and ovalbumin-loaded TMC nanoparticles. This indicates that the size of the nanocarrier plays an important role for permeation through skin in transcutaneous immunization, whereas in case of ID immunization, the skin barrier is not involved for nanocarrier transport and hence both nanoconjugates and nanoparticles perform equally well [75]. Tafaghodi et al. coated hepatitis B surface antigen with chitosan and TMC separately. Nasal as well as IM immunization in mice with both the nanoparticles induced higher serum antibody levels than those obtained with intramuscularly administered alum adsorbed hepatitis B surface antigen. Nasal and IM immunization in mice with both nanoparticles also generated a cellular immune response, whereas the alum adsorbed hepatitis B surface antigen failed to do so. Nasal immunization also resulted in induction of secretory IgA in nasal and vaginal secretions, the latter would be valuable in curbing sexually transmitted hepatitis B infection [76]. Balet al. showed that the immune response obtained with chitosan nanoparticles differs depending on the adjuvant used and the route of administration. They developed TMC nanoparticles containing both ovalbumin and an immunostimulant and studied their immunostimulatory efficacy on nasal and ID vaccination. Five immunostimulants were employed: lipopolysaccharide (LPS), PAM3CSK4, CpG DNA, muramyl dipeptide (MDP), and cholera toxin B subunit. Detectable sIgA levels were not generated following ID vaccination of any of the nanoparticles. Nasal vaccination with nanoparticles containing ovalbumin/MDP or ovalbumin/ LPS resulted in higher serum IgG and IgA levels in nasal secretions compared to non-adjuvanted TMC-ovalbumin nanoparticles. Nasal vaccination was unable to induce significant IgG2a levels with any of the nanoparticles. ID vaccination with the nanoparticles containing ovalbumin/CpG or ovalbumin/LPS demonstrated higher serum IgG levels than non-adjuvanted TMCovalbumin nanoparticles. ID vaccination with CpG adjuvanted ovalbumin TMC nanoparticles resulted in induction of IgG2a levels. The differences observed in the immune responses could be explained on the basis of the expression of the PRRs such as TLRs and NODlike receptors depending on the dendritic cell subset and localization. MDP, which showed efficacy on nasal administration, binds to NOD 2, which along with other NOD like receptors is found in the dendritic cells in the nose. Also, the PRRs for LPS and CpG (both of which proved to be most effective on ID vaccination), namely TLR4 and TLR9, are present on keratinocytes, dendritic cells in the dermis, and Langerhans cells [77]. Zhao et al. entrapped a lentogenic live virus vaccine for Newcastle disease virus in chitosan nanoparticles. The nanoparticles were 371 nm in size. Oral and nasal vaccination with these nanoparticles conferred 100% protection in chickens after challenge with a highly virulent strain of Newcastle disease virus, whereas the inactivated Newcastle disease vaccine and live virus vaccine could not [78]. 4.2 PLGA PLGA is a biodegradable polymer approved by the FDA. These polymers are aliphatic polyesters containing different ratios of lactic acid and glycolic acid. PLGA undergoes degradation by bulk erosion, during which water diffuses faster into the polymeric matrix in comparison to the rate of covalent bond breakage. In this case, polymer degrades throughout the matrix until a particular molecular weight is attained at which the degradation products are tiny enough to be dissolved. The polymeric structure is highly hydrated and porous at this Unauthenticated Download Date | 6/17/17 12:05 AM point, leading to release of entrapped moiety. As a result, there is a lag period for release of large molecular weight substances from PLGA matrix. Degradation by bulk erosion is beneficial for vaccine delivery since it does not allow premature release of antigen or immunostimulant before the nanoparticles have reached the APCs, thus limiting the entry of immunostimulant and antigen into systemic circulation. After uptake of PLGA nanoparticles by APCs, endosome internalization of nanoparticles occurs, the acidic environment of which expedites the degradation of PLGA to release antigen or immunostimulant. The rate of PLGA degradation is governed by its molecular weight, hydrophilicity, and crystallinity. PLGA withsmaller molecular weight and greater hydrophilicity tends to degrade faster. Hydrophilicity can be adjusted by adjusting the ratio of monomers: lactic acid: glycolic acid. Glycolic acid is more hydrophilic, thus its usage in higher quantities leads to greater hydrophilicity and faster degradation. The more crystalline the polymer, the slower the degradation. Crystallinity is governed by stereochemistry, wherein, L-lactic acid is crystalline, the use of which results in slower degradation. D,L-lactic acid is always employed to obtain a uniform dispersion of the antigen in the polymeric matrix. PLGA withlactic acid: glycolic acid in the ratio 50:50 is least crystalline, resulting in rapid degradation with a half life of about two weeks. Another important advantage of PLGA nanoparticles for vaccine delivery is their ability to cause cross-presentation of antigen resulting in both humoral and cell-mediated immunity [79]. Shen et al. have demonstrated this phenomenon using ovalbuminloaded PLGA nanoparticles. The nanoparticles induced T cell IL-2 secretion in bone marrow derived dendritic cells at 1000 times lower concentration compared to antigen administered in the soluble form, and 10 times lower compared to antigen-coated latex beads. The nanoparticles showed sustained antigen presentation via the MHC class I pathway for 72 hours [80]. PLGA hydrolyses into lactic and glycolic acid, both of which are biocompatible and easily bio-transformed by the body [81]. A grave concern associated with the use of PLGA nanoparticles for vaccine delivery is the creation of an acidic microenvironment on degradation, which may endanger the integrity and immunogenic potential of the vaccine [62]. Additionally, the use of organic solvents in the generation of PLGA nanoparticles may destroy the immunogenicity of the antigen [82]. PLGA nanoparticles have been formulated with a variety of antigens for both parenteral and mucosal routes. In certain cases, both antigen and immunostimulant have been co-formulated in PLGA nanoparticles in order to achieve a potent antigen-specific immune response. Also, targeted delivery Nanocarriers for vaccine delivery 27 of PLGA nanoparticles to dendritic cells has been achieved resulting in potent immune activation. Bharali et al. fabricated methoxypolyethylene glycolPLGA nanoparticles containing recombinant hepatitis B surface antigen which, on intraperitoneal immunization in mice, showed a higher and faster antibody generation compared to plain antigen [83]. Muttil et al. formulated recombinant hepatitis B surface antigen in PLGA/ PEG nanoparticles and spray dried the formulation. Pulmonary immunization in guinea pigs elicited systemic immunity comparable to parenteral immunization and mucosal immune response in the lungs [84]. Elamanchili et al. co-formulated the cancer associated antigen MUC1 mucin peptide (BLP25) and MPLA in PLGA nanoparticles and showed better stimulation of naive T cells compared to soluble antigen and MPLA [85]. Raghuwanshi et al. fabricated PLGA based nanoparticles for targeted delivery to dendritic cells. These were made by blending recombinant fusion protein with biotinylated PEG-PLGA nanoparticles containing ovalbumin. The recombinant protein contained strepatividin bound to single chain antibody against DEC-205 receptors found on dendritic cells. In vivo immunization in mice using the targeted delivery system in combination with a dendritic cell maturation agent also showed an enhanced immune response against ovalbumin. Co-administration of dendritic cell maturation agent is necessary as DEC-205 targeting in its absence leads to antigen-specific tolerance [86]. Hamdy et al. developed mannan conjugated PLGA nanoparticles for targeted dendritic cell delivery via mannose receptors. The nanoparticles showed increased CD4+ and CD8+ T cell immune response against ovalbumin in comparison to their non-targeted counterparts. Mannan, in addition to being a targeting ligand, is also reported to possess dendritic cell activation ability possibly due to TLR agonism. The nature of mannan – oxidized or reduced has been shown to guide the type of immune response -CD4+ or CD8+. Oxidized mannan causes endosomal escape of antigen into the cytoplasm resulting in a CD8+ response, whereas reduced mannan stays in the endosome, is degraded by the lysosomal enzymes and results in a CD4+ response [87]. 4.3 PLA Mattheolabakis et al. formulated ovalbumin-containing PLA nanoparticles (150 nm size) using the double emulsion technique for transcutaneous immunization. The nanoparticles demonstrated equivalent antibody generation to ovalbumin in solution; however, the cytokine production (IL-2, IFN-γ) was enhanced by administration of Unauthenticated Download Date | 6/17/17 12:05 AM 28 Priyanka Prabhu and Vandana Patravale ovalbumin in nanoparticles along with cholera toxin [88]. Jain et al. synthesized block co-polymer of PEG-PLA (PEGPLA-PEG) and fabricated its nanoparticles for mucosal delivery of hepatitis B surface antigen. The aim was to avoid the creation of an acidic milieu on degradation of PLA by making it more hydrophilic. The block copolymers assemble in water to give rise to a core shell structure with the shell made of PEG and the core made of PLA. The system demonstrated a triphasic release pattern beneficial for vaccine delivery. The nanoparticles showed comparable humoral immune response after single administration as the alum adsorbed hepatitis B surface antigen did after boosters. Additionally, the PEG-PLA-PEG nanoparticles also produced a mucosal immune response not only in the nasal mucosa but also in salivary, intestinal, and vaginal areas which was superior to that obtained after the administration of PLA nanoparticles. Also, the immune response generated was a mixed Th1/Th2 response [89]. Florindo etal fabricated PLA nanospheres encapsulating recombinant Streptococcus equi. M-like protein and S. equi protein extract for IM immunization in horses. The research group also incorporated immunostimulant molecules such as spermine, oleic acid, alginate and glycol-chitosan. The nanoparticles demonstrated a more effective humoral immune response than soluble protein alone or mixed with CpG. Nanospheres made using glycol chitosan showed a mixed Th1/Th2 response shown by the generation of high IgG1 and IgG2a titers and induction of IL-2 and IFN-γ levels. The nanospheres made using glycol chitosan had a positive surface charge which is known to enhance uptake by APCs [90]. Pavot et al. studied the immunostimulating potential of Nod1 and Nod2 ligands encapsulated within PLA nanoparticles. The nanoparticles showed excellent uptake by dendritic cells in vitro and showed upregulation of surface molecules and pro-inflammatory cytokine generation. Co-administration of nanoparticle encapsulated Nod ligands and PLA nanoparticles (200 nm) adsorbed with Gag p24 HIV-1 antigen subcutaneously in mice showed a 100-fold higher antibody generation as compared to alum adsorbed antigen [91]. 4.4 Polyanhydride nanoparticles Ulery et al. developed biodegradable polyanhydride nanoparticles (200 nm) for single shot intranasal vaccination against pneumonic plague. Yersinia pestis, the pathogen responsible for pneumonic plague, enters the human body through the respiratory tract, and hence intranasal vaccination, which can induce both local mucosal and systemic immunity, would be desirable. The developed nanoparticles induced higher F1-V specific antibody titers, the IgG1 antibodies showed higher avidity for F1-V, and the antibody response remained upto 23 weeks after vaccination in comparison to recombinant protein F1-V alone or MPLA mixed with F1-V. The nanoparticles were able to confer long term protection in mice against a lethal Yersinia pestis challenge [92]. Salman et al. developed mucoadhesive polyanhydride nanoparticles coated with mannose or Salmonella enteritidis derived flagellin for oral vaccination. Polyanhydride nanoparticles coated with either of the ligands (300-400 nm) were found to generate higher IgG1 and IgG2a antibody responses compared to the plain ovalbumin-loaded polyanhydride nanoparticles after single oral and SC administration. Oral vaccination of both mannosylated and flagellin coated nanoparticles showed higher intestinal secretory IgA antibody generation compared to SC administration. Mannose binds to mannose binding lectins (C-type lectin receptors) present on the surface of dendritic cells and other gut lymphoid cells enhancing uptake of the nanoparticles by APCs. The flagellin coating serves a dual purpose of acting as a TLR5 ligand and as a mucoadhesive. TLR-5 agonist binding leads to maturation and activation of dendritic cells resulting in a stronger immune response [93]. 