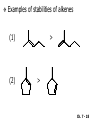Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Woodward–Hoffmann rules wikipedia , lookup
Asymmetric hydrogenation wikipedia , lookup
Bottromycin wikipedia , lookup
Fischer–Tropsch process wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Cracking (chemistry) wikipedia , lookup
Kinetic resolution wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
Vinylcyclopropane rearrangement wikipedia , lookup
Elias James Corey wikipedia , lookup
Enantioselective synthesis wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
Diels–Alder reaction wikipedia , lookup
1,3-Dipolar cycloaddition wikipedia , lookup
Petasis reaction wikipedia , lookup
Ene reaction wikipedia , lookup
Wolff rearrangement wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
Discodermolide wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Tiffeneau–Demjanov rearrangement wikipedia , lookup
Hydrogenation wikipedia , lookup
Asymmetric induction wikipedia , lookup
Stille reaction wikipedia , lookup
George S. Hammond wikipedia , lookup
Strychnine total synthesis wikipedia , lookup
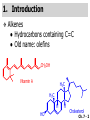
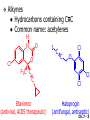
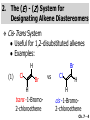
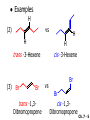
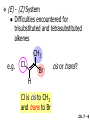
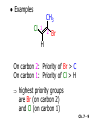
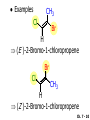
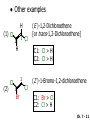
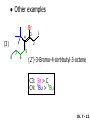
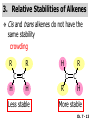
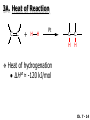
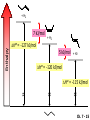
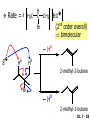
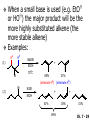
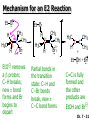
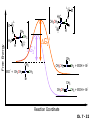
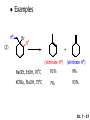
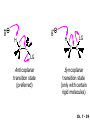
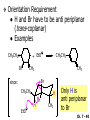
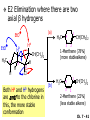
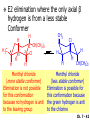
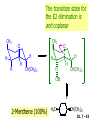
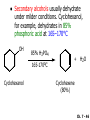
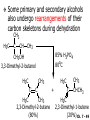
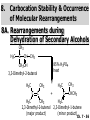
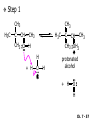
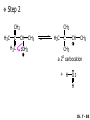
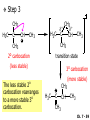
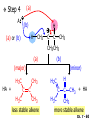
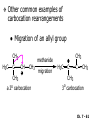
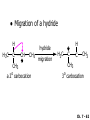
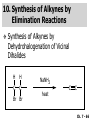
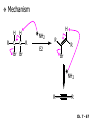
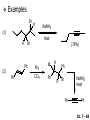
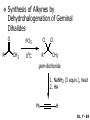
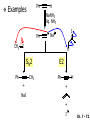
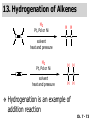
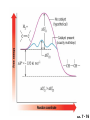
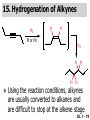
Chapter 7 Alkenes and Alkynes I: Properties and Synthesis. Elimination Reactions of Alkyl Halides Ch. 7 - 1 1. Introduction Alkenes ● Hydrocarbons containing C=C ● Old name: olefins CH2OH Vitamin A H3C H3C H HO H Cholesterol Ch. 7 - 2 Alkynes ● Hydrocarbons containing C≡C ● Common name: acetylenes H N Cl O O F3C C C Efavirenz (antiviral, AIDS therapeutic) I C Cl C O Cl Cl Haloprogin (antifungal, antiseptic) Ch. 7 - 3 2. The (E ) - (Z ) System for Designating Alkene Diastereomers Cis-Trans System ● Useful for 1,2-disubstituted alkenes ● Examples: H (1) Cl Br Br H trans -1-Bromo2-chloroethene vs Cl H H cis -1-Bromo- 2-chloroethene Ch. 7 - 4 ● Examples H (2) vs H H H cis -3-Hexene trans -3-Hexene (3) Br Br trans -1,3- Dibromopropene vs Br Br cis -1,3- Dibromopropene Ch. 7 - 5 (E) - (Z) System ● Difficulties encountered for trisubstituted and tetrasubstituted alkenes e.g. CH3 Cl Br cis or trans? H Cl is cis to CH3 and trans to Br Ch. 7 - 6 The Cahn-Ingold-Prelog (E) - (Z) Convention ● The system is based on the atomic number of the attached atom ● The higher the atomic number, the higher the priority Ch. 7 - 7 The Cahn-Ingold-Prelog (E) - (Z) Convention ● (E) configuration – the highest priority groups are on the opposite side of the double bond “E ” stands for “entgegen”; it means “opposite” in German ● (Z) configuration – the highest priority groups are on the same side of the double bond “Z ” stands for “zusammer”; it means “together” in German Ch. 7 - 8 ● Examples Cl CH3 1 2 Br H On carbon 2: Priority of Br > C On carbon 1: Priority of Cl > H ⇒ highest priority groups are Br (on carbon 2) and Cl (on carbon 1) Ch. 7 - 9 ● Examples Cl CH3 Br H ⇒ (E )-2-Bromo-1-chloropropene Br Cl CH3 H ⇒ (Z )-2-Bromo-1-chloropropene Ch. 7 - 10 ● Other examples (1) Cl H 2 1 Cl H (2) Cl C1: Cl > H C2: Cl > H 2 1 Br (E )-1,2-Dichloroethene [or trans-1,2-Dichloroethene] Cl (Z )-1-Bromo-1,2-dichloroethene C1: Br > Cl C2: Cl > H Ch. 7 - 11 ● Other examples Br 4 (3) 8 7 1 3 2 5 6 (Z )-3-Bromo-4-tert-butyl-3-octene C3: Br > C n t C4: Bu > Bu Ch. 7 - 12 3. Relative Stabilities of Alkenes Cis and trans alkenes do not have the same stability crowding R R C H H R C C H Less stable R C H More stable Ch. 7 - 13 3A. Heat of Reaction C C + H H Pt C C H H Heat of hydrogenation ● ∆H° ≃ -120 kJ/mol Ch. 7 - 14 + H2 Enthalpy 7 kJ/mol + H2 ∆H° = -127 kJ/mol 5 kJ/mol + H2 ∆H° = -120 kJ/mol ∆H° = -115 kJ/mol ≈ ≈ ≈ Ch. 7 - 15 3B. Overall Relative Stabilities of Alkenes The greater the number of attached alkyl groups (i.e., the more highly substituted the carbon atoms of the double bond), the greater the alkene’s stability. Ch. 7 - 16 R Relative Stabilities of Alkenes R R R > R R R H tetratrisubstituted substituted R H > R H > R H R R > H R disubstituted R H > H H H H H H > H H monounsubstituted substituted Ch. 7 - 17 Examples of stabilities of alkenes (1) (2) > > Ch. 7 - 18 4. Cycloalkenes Cycloalkenes containing 5 carbon atoms or fewer exist only in the cis form cyclopropene cyclobutene cyclopentene Ch. 7 - 19 Trans – cyclohexene and trans – cycloheptene have a very short lifetime and have not been isolated cyclohexene Hypothetical trans - cyclohexene (too strained to exist at r.t.) Ch. 7 - 20 Trans – cyclooctene has been isolated and is chiral and exists as a pair of enantiomers cis - cyclooctene trans - cyclooctenes Ch. 7 - 21 5. Synthesis of Alkenes via Elimination Reactions Dehydrohalogenation of Alkyl Halides H H C H C X H H base -HX H H H H Dehydration of Alcohols H H C H H H C OH H+, heat H H -HOH H H Ch. 7 - 22 6. Dehydrohalogenation of Alkyl Halides The best reaction conditions to use when synthesizing an alkene by dehydrohalogenation are those that promote an E2 mechanism H B: C E2 C C C + B:H + X X Ch. 7 - 23 6A. How to Favor an E2 Mechanism Use a secondary or tertiary alkyl halide if possible. (Because steric hinderance in the substrate will inhibit substitution) When a synthesis must begin with a primary alkyl halide, use a bulky base. (Because the steric bulk of the base will inhibit substitution) Ch. 7 - 24 Use a high concentration of a strong and nonpolarizable base, such as an alkoxide. (Because a weak and polarizable base would not drive the reaction toward a bimolecular reaction, thereby allowing unimolecular processes (such as SN1 or E1 reactions) to compete. Ch. 