4.5 Poly (γ-glutamic acid) Poly(γ-glutamic acid) is a bacterial capsular exopolymer synthesized by particular strains of Bacillus natto. Poly(γglutamic acid) undergoes degradation in the human body by γ- glutamyl transpeptidase. Poly(γ-glutamic acid) nanoparticles show uptake by dendritic cells followed by localization in lysosomal regions. Poly(γ-glutamic acid) induces dendritic cell maturation as evidenced by production of cytokines such as IL-12, TNF-α, and upregulation of maturation markers such as CD40, CD80, and CD86. Poly(γ-glutamic acid) nanoparticles induce dendritic cell maturation via the MyD88-mediated NF-κB signalling pathway [94]. SC vaccination with the influenza virus hemagglutinin generates only a virus-specific antibody response but fails to generate cellular immune responses, which are needed to tackle the intracellular virus burden. Okamoto et al. developed hemagglutinin-loaded polymeric nanoparticles made from poly(γ-glutamic acid)-graftL-phenylalanine copolymer for vaccination against influenza virus. SC immunization in mice resulted in augmented virus-specific antibody and cellular immune response generation compared to hemagglutinin alone and hemagglutinin with alum and induced protection in Unauthenticated Download Date | 6/17/17 12:05 AM mice against lethal influenza virus challenge [95]. Okamoto et al. have also demonstrated the potential of nasally administered influenza hemagglutinin along with poly(γglutamic acid) nanoparticles to produce cross-protection in mice against influenza virus. Intranasal immunization with A/New Caledonia 20/99 hemagglutinin vaccine plus poly(γ-glutamic acid) nanoparticles resulted in effective antibody and cell-mediated immune responses against both A/New Caledonia 20/99 virus strain and A/PR/8/34 strain. Intranasal immunization with a blend of hemagglutinin and poly(γ-glutamic acid) nanoparticles resulted in enhanced cytokine production (IL-4, IL-6, IFN-γ) compared to hemagglutinin alone or hemagglutinin administered with polyinosinic-polycytidylic acid (poly (I:C)) [94]. Wang et al. developed poly(γ-glutamic acid) nanoparticles containing ovalbumin. A good uptake of nanoparticles by dendritic cells was seen, accompanied by induction of their maturation. The physical mixture of poly(γ-glutamic acid) nanoparticles and ovalbumin also led to dendritic cell maturation. They also encapsulated HIV type-1 p24 in poly(γ-glutamic acid) nanoparticles. SC immunization with these nanoparticles demonstrated activation of antigen-specific IFN-γ secreting cells in spleen and led to serum antibody production. The serum antibody levels were comparable to those obtained with Complete Freund’s Adjuvant (CFA) [96]. Uto et al. developed ovalbumin-containing poly(γglutamic acid) nanoparticles. The nanoparticles resulted in production of both IgG1 and IgG2a antibodies in sera, antigen-specific IFN-γ levels in splenocyte, and cytotoxic T lymphocyte activation. Immunization with poly(γ-glutamic acid) nanoparticles containing CD8+ T cell epitope peptide of Listeria monocytogenes induced protection against a lethal challenge [97]. Uto et al. in another study showed that ovalbumin-containing poly(γ-glutamic acid) nanoparticles could produce greater CD8+T cell proliferation than ovalbumin administered with CFA. Also, the nanoparticles were devoid of any local side effects on SC injection, which is a major drawback associated with CFA [98]. Uto et al. have also demonstrated that after SC immunization of ovalbumincontaining poly(γ-glutamic acid) nanoparticles in mice, the nanoparticles were taken up by dendritic cells followed by their maturation and migration to regional lymph nodes and effective antigen presentation via the MHC class I pathway resulting in potent activation of ovalbumin-specific CD8+ T cells compared to plain ovalbumin and alum adsorbed ovalbumin [99]. 4.6 Polyphosphazene Polyphosphazenes are synthetic polymers comprising a backbone of alternating phosphorus and nitrogen atoms, with Nanocarriers for vaccine delivery 29 organic side groups tethered onto each phosphorus atom. Polyphosphazenes can be altered so as to contain ionic groups resulting in water solubility, and will undergo degradation via hydrolysis. Poly[di(carboxylatophenoxy)phosphazene] (PCPP) is the most extensively explored polyphosphazene for vaccine delivery [100]. Polyphosphazene forms noncovalent complexes with antigens resulting in improved stability of antigen and multimeric antigen presentation [101]. PCPP adjuvanted vaccines have been evaluated in clinical trials where they were shown to be safe and efficacious [102]. An aqueous formulation of PCPP when used in combination with influenza antigens showed 10-fold higher antibody levels than plain influenza antigen. PCPP has demonstrated good adjuvant activity with a plethora of other antigens including tetanus toxoid, hepatitis B surface antigen, HSV type 2 glycoprotein D, cholera, bovine serum albumin, and porcine serum albumin. IM immunization of rhesus monkeys with recombinant HIV-1 and PCPP led to the induction of long-lasting antibody levels for upto 43 weeks. A new polyphosphazene, poly[di(sodiumcarboxylat oethylphenoxy)phosphazene] (PCEP), when administered subcutaneously in mice demonstrated superior adjuvant activity to PCPP. Eng et al. administered influenza antigen alongwith both soluble PCPP and PCEP and showed that PCEP induced a stronger Th1 type response. Both PCPP and PCEP induced IgG and IgA antibodies in lung and vaginal washes. Another important advantage was the ability of PCPP and PCEP to induce long-lasting immunity for upto 8 weeks after single intranasal immunization [100]. Mapletoft et al. delivered bovine respiratory synctial virus (BRSV) co-formulated in combination with CpG oligodeoxynucleotide and polyphosphazene by the nasal route. The formulation induced both mucosal and systemic immune responses in mice and resulted in decreased viral replication on challenge [101]. Kovacs-Nolan et al. mixed indolicidin and CpG oligodeoxynucleotide 1826 with polyphosphazene and showed an enhanced ovalbuminspecific antibody response and cell mediated immune response on SC immunization. Indolicidin is a peptide containing 13 amino acids, obtained from cytoplasmic granules present in bovine neutrophils, and is known to stimulate chemokine IL-8 expression in bronchial epithelial cells [103]. Kovacs-Nolan et al. also used the combination of indolicidin and CpG oligodeoxynucleotide 1826 with polyphosphazene to induce immunity against BRSV infection in mice after SC immunization. The combination of adjuvants not only induced high virus neutralizing antibody levels but also resulted in enhanced IFN-γ secretion and protected mice against subsequent BRSV challenge [104]. Andrianov et al. demonstrated enhanced thermal stability and a dose sparing effect of PCPP on avian influenza antigen. Improved thermal Unauthenticated Download Date | 6/17/17 12:05 AM 30 Priyanka Prabhu and Vandana Patravale stability would be a boon for vaccine stocking in case of pandemic episodes and it would decrease the dependence on costly cold chain facilities for storage and distribution. PCPP resulted in a four-fold higher half life of the vaccine at 40o C. Also, IM immunization of the PCPP adjuvanted vaccine conferred 100% protection in ferrets at a 10-fold lower antigen dose than the non-adjuvanted vaccine [102]. Andrianov et al. also have formulated microneedles for ID immunization using PCPP. PCPP possesses ideal properties for use as film forming agent in microneedle technology owing to its high viscosity, water solubility, antigen stabilizing ability, and acceptability for human use. Unlike carboxymethylcellulose, PCPP does not require surfactants for fabrication of coated microneedles. PCPP-hepatitis B surface antigen coated microneedles containing 10 µg antigen when administered once ID elicited 10 times higher antibody levels than IM administered microneedles containing 20 µg antigen, 3-fold higher antibody levels than plain antigen administered IM, and 100-fold higher antibody levels than ID administration of non-adjuvanted antigen [105]. These studies all exemplify the immense potential of PCPP as a vaccine adjuvant owing to its biodegradability, efficacy and safety. It can certainly be utilized to formulate nanoparticles to accomodate both antigen and immunostimulant to further augment the immune response elicited. 4.7 Polystyrene Polystyrene is categorized as biocompatible by the FDA. Minigo et al. fabricated polystyrene nanoparticles for DNA vaccine delivery using poly-L-lysine as a cationic linker. Poly-L-lysine served to enhance bonding between negatively charged DNA (encoding for chicken egg ovalbumin) and polystyrene nanoparticles. ID vaccination in C57BL/6 mice demonstrated ovalbuminspecific antibody generation, cellular immune response production, and reduced tumour formation post challenge with ovalbumin expressing tumour. Nanoparticles of 50 nm were found to generate the most effective immune response. Additionally, polystyrene nanoparticles and DNA admixed without cationic linker were unable to induce immunity, indicating that the enhanced immune response was not just due to dose sparing of DNA (by avoiding its degradation) [106]. 4.8 Polypropylene sulphide (PPS) Stano et al. fabricated PPS nanoparticles (50 nm) and conjugated them with thiolated ovalbumin for nasal vaccination. The disulfide linkage connecting the antigen and the nanoparticle is cleaved in the reductive environment of the phagosome or endosome to release the antigen. The nanocarriers demonstrated permeation through nasal mucosa and were transported through the M cells followed by uptake into APC in NALT. Humoral immune responses in the airways and cytotoxic T lymphocyte responses in spleen and lung were generated. Flagellin was also conjugated as a TLR agonist, which led to the generation of mucosal immunity in vaginal and rectal mucosae due to CMIS [107]. Hirusoe et al. have demonstrated that the reduction labile linkage (disulfide) between the antigen and PPS nanoparticle is beneficial for successful presentation of exogenous antigen via the MHC class I pathway since it led to effective CD8+ T cell stimulation both in vitro and in vivo compared to the presence of a non-reductive linkage (vinyl sulfone) [108]. 4.9 Gantrez AN Gantrez AN is a copolymer composed of methyl vinyl ether and maleic anhydride and can simply react with amino groups. Gomez et al. fabricated Gantrez nanoparticles containing ovalbumin in encapsulated or coated form. The nanoparticles exhibited superior immune response after single ID administration compared to free ovalbumin and alum adsorbed ovalbumin. Gantrez nanoparticles containing encapsulated ovalbumin and a low amount of the cross linker 1,3-diaminopropane elicited a predominant Th1 response [109]. Salman et al. developed ovalbumin-containing thiamine-coated Gantrez nanoparticles to target Peyer’s patches for effective oral vaccination. Thiamine is known to interact with particular receptors in the intestine. Thiamine-coated nanoparticles were prepared by the reaction between the amino groups of thiamine and the anhydride group of Gantrez ® AN. Oral vaccination with the thiamine-coated nanoparticles effectively induced both systemic immunity (IgG1 and IgG 2a), and secretory antibody IgA (4 titers higher) in comparison with uncoated nanoparticles [110]. 4.10 Polyethyleneimine (PEI) Chen et al. utilized PEI based nanocarriers for cross presentation of ovalbumin. Ovalbumin was bound to PEI through electrostatic interactions. PEI shows a well established “proton sponge effect” wherein the nonprotonated amine groups of PEI undergo protonation in the acidic pH of endosomes, resulting in osmotic swelling and bursting of endosomes. As a result, ovalbumin is released from the endosome into the cytosol for MHC class I presentation. The nanoparticles were shown to present Unauthenticated Download Date | 6/17/17 12:05 AM ovalbumin via the MHC class I pathway in mouse bone marrow derived dendritic cells, and these dendritic cells were found to stimulate IL-2 secretion in RF33.70 cells [111]. 4.11 Dendrimers Dendrimers are defined as synthetic and extensively branched macromolecules composed of an inner core surrounded by many branches attached to it similar to a tree. Dendrimers are fabricated using polymers such as polyethylenimine, polyamidoamine (PAMAM), poly-(N-isopropylacrylamide), poly-(L-glutamic acid), polypropyleneimine, and polyethylene glycol [12]. PAMAM dendrimers are spherical monodisperse polymers with ahighly branched and symmetric structure. Their commercially available form has activated functional groups, thus permitting the attachment of different antigens, targeting ligands through simple chemical reactions. Dendrimers with 3 or 4 generations are ideal with regards to size and multivalency for DC-SIGN targeting [53]. Sheng et al. developed mannosylated dendrimers to achieve avid binding of an antigen delivery system to mannose receptors on dendritic cells. Mannosylated PAMAM dendrimer was linked to ovalbumin by controlled chemical synthesis. ID immunization in mice showed higher production of ovalbumin-specific CD4+ T cell and CD8+ T cell responses and higher serum IgG levels compared to plain ovalbumin. Delivery of ovalbumin using mannosylated dendrimer showed cross-presentation of ovalbumin in vivo. Immunized mice, when subcutaneously challenged with B16ovalbumin tumours, demonstrated late onset, retarded tumour growth and superior survival compared to mice immunized with plain ovalbumin [112]. Zaman et al. developed a polyacrylate ester dendrimer for intranasal delivery of J14 peptide to induce immunity against infection due to Streptococcus pyogenes. J14 peptide was chemically tethered onto the polyacrylate ester based dendrimer to form a self-assembling nano-sized (21 nm) vaccine delivery platform. Intranasal vaccination in mice showed production of J14 specific serum IgG antibody [113]. 5 Lipidic nanocarriers Lipidic nanocarriers are biocompatible and hence preferred over polymeric nanocarriers for vaccine delivery. This section highlights the various lipidic nanocarriers that have been explored for antigen delivery. Nanocarriers for vaccine delivery 31 5.1 Liposomes Liposomes are vesicular structures composed of an aqueous core surrounded by a lipid bilayer. Liposomes offer several beneficial features for vaccine delivery. The bilayered character of liposomes affords inclusion of both hydrophilic and hydrophobic constituents, and they offer protection to the antigen against enzymatic degradation. Additionally, their composition, charge and size could be altered so as to modulate their pharmacokinetic properties and their particulate character results in good uptake by APCs. Notably, they are biocompatible and biodegradable [114]. However, they also possess some drawbacks including the expense of raw materials, susceptibility of phospholipids to oxidative breakdown, and requirement for special storage conditions [12]. Liposomes have also been explored to enhance the immunostimulant activity of several immunostimulant molecules such as TLR ligands. Incorporation of MPLA into cationic liposomes further enhanced its immunostimulant potential. CAF01 is a promising cationic liposome-based adjuvant. It has a size of 500 nm and a charge of + 50 mV. It comprises dimethyldioctadecyl ammonium bromide and trehalose 6’6-dibehenate (TDB) in a 5:1 ratio. TDB is a synthetic analogue of mycobacterial cord factor, with a lower toxicity, and is an agonist of C-type lectin receptor. CAF01 systems have been explored for vaccination against malaria, tuberculosis, and chlamydia. CAF01 liposomes are undergoing clinical trials for tuberculosis vaccine. The CAF01 system has been shown to generate both humoral and cellular immune responses. CAF01 has also been explored in nasal influenza vaccination. Incorporation of TLR3 ligand poly(I:C) in CAF01 has also been studied. The CAF01 system allows both entrapment and adsorption of antigens. It can be lyophilized and reconstituted without hampering its immunogenic character, and can also be subjected to sterile filtration and γ-radiation based sterilization [114]. Liposomes have also been investigated for nasal vaccination. Bioadhesive liposomes have been prepared in order to extend the nasal retention time of liposomes. Chitosan adsorption to the surface of liposomes not only confers bioadhesive properties to the liposome but also induces a positive surface charge, enhancing uptake by nasal mucosa [115]. Chiou et al. fabricated a mixture of liposomes containing inactivated avian influenza virus and xanthan gum or tremella gum. The bioadhesive liposomes elicited a superior immune response in intranasally immunized chickens as compared to nonbioadhesive liposomes. Apart from their mucoadhesive property, xanthan gum and tremella are also reported to possess some immunostimulant function [116]. Unauthenticated Download Date | 6/17/17 12:05 AM 32 Priyanka Prabhu and Vandana Patravale A number of factors are known to significantly impact the nature and intensity of the immune response elicited by liposomes, and are listed below [117]. Method of antigen association: Antigen can either be physically or chemically associated with the liposome. One can encapsulate the antigen inside the liposome, covalently conjugate the antigen with liposome either before or after vesicle formation, or adsorb the antigen onto the surface via non-covalent interactions. Surface association of the antigen results in an enhanced antibodymediated response since B cell receptors can recognize the antigen on the surface of liposomes. Covalent conjugation of antigen to lipid or liposomes has been shown to enhance MHC class I presentation and subsequently an enhanced cellular immune response. Lipid composition: Some lipids show immunostimulant activity. Incorporation of these lipids would enhance the vaccine adjuvant potential of liposomes. A number of these lipids are described. Alltrans retinoic acid, which is a metabolite of vitamin A, has been shown to possess immunomodulatory activity via retinoic acid receptor agonism. Lauric acid has been shown to stimulate cytokine secretion and expression of costimulatory molecules on dendritic cells via TLR4 signalling. Palmitic acid has been shown to activate NLRP3-ASC inflammasome. Lysophosphatidylcholine is known to enhance CD86 expression, chemokine secretion, and upregulation of MHC class II molecules. DiC14-amidine, which is a cationic transfection agent, has been shown to be a TLR4 agonist. Charge: Cationic charge favours uptake by APCs owing to the negatively charged cell membrane. Bilayer fluidity: The more rigid the lipidic bilayer, the greater the immunostimulant activity of the liposome. As a result, lipids with higher gel-liquid crystal transition temperature show greater immunogenicity compared to those with lower gel-liquid crystal transition temperature. Size: 250-700 nm vesicles may result in a stronger Th1 response. Lamellarity: Multilamellar vesicles may result in an increased Th2 response. Fusogenicity: Inclusion of certain fusogenic lipids (dioleyl phosphatidyl ethanolamine) in the liposomal bilayer confers onto the liposome the property of fusing with the plasma membrane or the endosomal membrane. Fusion with the plasma membrane aids in enhanced uptake of liposomes by the APCs, while fusion with the endosomal membrane results in cross presentation of antigen. Fusogenic liposomes are extremely beneficial in delivery of DNA vaccines wherein it is imperative to release the vaccine cargo in the cytosol. Addition of immunostimulant: Addition of immunostimulant molecules such as TLR ligands, and NLR ligands enhances the immunogenic potential of liposomes. Tiwari et al. developed in situ gelling liposomes (719 nm) containing hepatitis B surface antigen for nasal vaccination. Liposomes were prepared by hydration of the lipid film with antigen solution containing polyacrylic acid. Polyacrylic acid shows pH dependent gelling and mucoadhesive properties, which leads to prolonged retention in the nasal cavity and enhanced mucosal uptake. The unentrapped polyacrylic acid containing antigen solution was used as continuous phase for suspending the gel core liposomes before administration. Intranasal immunization of these liposomes resulted in potent systemic and mucosal antibody responses as well as cellular immune response. IM administration of alum adsorbed antigen failed to generate both mucosal IgA and cellular immune responses. The liposomes showed a triphasic release behaviour in which the first phase involved burst release of antigens present on the surface of the hydrogel. The second phase involved release of the antigen via diffusion from the gel and liposomes. The third phase involved release of antigen from polyacrylic acid gel inside the liposomes, followed by its diffusion through phospholipid bilayer [118]. Tiwari et al. in another study formulated transmission-blocking malaria antigen Pfs25 in gel core liposomes fabricated from polyacrylic acid. IM immunization resulted in higher serum antibody levels than plain liposomes and alum adsorbed antigen. Also, the co-administration of CpG oligodeoxynucelotide in liposomes led to a more potent immune response compared to plain gel core liposomes. In conclusion, gel core liposomes offer great potential for single-shot vaccination, which is desirable from a patient compliance perspective [119]. Copland et al. developed mannosylated liposomes (260 nm) containing tetanus toxoid and studied the effect of mannosylation on uptake, maturation, and induction of T cell proliferation by dendritic cells. Mannosylated liposomes showed the highest expression of CD80, CD86, MHC class II molecules, and dendritic cell maturation marker CD83, and the greatest T cell proliferation. This was followed by plain liposomes, then plain antigen solution [120]. Yuba et al. fabricated liposomes encapsulating ovalbumin thatwere functionalized with succinylated poly (glycidol) and 3-methylglutarylated poly (glycidol). Functionalization with these polymers having carboxylic groups endowed the liposomes with the ability to fuse with the endosomes and release the antigen load into the cytosol for MHC class I presentation thereby eliciting Unauthenticated Download Date | 6/17/17 12:05 AM a strong cell mediated immune response. Liposomes modified with 3-methylglutarylated poly (glycidol) showed greater pH responsive fusogenic potential owing to the more hydrophobic nature of the polymer. Intranasal immunization led to superior cell-mediated immune response compared to unmodified liposomes and cellular immunity comparable to that of CFA [121]. 5.2 Archaeosomes Archaeosomes are vesicles enclosing lipid bilayer/s of total polar lipids extracted from microbes of the class Archaea. Archaeosomes exhibit greater uptake by macrophages and other APCs compared to liposomes. Apart from their uptake by APCs, they also promote recruitment and activation of APCs and carry the antigen to both MHC class I and MHC class II pathways for presentation of antigen to T cells, resulting in induction of potent humoral and cellular immune responses. Polar lipids obtained from Archaea differ from the lipids obtained from Eukarya and Bacteria. Phospholipids obtained from Eukarya and Bacteria are generally composed of unbranched and commonly unsaturated fatty acyl chains of different lengths, and are tethered through ester linkages to the sn-1,2 glycerol carbons of the glycerol backbone. Polar lipids of Archaea are composed of regularly branched 5-carbon repeating units, resulting in the formation of isopranoid chains which are commonly saturated and tethered via ether linkages to the sn-2,3 glycerol carbon atoms. Archaeosomes offer various advantages as compared to conventional liposomes. Due to the saturated character of the lipid chains they can be fabricated and stored in the presence of air/oxygen. They give rise to bilayer membranes with greater stability and lower permeability than conventional liposomes. They show 3 to 53 times greater uptake by phagocytes than conventional liposomes. They are selfadjuvanting vaccine delivery systems and do not need co-administration or co-formulation of immunostimulant molecules, unlike liposomes. They are safe and exhibit a good shelf life of more than 2 years [122]. Also, their immunostimulatory properties are independent of the inflammation in the host caused by infection [123]. Li et al. formulated archaeosomes composed of polar lipid fraction E of Sulfobus acidocaldarius for oral immunization with ovalbumin. The archaeosomes demonstrated excellent stability in simulated gastric fluids and intestinal fluids. The archaeosomes showed better systemic IgG and mucosal IgA response, and induced ovalbumin specific T cell proliferation than conventional liposomes [124]. Higa et al. have fabricated ultradeformable archaeosomes using total polar lipids Nanocarriers for vaccine delivery 33 from Halorubrum tebenquichense. Sodium cholate and soybean phosphatidylcholine were employed to make ultradeformable archaeosomes. Topical application of the developed ultradeformable archaeosomes showed 10-100 times higher serum IgG levels than ultradeformable liposomes applied topically [123]. Patel et al. have developed a vaccine delivery platform using archaeosomes for mucosal vaccination, called AMVAD (Archaeal lipid mucosal vaccine adjuvant and delivery). It is prepared by the interaction of archaeosomes sized between 100-200 nm with multivalent cations such as calcium. The difference between AMVAD and cochleates is the absence of aqueous volume and multilamellar nature of the latter. Antigen loading in the AMVAD system may be carried out in two ways-either the antigen entrapped archaeosomes may be interacted with calcium or soluble antigen is included in the buffer and mixed with preformed archaeosomes followed by blending with cations. Intranasal immunization with the AMVAD system showed good antigen-specific IgA antibody levels in the nasal mucosa, vaginal mucosa, serum, faeces, and bile, and potent serum IgG1 and IgG2a levels were produced. The AMVAD system also demonstrated generation of CD8+ cytotoxic T lymphocyte response [125]. 