7 - 25 Sodium ethoxide in ethanol (EtONa/EtOH) and potassium tertbutoxide in tertbutyl alcohol (t-BuOK/tBuOH) are bases typically used to promote E2 reactions Use elevated temperature because heat generally favors elimination over substitution. (Because elimination reactions are entropically favored over substitution reactions) Ch. 7 - 26 6B. Zaitsev’s Rule (1) Examples of dehydrohalogenations where only a single elimination product is possible EtONa EtOH, 55 C Br EtONa (2) (3) (79%) o Br ( ) n (91%) o EtOH, 55 C Br t -BuOK o t -BuOH, 40 C ( ) n (85%) Ch. 7 - 27 Rate = k H3C H C CH3 EtO (2nd order overall) ⇒ bimolecular Br ̶ H B Ha a Hb 2-methyl-2-butene Br ̶ H b 2-methyl-1-butene Ch. 7 - 28 ⊖ When a small base is used (e.g. EtO ⊖ or HO ) the major product will be the more highly substituted alkene (the more stable alkene) Examples: (1) Ha Hb + EtOH 70oC Br (2) NaOEt Br 69% 31% (eliminate Ha) (eliminate Hb) KOEt + EtOH 51% + 18% 69% 31% Ch. 7 - 29 Zaitsev’s Rule ● In elimination reactions, the more highly substituted alkene product predominates Stability of alkenes Me Me C Me C > Me > Me Me C Me Me Me C H C H C > Me C H > H Me C Me H C H H C H Ch. 7 - 30 Mechanism for an E2 Reaction Et O CH3 α C C CH3 H 3C β Br H Et O H EtO⊖ removes a β proton; C−H breaks; new π bond forms and Br begins to depart H H 3C C CH3 C CH3 H Br δ− δ− H3C H C C + Et OH Partial bonds in the transition state: C−H and C−Br bonds break, new π C−C bond forms CH3 CH3 + Br C=C is fully formed and the other products are EtOH and Br⊖ Ch. 7 - 31 δ− O Et H 3C Et O H Free Energy H 3C CH3CH2 δ− C CH3 C CH3 H Br δ− C Br δ− ∆G2‡ H C H H ∆G1‡ CH3 CH3 EtO- + CH3CH2 C CH3CH2 C CH2 + EtOH + Br CH3 Br CH3 CH3CH C CH3 + EtOH + Br Reaction Coordinate Ch. 7 - 32 6C. Formation of the Less Substituted Alkene Using a Bulky Base Hofmann’s Rule ● Most elimination reactions follow Zaitsev’s rule in which the most stable alkenes are the major products. However, under some circumstances, the major elimination product is the less substituted, less stable alkene Ch. 7 - 33 ● Case 1: using a bulky base EtO (small) CH3CH CHCH3 + CH3CH2CH CH2 (80%) (20%) CH3CH2CHCH3 Br t BuO (bulky) ⊖ EtO (small base) H CH3CH CHCH3 (30%) + CH3CH2CH CH2 (70%) H H H H C C C C H H Br H tBuO⊖ H (bulky base) Ch. 7 - 34 ● Case 2: with a bulky group next to the leaving halide less crowded β-H H3C Me H Br H C C C Me H C H Me H more crowded β-H EtO Me H H3C C C Me H C CH2 Me (mainly) Ch. 7 - 35 Zaitsev Rule vs. Hofmann Rule ● Examples (1) Ha Hb Br + a (eliminate H ) (eliminate Hb) NaOEt, EtOH, 70oC 69% 31% t t o KO Bu, BuOH, 75 C 28% 72% Ch. 7 - 36 ● Examples Hb (2) Br Ha + a (eliminate H ) b (eliminate H ) o NaOEt, EtOH, 70 C 91% 9% t o t KO Bu, BuOH, 75 C 7% 93% Ch. 7 - 37 6D. The Stereochemistry of E2 Reactions The 5 atoms involved in the transition state of an E2 reaction (including the base) must lie in the same plane The anti coplanar conformation is the preferred transition state geometry ● The anti coplanar transition state is staggered (and therefore of lower energy), while the syn coplanar transition state is eclipsed Ch. 7 - 38 B B H C LG H C C C LG Anti coplanar transition state (preferred) Syn coplanar transition state (only with certain rigid molecules) Ch. 7 - 39 Orientation Requirement ● H and Br have to be anti periplanar (trans-coplanar) ● Examples + EtO CH3CH2 Br CH3CH2 CH3 CH3 Br since: CH3CH2 EtO H H H CH3 Only H is anti periplanar to Br Ch. 7 - 40 E2 Elimination where there are two axial β hydrogens (a) EtO