5.3 Niosomes Non-ionic amphiphilic molecules self-assemble in water to give rise to closed bilayer structures called niosomes. The key advantages of niosomes include their biocompatibility, superior stability, economical ingredients, nonimmunogenicity, and amenability to easy large scale production, non-toxicity, and simple storage conditions. Niosomes have been explored for vaccine delivery via the SC, intraperitoneal, IM, oral, nasal, topical, and transcutaneous routes [126]. Maheshwari et al. fabricated niosomes containing hepatitis B surface antigen and cholera toxin B as an immunostimulant for transcutaneous vacccination. Cholera toxin B activated Langerhans cells in the epidermis followed by antigen presentation in the draining lymph node and generation of systemic immunity. The co-administration of cholera toxin B with hepatitis B surface antigen-loaded niosomes elicited both Th1 and Th2 type immune responses [127]. Vyas et al. developed niosomes composed of Span 85 and cholesterol loaded with DNA encoding hepatitis B surface antigen using the reverse-phase evaporation technique. Topical application of these niosomes in Balb/c mice showed the ability of niosomes to elicit equivalent systemic antibody generation and cytokine levels (IL-2 and IFN-γ indicating Th1 response) as IM recombinant hepatitis B surface Unauthenticated Download Date | 6/17/17 12:05 AM 34 Priyanka Prabhu and Vandana Patravale antigen and topically applied liposomes. Immunization of mice with recombinant hepatitis B surface antigen has been shown to generate only a humoral response owing to the presentation of exogenous antigen by the B cells via the MHC class II pathway to the Th2 cells. However, when DNA vaccine is given, a portion of the hepatitis B surface antigen produced in vivo is presented by the B cells via the MHC class II pathway to the Th2 cells and the rest is lysed in the APCs and presented via the MHC class I pathway to Th1 cells [128]. Rentel et al. developed two different niosomes containing ovalbumin for peroral administration, one composed of 70% stearate sucrose ester and 30% palmitate sucrose ester, and the other composed of 30% stearate sucrose ester and 70% palmitate sucrose ester. The former, which was more hydrophobic than the latter, was only effective in generating humoral immunity. The observed results may be due to limited uptake of the hydrophilic niosomes by the Peyer’s patches resulting in poor immune response generation [129]. Jain et al. developed mannose functionalized niosomes for oral delivery of DNA encoding hepatitis B surface antigen. Niosomes were coated with O-palmitoyl mannan to shield them from bile salts and GIT enzymes and also to enhance uptake by macrophages and dendritic cells present near Peyer’s patches. The mannosylated niosomes elicited both systemic and mucosal antibody responses and also produced cellular immunity (high levels of IL-2 and IFN-γ) on oral administration [130]. Cortesi et al. developed niosomes (320 nm) by lipid film hydration, containing secretory recombinant form of HSV type 1 glycoprotein B. Intranasal immunization of these niosomes in mice showed generation of both IgG 2a antibody in serum and IFN- γ generation in CD4+ splenocytes, and also resulted in 90% protection from a vaginal challenge with HSV [131]. 5.4 Emulsions Emulsions are biphasic systems comprising an oily phase and aqueous phase emulsified with the aid of surface active agents. There are three types of emulsions depending on the nature of the dispersed and continuous phase. Oil-in-water emulsions contain oil in the dispersed phase and water as the continuous phase, and water-inoil emulsions contain water in the dispersed phase and oil as the continuous phase. Multiple emulsions contain small droplets of the continuous phase dispersed in the internal dispersed phase. These can be oil-in-water-in oil or water-in-oil-in water. Emulsions have been used for vaccine adjuvant purposes for a long time. The most potent vaccine adjuvant, which is often considered as the gold standard for vaccine adjuvants, is Freund’s Adjuvant. CFA is a water-in-oil emulsion composed of mineral oil, mannide monooleate and heat killed mycobacteria. Incomplete Freund’s Adjuvant is devoid of the heat killed mycobacteria. However, the ability of Freund’s Adjuvant to cause granulomatous reaction at the injection site owing to the non-metabolizing property of the oil precludes its use in humans. MF59 is an oil-in-water emulsion that is licensed for use in an influenza vaccine (Fluad®), and contains squalene, Polysorbate 80, and sorbitan trioleate. Humoral immunity is generally induced by water-in-oil emulsions whereas oil-in-water or or water-in-oil-in-water multiple emulsions lead to a cellular immune response. The nature of the emulsion may also impact on the longevity of the resulting immune response [132]. Shahiwala et al. fabricated water-in-oil-in-water multiple emulsions containing ovalbumin and modified them with chitosan. The emulsions produced higher levels of both serum IgG and mucosal IgA than ovalbumin solution. Oral immunization was more effective in inducing both serum IgG and mucosal IgA levels compared to nasal immunization. Intranasal immunization led to higher serum IgG levels than mucosal IgA levels. Squalane oil employed as the oily phase resulted in immunostimulatory effects, and the presence of water in the outer continuous phase provided a low viscosity solution for nasal instillation. Chitosan was included with the aim of increasing the nasal residence time of the formulation [133]. Bielinska et al. formulated a water-in-oil nanoemulsion for nasal vaccination using Bacillus anthracis protective antigen. The nanoemulsion was found to be noninflammatory to the nasal mucosa of animals and conferred protection to guinea pigs against an ID challenge with Bacillus anthracis spores. The nanoemulsion vaccine only required single administration in guinea pigs and twice in mice compared to the commercial vaccine which requires six administrations over 18 months. The developed nanoemulsion showed potential in overcoming the drawbacks associated with the presently available anthrax vaccine, which is invasive, requires multiple injections, and is adjuvanted by alum leading to adverse effects [134]. 5.5 Transfersomes Transfersomes are ultra-deformable vesicles composed of an aqueous core surrounded by a complex lipid bilayer. Incorporation of membrane softeners endows transfersomes with an ultra-flexible character permitting them to squeeze across the intercellular lipid barrier of the stratum corneum. They show enhanced permeation through skin compared to traditional liposomes and niosomes [135]. Unauthenticated Download Date | 6/17/17 12:05 AM Transfersomes have been explored for transcutaneous delivery of several antigens. Mahor et al. developed cationic transfersomes from cationic lipid N-[1-(2, 3-dioleyloxy) propyl]-N,N,N-trimethylammonium chloride (DOTMA) and sodium deoxycholate loaded with hepatitis B surface antigen. Topical application of the cationic transfersomes resulted in higher serum antibody levels and cytokine levels (IL-2 AND IFN-γ) than topically applied naked DNA, and equivalent antibody levels and cytokine levels (IL-2 AND IFN-γ) as IM administered recombinant hepatitis B surface antigen [136]. Gupta et al. fabricated transfersomes containing tetanus toxoid. Topical immunization resulted in comparable antibody levels to those of IM administered tetanus toxoid [137]. Gupta et al. in another study demonstrated that topical immunization with tetanus toxoid-loaded transfersomes resulted in generation of equivalent antibody levels to those of SC administered alum adsorbed tetanus toxoid [138]. Xu et al. developed transfersomes containing DNA encoding respiratory syncytial virus (RSV) surface glycoproteins for topical immunization in mice against RSV infection. IM administration of DNA only resulted in serum antibody induction; no mucosal IgA antibody was produced, nor was virus-specific cellular immunity induced. Topical immunization with DNA-loaded transfersomes resulted in RSV-specific mucosal and systemic antibody responses and a higher number of IFN-γ producing cells. Additionally, topical immunization protected mice against RSV challenge as evidenced by the lesser lung abnormalities seen in the animals compared to IM immunization [139]. 5.6 Bilosomes Liposomes and niosomes show poor activity on oral administration due to their instability in the GIT. Bile salts cause dissolution and enzymatic breakdown. Inclusion of bile salts in the membrane of these vesicles confers stability to the vesicles against bile acids. Vesicles in which bile salts are included are termed as bilosomes [140]. Bilosomes possess greater chemical stability, GIT stability, and require lower doses of antigens compared to liposomes and niosomes. In addition, unlike liposomes, there is no requirement for special storage conditions [12]. Arora et al. fabricated mannan functionalized bilosomes for oral hepatitis surface B antigen vaccination. Mannan coating not only stabilizes the vesicles in GIT but also acts a targeting moiety for mannose receptors present on dendritic cells and macrophages. Mannan coated bilosomes showed higher antigen-specific IgG levels and secretory IgA levels in intestinal, nasal, vaginal, and salivary secretions compared to plain bilosomes [140]. Nanocarriers for vaccine delivery 35 Mann et al. fabricated tetanus toxoid-loaded bilosomes for oral vaccination. The bilosomes showed both systemic and mucosal immune responses [141]. Singh et al. fabricated bilosomes loaded with BSA and conjugated them with cholera toxin B subunit to increase affinity for M cells present in Peyer’s patches. Single shot oral immunization of these bilosomes demonstrated similar immune responses to those obtained with parenteral immunization of BSA emulsified with CFA. Also, unlike CFA, the bilosomes were free from any side effects [142]. Shukla et al. developed bilosomes loaded with hepatitis B surface antigen using the lipid film cast technique. Oral immunization with these bilosomes showed generation of both systemic IgG antibody and mucosal sIgA antibody in saliva, vaginal, and intestinal secretions [143]. 5.7 Immunostimulating complexes (ISCOMs) ISCOMs are antigen-containing cage-like nanostructures which are fabricated from phospholipids, cholesterol, and Quil A saponin from Quillaja saponaria bark. They show a spherical shape and are negatively charged with a size of around 40 nm [29]. ISCOMs possess glucuronic acid on their surface, thus conferring a negative charge and facilitating the association of positively charged antigens via electrostatic interactions [144]. They offer dual benefits as they resemble viruses in terms of size and surface protein orientation and also possess the strong immunostimulant potential of Quillaja saponin [3]. Quil A saponin possesses haemolytic properties, rendering it toxic for human use. Mixing it with cholesterol, however, results in loss of this characteristic [145]. ISCOMs devoid of antigen are called ISCOM matrices. Hydrophobic antigen loading occurs via direct encapsulation or anchoring with the lipidic portions whereas hydrophilic antigens are loaded through special procedures. ISCOMs were formerly used parenterally, but now have also been explored successfully for oral and intranasal immunization. ISCOMs undergo uptake by APCs via endocytosis and activate the APCs [29]. ISCOMs enhance the expression of MHC class II molecules and promote the secretion of pro-inflammatory cytokines [144]. ISCOMs after their internalization and processing inside the APCs are known to induce the production of IFN-γ and IL-2 leading to the generation of a potent Th1 response [29]. A commercially available ISCOM based veterinary vaccine used for equine influenza is Equilis ®Prequenza by Merck Animal Health [82,146]. 5.8 Cochleates Cochleates are solid particles containing large continuous lipid bilayer sheets rolled up in a spiral structure. They are Unauthenticated Download Date | 6/17/17 12:05 AM 36 Priyanka Prabhu and Vandana Patravale prepared by fusing small negatively charged unilamellar liposomes in the presence of calcium. Cochleate formation takes place owing to the hydrophobic lipid bilayer sheets which fold up to minimize contact with water. Cochleates possess higher stability than liposomes against lipase, pH and temperature. Phospholipids used to fabricate cochleates include phosphatidyl serine, dioleoylphosphatidylserine, phosphatidic acid, phosphatidylinositol, phosphatidyl glycerol, phosphatidylcholine, phosphatidylethanolamine, diphosphatidylglycerol, dioleoyl phosphatidic acid, distearoyl phosphatidylserine, dimyristoyl phosphatidylserine, and dipalmitoyl phosphatidylgycerol [147]. Advantages of cochleates for vaccine delivery include their biodegradability, biocompatibility, simple fabrication, prolonged release of antigen, and ability to protect the encochleated antigen from degradation, ability to be lyophilized and stored post lyophilisation [148]. Kheiri et al. developed influenza membrane glycoprotein-loaded cochleates for oral and IM immunization. Both oral and IM immunization led to high serum IgG levels, with the IM route giving higher IgG levels. Oral immunization resulted in generation of IgA in saliva, whereas IM administration only led to IgG in saliva. Oral immunization also conferred protection to mice against lung and tracheal infection post nasal influenza challenge [149]. 5.9 Solid Lipid Nanoparticles (SLN) SLN are composed of a solid lipid matrix or a matrix made up of a blend of solid lipids. SLN offer a number of beneficial features for vaccine delivery, including biocompatibility, simple manufacture, sustained release of antigen, protection of antigen from degradation, and amenability to ligand functionalization for targeted delivery to APCs [150]. Mishra et al. developed hepatitis B surface antigen-loaded mannose functionalized SLN for SC vaccination. SLN were fabricated using the solvent injection technique. They demonstrated lymph node and spleen uptake and also elicited sustained antibody production. Antibody titers obtained were equivalent to those obtained after IM hepatitis B surface antigen immunization [17]. 5.10 Miscellaneous lipidic nanocarriers Arias et al. developed yellow carnauba wax nanoparticles onto which HIV-gp140 glycoprotein was adsorbed. The nanoparticles resulted in superior T cell proliferation in comparison to plain HIV gp-140. ID immunization resulted in high levels of antigen-specific IgG antibody, and intranasal immunization led to generation of serum and vaginal IgG and IgA antibodies. The study is beneficial for HIV vaccine design wherein it is necessary to activate the mucosal immune response in the female genital tract [151]. Sloat et al. developed lipidic nanoparticles (200 nm) made from glyceryl monostearate and lecithin. BSA and Bacillus anthracis protective antigen were covalently conjugated onto the surface of these nanoparticles for SC vaccination. BSA conjugated nanoparticles showed higher IgG and IgM antibody generation compared to alum adsorbed antigen, and antibody levels comparable to those obtained with Incomplete Freund’s Adjuvant. Mice immunized with B. Anthracis protective antigen conjugated nanoparticles showed protection against lethal anthrax challenge [152]. Moon et al. developed multilamellar lipidic vesicles composed of recombinant Plasmodium vivax circumsporozoite antigen VMP001 encapsulated within the aqueous core in addition to being attached to the outer membrane. SC immunization with these vesicles and MPLA led to induction of high antibody titres which persisted for more than a year at a 10-fold lower antigen dose compared to alum adsorbed antigen and soluble antigen administered with MPLA. The presence of antigen anchored onto the surface of vesicles resembled the multivalent presentation of epitopes offered by invading pathogens leading to B cell receptor crosslinking, germinal cell formation and increased expansion of antigen-specific follicular helper T cells [153]. 6 Inorganic nanocarriers 6.1 Silica nanoparticles Porous silica nanoparticles are endowed with numerous beneficial features for vaccine delivery. Firstly, their large surface area and huge pore volume provides high loading of vaccine. Secondly, the insoluble silica allows slow release of antigen creating a depot phenomenon. Thirdly, their rigid architecture provides protection to entrapped antigen against degradation. Fourthly, they show excellent chemical stability, biocompatibility, less toxicity, and their pore diameter and particle size can be adjusted. Wang et al. developed porous silica nanoparticles containing BSA for oral vaccination. Their results suggested dependence of the immune response on the size and architecture of silica nanoparticles. Nanoparticles of 430 nm and containing large pores showed prolonged release of antigen due to increased diffusional pathlength and induced both systemic and Unauthenticated Download Date | 6/17/17 12:05 AM mucosal immunity more effectively compared to the nanoparticles of 130 nm and 1-2 µm. Also, nanoparticles of around 500 nm are taken up to a greater extent by M cells and enterocytes, which may have contributed to the superior adjuvanticity of the 430 nm nanoparticles. The nanoparticles also induced a mixed Th1/Th2 response [154]. Carvalho et al. developed mesoporous SB-15 silica nanoparticles and compared their vaccine adjuvant potential with alum and Incomplete Freund’s Adjuvant by both IM and oral routes. BSA was encapsulated or adsorbed onto SBA-15. SBA-15 nanoparticles with BSA generated both Th1 and Th2 responses. SBA-15 particles are taken up by macrophages by phagocytosis and then stimulate production of IL-1β via NALP3 inflammasome activation [155]. Guo et al. fabricated hollow mesoporous silica nanoparticles containing porcine circovirus type 2 ORF2 protein for IM vaccination. Controlled release of protein was seen from the nanoparticles over a 2 week period. Also, the nanoparticles elicited higher antibody titres and T lymphocyte proliferation compared to porcine circovirus type 2 ORF2 protein administered alone [156]. Gordon et al. fabricated temperature sensitive chitosan hydrogels loaded with silica nanoparticles for SC ovalbumin delivery. Thermoresponsive hydrogels are promising for vaccine delivery as they are easy to inject in solution form and gel in vivo at body temperature providing prolonged delivery of antigen from the gel. Chitosan solutions gel in response to pH, however, inclusion of polyol salts in chitosan solutions results in temperature based gelation. The study also compared the efficacy of Quil A adjuvant and antigen delivered in silica nanoparticles loaded within hydrogels with that of soluble ovalbumin and Quil A delivered in chitosan hydrogels. The chitosan hydrogel released 35% of soluble ovalbumin, whereas the chitosan hydrogels loaded with silica nanoparticles containing ovalbumin showed a release of 16% in 14 days. Chitosan hydrogel loaded antigen irrespective of the form of antigen (soluble v/s particulate), or the presence or absence of Quil A as an adjuvant, generated both humoral and cellmediated immune responses. Silica nanoparticulate based delivery of antigen and adjuvant in hydrogel proved to be more effective in generating CD4+ T cell proliferation in mice when compared to soluble antigen and adjuvant in chitosan hydrogel [157]. 6.2 Calcium Phosphate Nanoparticles Inorganic nanoparticles possess certain benefits over organic nanoparticles, including their lower propensity to microbial degradation, superior resistance to bile salts and lipases, non-toxicity, very good storage stability, low Nanocarriers for vaccine delivery 37 cost, and ability to be fabricated at lower temperatures [158]. Calcium phosphate has been utilized in Europe for vaccines against tetanus and diphtheria antigens [159]. Calcium phosphate offers several beneficial features for vaccine delivery, including good biocompatibility, biodegradability, non-toxicity, easy manufacture, economical, indigenous material present in bones and teeth, and high affinity for antigens, DNA, and proteins [158]. Calcium phosphate nanoparticles are taken up by the cells and then dissolved in lysosomes; calcium ions are then quickly removed from the cell [160]. Behera et al. developed calcium phosphate nanoparticles (less than 200 nm) for parenteral immunization in fish by adsorbing the S-layer protein (a protein from Aeromonas hydrophila, a pathogen infecting fish) as an antigen. The nanoparticles demonstrated an adjuvant effect similar to that obtained with antigen emulsified with Freund’s Incomplete Adjuvant, but without the associated adverse effects [158]. He et al. compared the vaccine adjuvant effect of calcium phosphate nanoparticles with alum adjuvant using HSV type 2 and Epstein-Barr virus as antigens. Calcium phosphate nanoparticles were shown to be better adjuvants than alum, with negligible inflammation at the site of vaccination. Also, mice immunized with calcium phosphate nanoparticles showed 100% protection against intravaginal challenge with HSV type 2. Calcium phosphate nanoparticles induced an IgG2a antibody response unlike alum which only gives a Th2 type response [159]. He et al. in another study have shown the efficacy of calcium phosphate nanoparticles for mucosal immunization against HSV type 2. Intranasal and intravaginal administration of calcium phosphate nanoparticles generated both mucosal and systemic immunity in mice and enhanced their survival after viral challenge [161]. Sokolova et al. fabricated calcium phosphate nanoparticles containing TLR ligands: poly (I:C) and CpG, and antigen hemagglutinin. The nanoparticles caused dendritic cell activation as evidenced by the upregulation of surface molecules and cytokine generation (IL-12, IL-2, TNF-α), and also stimulated T cell proliferation [160]. 6.3 Gold nanoparticles Gold nanoparticles offer some attractive features for vaccine delivery. Their size can be simply controlled from around 1 nm to 100 nm or more. They also allow anchoring of peptide antigens or nucleic acids onto their surface using simple chemistry. They are biocompatible and safe [162]. Gold nanoparticles have been explored in clinical trials for hepatitis B and malaria vaccines. DNA vaccines Unauthenticated Download Date | 6/17/17 12:05 AM 38 Priyanka Prabhu and Vandana Patravale were coated onto gold nanoparticles and delivered to the skin epidermis using a gene gun [44]. Barhate et al. developed chitosan modified gold nanoparticles (40 nm) loaded with tetanus toxoid and Quillaja saponaria extract. The nanoparticles were able to shield tetanus toxoid from gastric degradation in vitro. Oral vaccination in mice demonstrated 28 times and 6.5 times higher IgG immune response compared to tetanus toxoid administered alone and with Quillaja saponaria extract respectively. Induction of IgA antibody was also observed in faeces and intestinal washes. Quillaja saponaria extract is reported to induce dendritic cell maturation through interaction with the TLR 4 on dendritic cells. Additionally, chitosan, being mucoadhesive, enhances uptake by the M cells of Peyer’s patches [163]. Bastus et al. conjugated gold nanoparticles to amyloid growth inhibitory peptide and sweet arrow peptide. Bone marrow macrophages showed uptake of these nanoparticle conjugated peptides although they are otherwise unable to uptake the peptide alone. Uptake occurred via TLR-4. Macrophage activation occurred and pro-inflammatory cytokine production (IL-6, TNF-α, IL-1 β) was also stimulated [164]. 6.4 Silver Nanoparticles Jazayeri et al. developed silver nanoparticles (12 nm) for oral DNA vaccination using green technology. The DNA used was plasmid DNA encoding the hemagglutinin gene of the avian influenza virus. Single shot oral immunization in chickens resulted in the induction of both antibody and cellular immune responses and increased cytokine levels. Also, the silver nanoparticles were found to be noncytotoxic [165]. 7 Miscellaneous nanocarriers 7.1 Carbon Nanoparticles Wang et al. fabricated carbon nanoparticles (470 nm) with larger pores than conventional mesoporous carbon particles for BSA antigen delivery. Mesoporous carbon particles offer several advantages for vaccine delivery due to their chemical stability and biocompatibility. The idea was to increase the antigen loading of conventional mesoporous carbon particles which have a low pore size of less than 10 nm. The researchers used a lower amount of carbon source (sucrose) and fabricated mesoporous carbon nanoparticles with a pore size of 40-60 nm. These carbon nanoparticles not only offer higher loading of antigen, but also aid in prevention of antigen aggregation, and offer simple loading of antigen by mixing with antigen solution at 4 o C, thus eliminating the harsh conditions of heat and organic solvents which may be used to load antigen in other carriers. Also, the rigid architecture of the nanostructure offers protection to antigen in the hostile environment of the GIT. Oral immunization in mice resulted in serum antibody levels almost equivalent to those obtained with parenteral administration of BSA in CFA and mucosal IgA antibodies in intestinal, vaginal, and salivary secretions. The success of these nanoparticles may also be a result of their hydrophobic nature and optimal particle size which is known to enhance M cell uptake in oral antigen delivery [166]. Carbon nanotubes are nanodimensional allotropes of carbon. They may be single walled carbon nanotubes (SWCT) or multiwalled carbon nanotubes, the latter being of a larger thickness and greater length [12]. Carbon nanotubes offer several advantages for vaccine delivery. They are able to internalize into cells by a myriad of mechanisms, their high aspect ratio may afford better immunogenicity of the associated antigen, and their large surface area aids in chemical modification. Villa et al. conjugated Wilm’s tumour protein to solubilised SWCT. The protein is upregulated in many cancers but suffers from poor binding affinity to MHC class II molecules. Peptide conjugated SWCTs showed good uptake by dendritic cells and macrophages in vitro. SC immunization of these carbon nanotubes, along with the adjuvant TiterMax®, resulted in induction of antigen-specific IgG levels, however mere mixing of peptide and adjuvant failed to generate significant antibodies [167]. 7.2 Nanodecoy Nanodecoy systems comprise a ceramic core and an outer shell of carbohydrate. They offer many beneficial features for vaccine delivery. They are biocompatible and biodegradable, and their external hydrophilic carbohydrate layer helps to retain the protein conformation and prevents aggregation of the nanostructure. Goyal et al. self-assembled hydroxyapatite and cellubiose followed by coating with hepatitis B surface antigen to form nanodecoy systems. SC immunization in mice showed enhanced serum antibody generation and higher levels of IFN-γ and IL-2 compared to alum adsorbed hepatitis B surface antigen and plain hepatitis B surface antigen [168]. Goyal et al.in another study, fabricated aquasomes (200 nm) for BSA delivery. Self-assembly of the hydroxyapatite core was enabled by a co-precipitation method followed by coating with trehalose and cellubiose, BSA was then adsorbed onto these systems. SC immunization in mice Unauthenticated Download Date | 6/17/17 12:05 AM showed higher serum antibody levels compared to alum adsorbed BSA and BSA alone. Aquasomes caused a mixed Th1/Th2 response with a high ratio of IgG2a to IgG1. Polyhydroxyl oligomers such as trehalose and cellubiose which are epitaxially adsorbed onto the hydroxyapatite core serve a dual purpose: firstly they act as ligands for mannose receptors present on APCs, and secondly, they stabilize the structure, both resulting in enhanced antigen uptake and presentation [169]. 7.3 Vaults Vaults are defined as large ribonucleoproteins present in eukaryotic cells such as dendritic cells, endometrium, and lung. Vaults are barrel shaped, containing numerous copies of three species of proteins and many copies of small untranslated RNA. The major vault protein (MVP), which is found in 96 copies per vault, is responsible for the barrel shape of vaults. The MVP sequence is able to encode all information required to achieve self-assembly of protein giving rise to a recombinant vault nanoparticle with bacilovirus system. Recombinant vaults possess a huge internal cavity to house many immunogenic proteins. The vault size mimics that of microbes, thus facilitating their uptake by dendritic cells. Champion et al. fabricated hollow vault nanocapsules encapsulating major outer membrane protein of Chlamydia muridarum for nasal vaccination. Vaults were found to activate inflammasome and increase IL-β1 secretion without TLR involvement. They were able to induce immunity without causing inflammation (without causing production of proinflammatory cytokines), and reduce bacterial burden in mice following vaginal challenge [170]. Kar et al. formulated ovalbumin-containing vault nanocapsules for SC vaccination. They resulted in potent ovalbuminspecific CD4+ memory T cell and CD8+ memory T cell generation in vaccinated mice [171]. 8 Conclusion The burgeoning field of vaccine delivery has made noteworthy advancements in recent years, fuelled by the improved understanding of both innate and adaptive immune mechanisms coupled with the development of a myriad of novel nanoparticulate antigen delivery systems. However, there are several unmet needs in vaccinology including a paucity of adjuvants to mediate the cellular arm of the immune response and lack of effective adjuvants for non-invasive vaccination. Also, development and approval of novel vaccine adjuvants is a time-consuming Nanocarriers for vaccine delivery 39 process. Adjuvants cannot be registered alone; they must be registered as an adjuvant/antigen combination. Apart from the cardinal attributes of efficacy, safety, and stability, it is necessary for vaccine adjuvants to be prepared on a large scale in a simple and cost effective way to facilitate mass vaccination especially in the developing countries of the world. There is also a need for humanized animal models to closely mimic the human immune system while evaluating potential vaccine adjuvants. Nanocarriers possess immense potential in successful vaccine delivery due to their ability to protect the antigen, provide its controlled release and target it specifically to the APCs. Nanocarriers such as mucoadhesive nanoparticles and vesicular nanocarriers also have the ability to afford mucosal and topical vaccination respectively. Some issues in nanovaccinology still need to be studied in depth including the uptake mechanisms of nanocarriers through APCs, their uptake in other parts of the body apart from APCs and the resulting effects, and their elimination from the body, amongst other studies. Future development in vaccine adjuvants would involve combination of immunostimulants with traditional delivery vehicles to achieve potent vaccine adjuvanticity and minimize the systemic toxicity associated with immunostimulant molecules. Also, vaccine delivery systems utilizing core materials such as chitosan which are endowed with inherent immunostimulant potential or self-adjuvanting systems such as archaeosomes or ISCOMs should be pursued. In conclusion, there are countless opportunities concealed within the field of nanovaccinology, which could pave the way for successful vaccines in the market, driven by collaborations between formulation scientists, immunologists, clinical research scientists, and regulatory authorities. Acknowledgements: The authors wish to thank University Grants Commission for fellowship. Conflict of interest: The authors have no conflict of interest. Received: June 30, 2013; Accepted: December 16, 2013. References [1] [2] [3] Leroux-Roels G., Unmet needs in modern vaccinology adjuvants to improve the immune response, Vaccine, 2010, 28S, C25–C36. Leleux J., Roy K., Micro and nanoparticle-based delivery systems for vaccine immunotherapy: an immunological and materials perspective, Adv. Healthcare Mater., 2013, 2, 72–94. Shahiwala A., Vyas T.K., Amiji M.M., Nanocarriers for systemic and mucosal vaccine delivery, Recent Pat. Drug Deliv. Formul., 2007, 1, 1-9. Unauthenticated Download Date | 6/17/17 12:05 AM 40 [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] Priyanka Prabhu and Vandana Patravale Peek L.J., Middaugh C.R., Berkland C., Nanotechnology in vaccine delivery, Adv. Drug Deliv. Rev., 2008, 60, 915-928. Hirschberg H.J.H.B., van de Wijdeven G.G.P., Kelder A.B., van den Dobbelsteen G.P.J.M., Kersten G.F.A., BioneedlesTM as vaccine carriers, Vaccine, 2008, 26, 2389-2397. Gander B., Trends in particulate antigen and DNA delivery systems for vaccines, Adv. Drug Deliv. Rev., 2005, 57, 321-323. Arca H.C., Gunbeyaz M., Senel S., Chitosan based systems for the delivery of vaccine antigens, Expert Rev Vaccines., 2009, 8, 937-953. Fahmy T.M., Demento S.L., Caplan M.J., Mellman I., Saltzman W.M., Design opportunities for actively targeted nanoparticle vaccines, Nanomedicine, 2008, 3, 343-355. Demento S.L., Siefert A.L., Bandyopadhyay A., Sharp F.A., Fahmy T.M., Pathogen associated molecular patterns on biomaterials: a paradigm for engineering new vaccines, Trends Biotechnol., 2011, 29, 294-306. Chadwick S., Kriegel C., Amiji M., Nanotechnology solutions for mucosal immunization, Adv Drug Deliv Rev., 2010, 62, 394–407. Babiuk L.A., Broadening the approaches to developing more effective vaccines, Vaccine, 1999, 17, 1587-1595. Jain S., Khomane K., Jain A.K., Dani P., Nanocarriers for transmucosal vaccine delivery, Current Nanoscience, 2011, 7, 160-177. Combadière B., Mahe B., Particle-based vaccines for transcutaneous vaccination, Comp. Immunol. Microbiol. Infect. Dis., 2008, 31, 293–315. Pitaksuteepong T., Nanoparticles: a vaccine adjuvant for subcutaneous administration, Naresuan Univ. J., 2005, 13, 53-62. Cui Z., Mumper R.J., Microparticles and nanoparticles as delivery systems for DNA vaccines, Crit. Rev. Ther. Drug Carrier Syst., 2003, 20, 103-137. Singh M., Chakrapani A., O’Hagan D., Nanoparticles and microparticles as vaccine-delivery systems, Expert Rev. Vaccines, 2007, 6, 797-808. Mishra H., Mishra D., Mishra P.K., Nahar M., Dubey V., Jain N.K., Evaluation of solid lipid nanoparticles as carriers for delivery of Hepatitis B Surface antigen for vaccination using subcutaneous route, J. Pharm. Pharm. Sci., 2010, 13, 495-509. Pal I., Ramsey J.D., The role of the lymphatic system in vaccine trafficking and immune response, Adv. Drug Deliv. Rev., 2011, 63, 909–92. Harandi A.M., Medaglini D., Shattock R.J., Vaccine adjuvants: a priority for vaccine research, Conference Report in: Vaccine, 2010, 28, 2363-2366. Vila A., Sànchez A., Janes K., Behrens I., Kissel T., Jato J.L.V., et al., Low molecular weight chitosan nanoparticles as new carriers for nasal vaccine delivery in mice, Eur. J. Pharm. Biopharm., 2004, 57, 123-131. Sharma S., Mukkur T.K., Benson H.A.E., Chen Y., Pharmaceutical aspects of intranasal delivery of vaccines using particulate systems, J. Pharm. Sci., 2009, 98, 812-843. Amorij J-P., Kersten G.F.A., Saluja V., Tonnis W.F., Hinrichs W.L.J., Slütter B., et al., Towards tailored vaccine delivery: needs, challenges and perspectives, J. Control Release, 2012, 161, 363–376. [23] Gupta P.N., Singh P., Mishra V., Jain S., Dubey P.K., Vyas S.P., Topical immunization: mechanical insight and novel delivery systems, Indian J. Biotechnol., 2004, 3, 9-21. [24] Singh R.P., Singh P., Mishra V., Prabakaran D., Vyas S.P., Vesicular systems for noninvasive topical immunization: rationale and prospects, Indian J. Pharmacol., 2002, 34, 301-310. [25] Lee M-Y., Shin M-C., Yang V.C., Transcutaneous antigen delivery system, BMB Rep.,2013, 46, 17-24. [26] Partidos C.D., Beignon A-S., Semetey V., Briand J-P., Muller S., The bare skin and the nose as non-invasive routes for administering peptide vaccines, Vaccine, 2001, 19, 2708-2715. [27] Moser M., Leo O., Key concepts in immunology, Vaccine, 2010, 28S, C2–C13. [28] Zepp F., Principles of vaccine design – Lessons from nature, Vaccine, 2010, 28S, C14-C24. [29] De Temmerman M-L., Rejman J., Demeester J., Irvine D.J., Gander B., De Smedt S.C., Particulate vaccines: on the quest for optimal delivery and immune response, Drug Discov. Today, 2011, 16, 569-582. [30] Perrie Y., Mohammed A.R., Kirby D.J., McNeil S.E., Bramwell V.W., Vaccine adjuvant systems: enhancing the efficacy of sub-unit protein antigens, Int. J. Pharm., 2008, 364, 272–280. [31] Coffman R.L., Sher A., Seder R.A., Vaccine adjuvants: putting innate immunity to work, Immunity, 2010, 33,492-503. [32] Esser M.T., Marchese R.D., Kierstead L.S., Tussey L.G., Wang F., Chirmule N. et al., Memory T cells and vaccines, Vaccine, 2003, 21, 419–430. [33] Gamvrellis A., Leong D., Hanley J.C., Xiang S.D., Mottram P., Plebanski M., Vaccines that facilitate antigen entry into dendritic cells, Immunol. Cell Biol., 2004, 82, 506–516. [34] Reddy S.T., Swartz M.A., Hubbell J.A., Targeting dendritic cells with biomaterials: developing the next generation of vaccines, Trends Immunol., 2006, 27, 573-579. [35] Storni T., Kundig T.M., Senti G., Johansen P., Immunity in response to particulate antigen-delivery systems, Adv. Drug Deliv. Rev., 2005, 57, 333– 355. [36] Espuelas S., Irache J.M., Gamazo C., Synthetic particulate antigen delivery systems for vaccination, Immunologia, 2005, 24, 208-223. [37] Pulendran B., Ahmed R., Translating innate immunity into immunological memory: implications for vaccine development, Cell, 2006, 124, 849-863. [38] Badiee A., Shargh V.H., Khamesipour A., Jaafari M.R., Micro/ nanoparticle adjuvants for antileishmanial vaccines: present and future trends, Vaccine, 2013, 31, 735-749. [39] Correia-Pinto J.F., Csaba, N., Alonso, M.J., Vaccine delivery carriers: insights and future perspectives, Int. J. Pharm., 2013, 440, 27-38. [40] Reed S.G., Bertholet S., Coler R.N., Friede M., New horizons in adjuvants for vaccine development, Trends Immunol., 2008, 30, 23-32. [41] Friede M., Aguado M.T., Need for new vaccine formulations and potential of particulate antigen and DNA delivery systems, Adv. Drug Deliv. Rev., 2005, 57, 325– 331. [42] Kurella S., Manocha M., Sabhnani L., Thomas B., Rao D.N., New Age Adjuvants and delivery systems for subunit vaccines. Indian J. Clin. Biochem., 2000, 15, 83-100. Unauthenticated Download Date | 6/17/17 12:05 AM [43] Guy B., The perfect mix: recent progress in adjuvant research, Nat. Rev. Microbiol., 2007, 5, 505-517. [44] Brunner R., Jensen-Jarolim E., Pali-Schöll I., The ABC of clinical and experimental adjuvants-A brief overview, Immunol. Lett., 2010, 128, 29-35. [45] Mbow M.L., Gregorio E.D., Valiante N.M., Rappuoli R., New adjuvants for human vaccines, Curr. Opinion Immunol., 2010, 22, 411–416. [46] Oyewumi M.O., Kumar A., Cui Z., Nano-microparticles as immune adjuvants: correlating particle sizes and the resultant immune responses, Expert. Rev. Vaccines, 2010, 9, 1095-1107. [47] Kanchan V., Panda A.K., Interactions of antigen-loaded polylactide particles with macrophages and their correlation with the immune response, Biomaterials, 2007, 28, 5344 -5357. [48] Jung T., Kamm W., Breitenbach A., Hungerer K-D., Hundt E., Kissel T., Tetanus toxoid loaded nanoparticles from sulfobutylated poly(vinyl alcohol)-graft-poly(lactide-co-glycolide): evaluation of antibody response after oral and nasal application in mice, Pharm. Res., 2001, 18, 352–360. [49] Caputo A., Brocca-Cofano E., Castaldello A., Voltan, R., Gavioli, R., Srivastava, I.K., et al., Characterization of immune responses elicited in mice by intranasal co-immunization with HIV-1 Tat, gp140 ΔV2Env and/or SIV Gag proteins and the nontoxicogenic heat-labile Escherichia coli enterotoxin, Vaccine, 2008, 26, 1214–1227. [50] Yan, S., Gu, W., Xu, Z.P., Re-considering how particle size and other properties of antigen–adjuvant complexes impact on the immune responses, J. Colloid Interface Sci., 2013, 395, 1-10. [51] Ferreira S.A., Gama F.M., Vilanova M., Polymeric nanogels as vaccine delivery systems, Nanomedicine: Nanotechnology, Biology, and Medicine, 2013, 9, 159-173. [52] Tacken P.J., Torensma R., Figdor C.G., Targeting antigens to dendritic cells in vivo, Immunobiology, 2006, 211, 599–608. [53] García-Vallejo J.J., Ambrosini M., Overbeek A., van Riel W.E., Bloema K., Unger W.W.J., et al., Multivalent glycopeptide dendrimers for the targeted delivery of antigens to dendritic cells, Mol. Immunol., 2013, 53, 387– 397. [54] Moon J.J., Huang B., Irvine D.J., Engineering nano- and microparticles to une Iimmunity, Adv. Mater., 2012, 24, 3724-3746. [55] Joshi M.D., Unger W.J., Storm G., van Kooyk Y., Mastrobattista E., Targeting tumor antigens to dendritic cells using particulate carriers, J. Control Release., 2012, 161, 25–37. [56] Nakamura T., Moriguchi R., Kogure K., Shastri N., Harashima H., Efficient MHC class I presentation by controlled intracellular trafficking of antigens in octaarginine-modified liposomes, Mol. Ther., 2008, 16, 1507–1514. [57] Suzuki R., Oda Y., Utoguchi N., Namai E., Taira Y., Okada N., et al., A novel strategy utilizing ultrasound for antigen delivery in dendritic cell-based cancer immunotherapy, J. Control. Release, 2009, 133, 198–205. [58] Foged C., Hansen J., Agger E.M., License to kill: Formulation requirements for optimal priming of CD8+ CTL responses with particulate vaccine delivery systems, Eur. J. Pharm. Sci., 2012, 45, 482–491. Nanocarriers for vaccine delivery 41 [59] Krishnamachari Y., Geary S.M., Lemke C.D., Salem A.K., Nanoparticle delivery systems in cancer vaccines, Pharm. Res., 2011, 28, 215-236. [60] Tyagi R.K., Garg N.K., Sahu T., Vaccination Strategies against malaria: novel carrier(s) more than a tour de force, J. Control Release, 2012, 162, 242-254. [61] Garcon N., Chomez P., Mechelen M.V., GlaxoSmithKline adjuvant systems in vaccines: concepts, achievements, and perspectives, Expert Rev. Vaccines., 2007, 6, 723-739. [62] Mallapragada S.K., Narasimhan B., Immunomodulatory biomaterials, Int. J. Pharm., 2008, 364, 265–271. [63] Singh M., O’Hagan D., The preparation and characterization of polymeric antigen delivery systems for oral administration, Adv. Drug Deliv. Rev., 1998, 34, 285–304. [64] Amidi M., Mastrobattista E., Jiskoot W., Hennink W.E., Chitosan-based delivery systems for protein therapeutics and antigens, Adv. Drug Deliv. Rev. 2010, 62, 59–82. [65] Senel S., Chitosan-based particulate systems for non-invasive vaccine delivery, Adv Polym Sci., 2011, 243, 111-138. [66] van der Lubben I.M., Verhoef J.C., Borchard G., Junginger H.E., Chitosan for mucosal vaccination, Adv Drug Deliv Rev., 2001, 52, 139-144. [67] van der Lubben I.M., Verhoef J.C., Borchard G., Junginger H.E., Chitosan and itsderivatives in mucosal drug and vaccine delivery, Eur J Pharm Sci., 2001, 14, 201–207. [68] Wen Z-S., Xu Y-L., Zou X-T., Xu Z-R., Chitosan nanoparticles act as an adjuvant to promote both Th1 and Th2 immune responses induced by ovalbumin in mice, Mar. Drugs, 2011, 9, 1038-1055. [69] Amidi M., Romeijn S.G., Borchard G., Junginger H.E., Hennink W.E., Jiskoot W., Preparation and characterization of protein-loaded N-trimethyl chitosan nanoparticles as nasal delivery system, J. Control Release, 2006, 111, 107 -116. [70] Dzung N.A., Ha N.T.N, Van D.T.H., Phuong N.T.L., Quynh N.T.N., Hiep D.M., et al., Chitosan nanoparticle as a novel delivery system for A/H1n1 influenza vaccine: safe property and immunogenicity in mice, World Academy of Science, Engineering and Technology, 2011, 60, 1839-1846. [71] Prego C., Paolicelli, P., Díaz B., Vicente S., Sánchez A., González-Fernández A., et al., Chitosan-based nanoparticles for improving immunization against hepatitis B infection, Vaccine, 2010, 28, 2607–2614. [72] Slutter B., Bal S.M., Que I., Kaijzel E., Lowik C., Bouwstra J., et al., Antigen-adjuvant nanoconjugates for nasal vaccination: an improvement over the use of nanoparticles?, Mol. Pharm., 2010, 7, 2207–2215. [73] Khatri K., Goyal A.K., Gupta P.N., Mishra N., Vyas S.P., Plasmid DNA loaded chitosan nanoparticles for nasal mucosal immunization against hepatitis B, Int J Pharm., 2008, 354, 235–241. [74] Sayın B., Somavarapu S., Li X.W., Sesardic D., Senel S., Alpar O.H., TMC–MCC (Ntrimethyl chitosan–mono-Ncarboxymethyl chitosan) nanocomplexes for mucosal delivery of vaccines, Eur. J. Pharm. Sci., 2009, 38, 362–369. [75] Bal S.M., Slütter B., Jiskoot W., Bouwstra J.A., Small is beautiful: N-trimethyl chitosan–ovalbumin conjugates for microneedle-based transcutaneous immunization, Vaccine, 2011, 29, 4025-4032. Unauthenticated Download Date | 6/17/17 12:05 AM 42 Priyanka Prabhu and Vandana Patravale [76] Tafaghodi M., Saluja V., Kersten G.F.A., Kraan H., Slütter B., Amorij J-P. et al., Hepatitis B surface antigen nanoparticles coated with chitosan and trimethyl chitosan: Impact of formulation on physicochemical and immunological characteristics, Vaccine, 2012, 30, 5341– 5348. [77] Bal S.M., Slütter B., Verheul R., Bouwstra J.A., Jiskoot W., Adjuvanted, antigen loaded N-trimethyl chitosan nanoparticles for nasal and intradermal vaccination: adjuvant- and site dependent immunogenicity in mice, Eur. J. Pharm. Sci., 2012, 45, 475–481. [78] Zhao K., Chen G., Shi X-M., Gao T-T., Li W., Zhao Y. et al., Preparation and efficacy of a live newcastle disease virus vaccine encapsulated in chitosan nanoparticles, PLoS One, 2012, 7, 1-11. [79] Hamdy S., Haddadi A., Hung R.W., Lavasanifar A., Targeting dendritic cells with nanoparticulate PLGA cancer vaccine formulations, Adv. Drug Deliv. Rev., 2011, 63, 943–955. [80] Shen H., Ackerman A.L., Cody V., Giodini A., Hinson E.R., Cresswell P. et al.,Enhanced and prolonged crosspresentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles, Immunology, 2006, 117, 78-88. [81] Kunda N.K., Somavarapu S., Gordon S.B., Hutcheon G.A., Saleem I.Y., Nanocarriers targeting dendritic cells for pulmonary vaccine delivery, Pharm. Res., 2013, 30, 325–341. [82] Saroja C.S., Lakshmi P.K., Bhaskaran S., Recent trends in vaccine delivery systems: Areview, Int. J. Pharm. Investig., 2011, 1, 64-74. [83] Bharali D.J., Pradhan V., Elkin G., Qi W., Hutson A., Mousa S.A. et al., Novel nanoparticles for the delivery of recombinant hepatitis B vaccine, Nanomedicine: Nanotechnology, Biology, and Medicine, 2008, 4, 311–317. [84] Muttil P., Prego C., Garcia-Contreras L., Pulliam B., Fallon J.K., Wang C. et al., Immunization of guinea pigs with novel hepatitis B antigen as nanoparticle aggregate powders administered by the pulmonary route, AAPS J, 2010, 12, 330-337. [85] Elamanchili P., Diwan M., Cao M., Samuel J., Characterization of poly(d,l-lactic-co glycolic acid) based nanoparticulate system for enhanced delivery of antigens to dendritic cells, Vaccine, 2004, 22, 2406–2412. [86] Raghuwanshi D., Mishra V., Suresh M.R., Kaur K., A simple approach for enhanced immune response using engineered dendritic cell targeted nanoparticles, Vaccine, 2012, 30, 7292–7299. [87] Hamdy S., Haddadi A., Shayeganpour A., Samuel J., Lavasanifar A., Activation of antigen-specific T cell-responses by mannan-decorated PLGA nanoparticles, Pharm. Res., 2011, 28, 2288–2301. [88] Mattheolabakis G., Lagoumintzis G., Panagi Z., Papadimitriou E., Partidos C.D., Avgoustakis K., Transcutaneous delivery of a nanoencapsulated antigen: Induction of immune responses, Int. J. Pharm., 2010, 385, 187–193. [89] Jain A.K., Goyal A.K., Gupta P.N., Khatri K., Mishra N., Mehta A. et al., Synthesis, characterization and evaluation of novel triblock copolymer based nanoparticles for vaccine delivery against hepatitis B, J. Control Release, 2009, 136, 161–169. [90] Florindo H.F., Pandit S., Gonçalves L.M.D., Videira M., Alpar O., Almeida A.J., Antibody and cytokine-associated immune [91] [92] [93] [94] [95] [96] [97] [98] [99] [100] [101] [102] [103] responses to S. equi antigens entrapped in PLA nanospheres, Biomaterials, 2009, 30, 5161–5169. Pavot V., Rochereau N., Primard C., Genin C., Perouzel E., Lioux T. et al., Encapsulation of Nod1 and Nod2 receptor ligands into poly (lactic acid) nanoparticles potentiates their immune properties, J. Control Release, 2013, 167, 60–67. Ulery B.D., Kumar D., Ramer-Tait A.E., Metzger D.W., Wannemuehler M.J., Narasimhan B., Design of a protective single-dose intranasal nanoparticle-based vaccine platform for respiratory infectious diseases, PLoS ONE, 2011, 6. Salman H.H., Irache J.M., Gamazo C., Immunoadjuvant capacity of flagellin and mannosamine-coated poly(anhydride) nanoparticles in oral vaccination, Vaccine, 2009, 27, 4784–4790. Okamoto S., Matsuura M., Akagi T., Akashi M., Tanimoto T., Ishikawa T. et al., Poly(γ-glutamic acid) nano-particles combined with mucosal influenza virus hemagglutinin vaccine protects against influenza virus infection in mice, Vaccine, 2009, 27, 5896–5905. Okamoto S., Yoshii H., Akagi T., Akashi M., Ishikawa T., Okuno Y. et al., Influenza hemagglutinin vaccine with poly(γ-glutamic acid) nanoparticles enhances the protection against influenza virus infection through both humoral and cell-mediated immunity, Vaccine, 2007, 25, 8270–8278. Wang X., Uto T., Akagi T., Akashi M., Baba M., Poly(γ-Glutamic Acid) Nanoparticles as an efficient antigen delivery and adjuvant system: potential for an AIDS vaccine, J. Med. Virol., 2008, 80, 11-19. Uto T., Wang X., Sato K., Haraguchi M., Akagi T., Akashi M. et al., Targeting of antigen to dendritic cells with Poly (γ-Glutamic acid) nanoparticles induces antigen-specific humoral and cellular immunity, J. Immunol., 2007, 178, 2979-2986. Uto T., Wang X., Akagi T., Zenkyu R., Akashi M., Baba M., Improvement of adaptive immunity by antigen-carrying biodegradable nanoparticles, Biochem. Biophys. Res. Commun., 2009, 379, 600–604. Uto T., Toyama M., Nishi Y., Akagi T., Shima F., Akashi M. et al., Uptake of biodegradable poly(γ-glutamic acid) nanoparticles and antigen presentation by dendritic cells in vivo, Results Immunol., 2013, 3, 1–9. Eng N.F., Garlapati S., Lai K., Gerdts V., Mutwiri G.K., Polyphosphazenes enhance mucosal and systemic immune responses in mice immunized intranasally with influenza antigens, The Open Vaccine Journal, 2009, 2, 134-143. Mapletoft J.W., Oumouna M., Kovacs-Nolan J., Latimer L., Mutwiri G., Babiuk L.A. et al., Intranasal immunization of mice with a formalin-inactivated bovine respiratory syncytial virus vaccine co-formulated with CpG oligodeoxynucleotides and polyphosphazenes results in enhanced protection, J. Gen. Virol., 2008, 89, 250–260. Andrianov A.K., Decollibus D.P., Marin A., Webb A., Griffin Y., Webby R.J., PCPP formulated H5N1 influenza vaccine displays improved stability and dose-sparing effect in lethal challenge studies, J Pharm. Sci., 2011, 100, 1436-1443. Kovacs-Nolan, J., Latimer, L., Landi, A., Jenssen, H., Hancock, R.E.W., Babiuk, L.A. et al., The novel adjuvant combination of CpG ODN, indolicidin and polyphosphazene induces potent antibody- and cell-mediated immune responses in mice, Vaccine, 2009, 27, 2055-2064. Unauthenticated Download Date | 6/17/17 12:05 AM [104] Kovacs-Nolan J., Mapletoft J. W., Lawman Z., Babiuk L. A., van Drunen Littel-van den Hurk S., Formulation of bovine respiratory syncytial virus fusion protein with CpG oligodeoxynucleotide, cationic host defence peptide and polyphosphazene enhances humoral and cellular responses and induces a protective type 1 immune response in mice, J. Gen. Virol., 2009, 90, 1892–1905. [105] Andrianov A.K., DeCollibus D.P., Gillis H.A., Kha H.H., Marin A., Prausnitz M.R. et al., Poly[di(carboxylatophenoxy) phosphazene]is a potent adjuvant for intradermal immunization, PNAS, 2009, 106, 18936- 18941. [106] Minigo G., Scholzen A., Tang C.K., Hanley J.C., Kalkanidis M., Pietersz G.A. et al., Poly-L-lysine-coated nanoparticles: a potent delivery system to enhance DNA vaccine efficacy, Vaccine, 2007, 25, 1316–1327. [107] Stano A., van der Vlies A.J., Martino M.M., Swartz M.A., Hubbell J.A., Simeoni E., PPS nanoparticles as versatile delivery system to induce systemic and broad mucosal immunity after intranasal administration, Vaccine, 2011, 29, 804–812. [108] Hirosue S., Kourtis I.C., van der Vlies A.J., Hubbell J.A., Swartz M.A., Antigen delivery to dendritic cells by poly(propylene sulfide) nanoparticles with disulfide conjugated peptides: Cross-presentation and T cell activation, Vaccine, 2010, 28, 7897–7906. [109] Gómez S., Gamazo C., Roman B.S., Vauthier C., Ferrer M., Irache J.M., Development of a novel vaccine delivery system based on Gantrez nanoparticles, J. Nanosci. Nanotechnol., 2006, 6, 3283-3289. [110] Salman H.H., Gamazo C., Agueros M., Irache J.M., Bioadhesive capacity and immunoadjuvant properties of thiamine-coated nanoparticles, Vaccine, 2007, 25, 8123–8132. [111] Chen J., Li Z., Huang H., Yang Y., Ding Q., Mai J. et al., Improved antigen cross presentation by polyethyleneiminebased nanoparticles, Int. J. Nanomed., 2011, 6, 77–84. [112] Sheng K-C., Kalkanidis M., Pouniotis D.S., Esparon S., Tang C.K., Apostolopoulos, V., et al., Delivery of antigen using a novel mannosylated dendrimer potentiates immunogenicity in vitro and in vivo, Eur. J. Immunol., 2008, 38, 424–436. [113] Zaman M., Skwarczynski M., Malcolm J.M., Urbani C.N., Jia Z., Batzloff M.R., et al., Self-adjuvanting polyacrylic nanoparticulate delivery system for group A streptococcus (GAS) vaccine, Nanomedicine: Nanotechnology, Biology, and Medicine, 2011, 7, 168–173. [114] Henriksen-Lacey M., Korsholm K.S., Andersen P., Perrie Y., Christensen D., Liposomal vaccine delivery Systems, Expert Opin. Drug Deliv., 2011, 8, 505-519. [115] Heurtault B., Frisch B., Pons F., Liposomes as delivery systems for nasal vaccination: strategies and outcomes, Expert Opin. Drug Deliv., 2010, 7, 829-844. [116] Chiou C.J., Tseng L.P., Deng M.C., Jiang P.R., Tasi S.L., Chung T.W. et al. Mucoadhesive liposomes for intranasal immunization with an avian influenza virus vaccine in chickens. Biomaterials, 2009, 30, 5862-5868. [117] Watson, D.S., Endsley A.N., Huang L., Design considerations for liposomal vaccines:Influence of formulation parameters on antibody and cell-mediated immune response to liposome associated antigens, Vaccine, 2012, 30, 2256– 2272. Nanocarriers for vaccine delivery 43 [118] Tiwari S., Goyal A.K., Mishra N., Vaidya B., Mehta A., Dube D. et al., Liposome in situ gelling system: novel carrier based vaccine adjuvant for intranasal delivery of recombinant protein vaccine, Procedia Vaccinol., 2009, 1, 148–163. [119] Tiwari S., Goyal A.K., Mishra N., Khatri K., Vaidya B., Mehta A. et al., Development and characterization of novel carrier gel core liposomes based transmission blocking malaria vaccine, J. Control Release, 2009, 140, 157–165. [120] Copland M.J., Baird M.A., Rades T., McKenzie J.L., Becker B., Reck F. et al., Liposomal delivery of antigen to human dendritic cells, Vaccine, 2003, 21, 883–890. [121] Yuba E., Kojima C., Harada A., Tana, Watarai S., Kono K., pH-Sensitive fusogenic polymer-modified liposomes as a carrier of antigenic proteins for activation of cellular immunity, Biomaterials, 2010, 31, 943–951. [122] Patel G.B., Chen W., Archaeosome immunostimulatory vaccine delivery system, Curr. Drug Deliv., 2005, 2, 407-421. [123] Higa L.H., Schilrreff P., Perez A.P., Irirarte M.A., Roncaglia D.I., Morilla M.J., et al. Ultradeformable archaeosomes as new topical adjuvants, Nanomedicine: Nanotechnology, Biology and Medicine, 2012, 8, 1319-1328. [124] Li Z., Zhang L., Sun W., Ding Q., Hou Y., Xu Y., Archaeosomes with encapsulated antigens for oral vaccine delivery, Vaccine, 2011, 29, 5260– 5266. [125] Patel G.B. Chen W., Archaeal lipid mucosal vaccine adjuvant and delivery system, Expert Rev. Vaccines, 2010, 9, 431–440. [126] Shilpa S., Srinivasan B.P., Chauhan M., Niosomes for vesicular carriers for delivery ofproteins and biologicals, Int. J. Drug Deliv., 2011, 3, 14-24. [127] Maheshwari C., Pandey R.S., Chaurasiya A., Kumar A., Selvam D.T., PrasadG.B.K.S. et al., Non-ionic surfactant vesicles mediated transcutaneous immunization against hepatitis B, Int. Immunopharmacol., 2011, 11, 1516–1522. [128] Vyas S.P., Singh R.P., Jain S., Mishra V., Mahor S., Singh P. et al., Non-ionic surfactant based vesicles (niosomes) for non-invasive topical genetic immunization against hepatitis B, Int. J. Pharm., 2005, 296, 80–86. [129] Rentel C-O., Bouwstra J.A., Naisbett B., Junginger H.E., Niosomes as a novel peroral vaccine delivery system, Int. J. Pharm., 1999, 186, 161–167. [130] Jain S., Singh P., Mishra V., Vyas S.P., Mannosylated niosomes as adjuvant–carrier system for oral genetic immunization against Hepatitis B, Immunol. Lett., 2005, 101, 41–49. [131] Cortesi R., Ravani L., Rinaldi F., Marconi P., Drechslere M., Manservigi M. et al., Intranasal immunization in mice with non-ionic surfactants vesicles containing HSV immunogens: A preliminary study as possible vaccine against genital herpes, Int. J. Pharm., 2013, 440, 229– 237. [132] Copland M.J., Rades T., Davies N.M., Baird M.A., Lipid based particulate formulations for the delivery of antigen, Immunol. Cell Biol., 83, 2005, 97-105. [133] Shahiwala A., Amiji M.M., Enhanced mucosal and systemic immune response with squalane oil-containing multiple emulsions upon intranasal and oral administration in mice, J. Drug Target., 2008, 16, 302-310. [134] Bielinska A.U., Janczak K.W., Landers J.J., Makidon P., Sower L.E., Peterson J.W. et al., Mucosal immunization with a novel nanoemulsion-based recombinant anthrax protective antigen vaccine protects against Bacillus anthracis spore challenge, Infect. Immun., 2007, 75, 4020–4029. Unauthenticated Download Date | 6/17/17 12:05 AM 44 Priyanka Prabhu and Vandana Patravale [135] Walve J.R., Bakliwal S.R., Rane B.R., Pawar S.P., Transfersomes: a surrogated carrier for transdermal drug delivery system, International Journal of Applied Biology and Pharmaceutical Technology, 2011, 2, 204-213. [136] Mahor S., Rawat A., Dubey P.K., Gupta P.N., Khatri K., Goyal A.K. et al., Cationic transfersomes based topical genetic vaccine against hepatitis B, Int. J. Pharm., 2007, 340, 13–19. [137] Gupta P.N., Mishra V., Singh P., Rawat A., Dubey P., Mahor S. et al., Tetanus toxoid loaded transfersomes for topical immunization, J. Pharm. Pharmacol., 2005, 57, 295-301. [138] Gupta P.N., Vyas S.P., Vishwidyalaya H.S.G., Transfersomes for vaccine delivery: a potential approach for topical immunization, Med, Chem. Res., 2004, 13, 414-426. [139] Xu J., Ding Y., Yang Y., Enhancement of mucosal and cellular immune response in mice by vaccination with respiratory syncytial virus DNA encapsulated with transfersome, Viral Immunol., 2008, 21, 483-489. [140] Arora D., Khurana B., Kumar M.S., Vyas S.P., Oral immunization against Hepatitis B Virus using mannosylated bilosomes, Int. J. Rec. Adv. Pharm. Res., 2011, 1, 45-51. [141] Mann J.F., Scales H.E., Shakir E., Alexander J., Carter K.C., Mullen A.B. et al., Oral delivery of tetanus toxoid using vesicles containing bile salts (bilosomes) induces significant systemic and mucosal immunity, Methods, 2006, 38, 90-95. [142] Singh P., Prabakaran D., Jain S., Mishra V., Jaganathan K.S., Vyas S.P., Cholera toxin B subunit conjugated bile salt stabilized vesicles (bilosomes) for oral immunization, Int. J. Pharm., 2004, 278, 379-390. [143] Shukla A., Khatri K., Gupta P.N., Goyal A.K., Mehta A., Vyas S.P., Oral immunization against hepatitis B using bile salt stabilized vesicles (bilosomes), J. Pharm. Pharm. Sci., 2008, 11, 59-66. [144] Csaba N., Garcia-Fuentes M., Alonso M.J., Nanoparticles for nasal vaccination, Adv. Drug Deliv. Rev., 2009, 61, 140–157. [145] Mastelic B., Ahmed S., Egan W.M., Giudice G.D., Golding H., Gust I. et al., Mode of action of adjuvants: implications for vaccine safety and design, Biologicals, 2010, 38, 594-601. [146] Heldens J.G., Pouwels H.G., Derks C.G., Van de Zande S.M., Hoeijmakers M.J., The first safe inactivated equine influenza vaccine formulation adjuvanted with ISCOM-matrix that closes the immunity gap, Vaccine, 2009, 27, 5530-5537. [147] Sankar V.R., Reddy Y.D., Nanocochleate- a new approach in lipid drug delivery, Int. J. Pharm. Pharm. Sci., 2010, 2, 220-223. [148] Ramasamy T., Khandasamy U., Hinabindhu R., Kona K., Nanocochleate - a new drug delivery system. FABAD J. Pharm. Sci., 2009, 34, 91–101. [149] Kheiri M.T., Feketeova E., Wang Z., Mannino R.J., GouldFogerite S., Protective immunity in mice following immunization with the cochleate-based subunit influenza vaccines, Iran Biomed. J., 2001, 5, 33-38. [150] Patidar A., Thakur D.S., Kumar P., Verma J., A review on novel lipid based nanocarriers, Int. J. Pharm. Pharm. Sci., 2010, 2, 30- 35. [151] Arias M.A., Loxley A., Eatmon C., Roey G.V., Fairhurst D., Mitchnick M. et al., Carnauba wax nanoparticles enhance strong systemic and mucosal cellular and humoralimmune responses to HIV-gp140 antigen, Vaccine, 2011, 29, 1258–1269. [152] Sloat B.R., Sandoval M.A., Hau A.M., He Y., Cui Z., Strong antibody responses induced by protein antigens conjugated onto the surface of lecithin-based nanoparticles, J. Control Release, 2010, 141, 93–100. [153] Moon J.J., Suh H., Li A.V., Ockenhouse C.F., Yadava A., Irvine D.J., Enhancing humoral responses to a malaria antigen with nanoparticle vaccines that expand Tfh cells and promote germinal center induction, PNAS, 2012, 109, 1080-1085. [154] Wang T., Jiang H., Zhao Q., Wang S., Zou M., Cheng G., Enhanced mucosal and systemic immune responses obtained by porous silica nanoparticles used as an oral vaccine adjuvant: Effect of silica architecture on immunological properties, Int. J. Pharm., 2012, 436, 351– 358. [155] Carvalho L.V., Ruiz R.D.C., Scaramuzzi K., Marengo E.B., Matos J.R., Tambourgi D.V., Fantini M.C.A., Sant’Annaa O.A., Immunological parameters related to the adjuvant effect of the ordered mesoporous silica SBA-15, Vaccine, 2010, 28, 7829–7836. [156] Guo H-C., Feng X-M., Sun S-Q., Wei Y-Q., Sun D-H., Liu X-T. et al., Immunization of mice by hollow mesoporous silica nanoparticles as carriers of porcine circovirus type 2 ORF2 protein, Virol. J., 2012, 9, 108. [157] Gordon S., Teichmann E., Young K., Finnie K., Rades T., Hook S., In vitro and in vivo investigation of thermosensitive chitosan hydrogels containing silica nanoparticles for vaccine delivery, Eur. J. Pharm. Sci., 2010, 41, 360–368. [158] Behera T., Swain P., Antigen adsorbed calcium phosphate nanoparticles stimulate both innate and adaptive immune response in fish, Labeo rohita H. Cell Immunol., 2011, 271, 350–359. [159] He Q., Mitchell A.R., Johnson S.L., Wagner-Bartak C., Morcol T., Bell S.J.D., Calcium phosphate nanoparticle adjuvant, Clin. Diagn. Lab. Immunol., 2000, 7, 899–903. [160] Sokolova V., Knuschke T., Kovtun A., Buer J., Epple M., Westendorf A.M., The use of calcium phosphate nanoparticles encapsulating Toll-like receptor ligands and the antigen hemagglutinin to induce dendritic cell maturation and T cell activation, Biomaterials, 2010, 31, 5627-5633. [161] He Q., Mitchell A., Morcol,T., Bell S.J.D., Calcium phosphate nanoparticles induce mucosal immunity and protection against herpes simplex virus type 2, Clin. Diagn. Lab. Immunol., 2002, 9, 1021-1024. [162] Lee I-H., Kwon H-K., An S., Kim D., Kim S., Yu M.K. et al., Imageable antigen- presenting gold nanoparticle vaccines for effective cancer immunotherapy in vivo, Angew. Chem. Int. Ed., 2012, 51, 8800 –8805. [163] Barhate G., Gautam M., Gairola S., Jadhav S., Pokharkar V., Quillaja saponaria extract as mucosal adjuvant with chitosan functionalized gold nanoparticles for mucosal vaccine delivery: Stability and immunoefficiency studies, Int. J. Pharm., 2013, 441, 636– 642. [164] Bastús N.G., Sánchez-Tilló E., Pujals S., Farrera C., Kogan M.J., Giralt E.et al., Peptides conjugated to gold nanoparticles induce macrophage activation, Mol. Immunol., 2009, 46, 743–748. [165] Jazayeri S.D., Ideris A., Zakaria Z., Shameli K., Moeini H., Omar A.R., Cytotoxicity and immunological responses following oral vaccination of nanoencapsulated avian influenza virus H5 DNA vaccine with green synthesis silver nanoparticles, J. Control Release, 2012, 161, 116-123. Unauthenticated Download Date | 6/17/17 12:05 AM [166] Wang T., Zou M., Jiang H., Ji Z., Gao P., Cheng G., Synthesis of a novel kind of carbon nanoparticle with large mesopores and macropores and its application as an oral vaccine adjuvant, Eur. J. Pharm. Sci., 2011, 44, 653–659. [167] Villa C.H., Dao T., Ahearn I., Fehrenbacher N., Casey E., Rey D.A. et al., Single-walled carbon nanotubes deliver peptide antigen into dendritic cells and enhance IgG responses to tumor-associated antigens, ACS Nano, 2011, 5, 5300-5311. [168] Goyal A.K., Rawat A., Mahor S., Gupta P.N., Khatri K., Vyas S.P., Nanodecoy system: A novel approach to design hepatitis B vaccine for immunopotentiation, Int. J. Pharm., 2006,309, 227–233. Nanocarriers for vaccine delivery 45 [169] Goyal A.K., Khatri K., Mishra N., Mehta A., Vaidya B., Tiwari S. et al., Aquasomes—a nanoparticulate approach for the delivery of Antigen, Drug Dev. Ind. Pharm., 2008, 34,1297–1305. [170] Champion C.I., Kickhoefer V.A., Liu G., Moniz R.J., Freed A.S., Bergmann L.L. et al., A vault nanoparticle vaccine induces protective mucosal immunity, PLoS ONE, 2009, 4. [171] Kar U.K., Jiang J., Champion C.I., Salehi S., Srivastava M., Sharma S.,et al., Vault nanocapsules as adjuvants favor cell-mediated over antibody-mediated immune responses following immunization of mice, PLoS ONE, 2012, 7. Unauthenticated Download Date | 6/17/17 12:05 AM