EtO H3C H b H 4 H H Ha 3 a 1 2 H3C 1 4 3 2 1-Menthene (78%) (more stablealkene) CH(CH3)2 H Cl Hb Both and hydrogens are anti to the chlorine in this, the more stable conformation CH(CH3)2 (b) H3C 1 4 3 CH(CH3)2 2 2-Menthene (22%) (less stable alkene) Ch. 7 - 41 E2 elimination where the only axial β hydrogen is from a less stable Conformer H H3C 1 4 H H 3 CH3 H 2 CH(CH3)2 Cl H Menthyl chloride (more stable conformer) Elimination is not possible for this conformation because no hydrogen is anti to the leaving group Cl H H H H CH(CH3)2 Menthyl chloride (less stable conformer) Elimination is possible for this conformation because the green hydrogen is anti to the chlorine Ch. 7 - 42 The transition state for the E2 elimination is anti coplanar CH3 CH3 Cl H H H H CH(CH3)2 Cl H H H H CH(CH3)2 OEt 2-Menthene (100%) H3C CH(CH3)2 Ch. 7 - 43 7. Acid-Catalyzed Dehydration of Alcohols Most alcohols undergo dehydration (lose a molecule of water) to form an alkene when heated with a strong acid C C H OH HA heat C C + H2O Ch. 7 - 44 The temperature and concentration of acid required to dehydrate an alcohol depend on the structure of the alcohol substrate ● Primary alcohols are the most difficult to dehydrate. Dehydration of ethanol, for example, requires concentrated sulfuric acid and a temperature of 180°C H H H C C H OH H conc. H2SO4 H 180oC H Ethanol (a 1o alcohol) H C + H2O C H Ch. 7 - 45 ● Secondary alcohols usually dehydrate under milder conditions. Cyclohexanol, for example, dehydrates in 85% phosphoric acid at 165–170°C OH 85% H3PO4 165-170oC Cyclohexanol + H2O Cyclohexene (80%) Ch. 7 - 46 ● Tertiary alcohols are usually so easily dehydrated that extremely mild conditions can be used. tert-Butyl alcohol, for example, dehydrates in 20% aqueous sulfuric acid at a temperature of 85°C CH3 H3C C OH CH3 tert-Butyl alcohol CH2 20% H2SO4 85oC H3C CH3 + H2O 2-Methylpropene (84%) Ch. 7 - 47 ● The relative ease with which alcohols will undergo dehydration is in the following order: R R C R OH R 3o alcohol > R C H OH H 2o alcohol > R C OH H 1o alcohol Ch. 7 - 48 Some primary and secondary alcohols also undergo rearrangements of their carbon skeletons during dehydration CH3 H3C C CH CH3 85% H3PO4 80oC CH3OH 3,3-Dimethyl-2-butanol H3C CH3 C C CH3 H3C 2,3-Dimethyl-2-butene (80%) CH3 H3C + C CHCH3 H2C 2,3-Dimethyl-1-butene (20%) Ch. 7 - 49 ● Notice that the carbon skeleton of the reactant is C C C C C C while that of the product is C C C C C C Ch. 7 - 50 7A. Mechanism for Dehydration of 2 o & 3 Alcohols: An E1 Reaction o Consider the dehydration of tert-butyl alcohol + H O ● Step 1 H CH3 H3C C CH3 H O H + H O H H3C H3C H C O H CH3 protonated alcohol Ch. 7 - 51 ● Step 2 H3C H3C H C O CH3 H H3C CH3 C CH3 + H O H a carbocation ● Step 3 H H H3C C C H CH3 + H CH2 O H H3C C CH3 H + H O H 2-Methylpropene Ch. 7 - 52 7B. Carbocation Stability & the Transition State Recall R R C H > R R 3o most stable C H > H R > 2o C H > H R > 1o C H > methyl least stable Ch. 7 - 53 Ch. 7 - 54 7C. A Mechanism for Dehydration of Primary Alcohols: An E2 Reaction protonated alcohol o 1 alcohol H C C H H O H + H A acid catalyst H H O fast slow r.d.s H + HA + C C H alkene H H C C O H H H +A conjugate base Ch. 7 - 55 8. Carbocation Stability & Occurrence of Molecular Rearrangements 8A. Rearrangements during Dehydration of Secondary Alcohols CH3 H3C C CH CH3 85% H3PO4 heat CH3OH 3,3-Dimethyl-2-butanol H3C C C CH3 H3C CH3 + CH3 H3C 2,3-Dimethyl-2-butenol (major product) C CHCH3 H2C 2,3-Dimethyl-1-butene (minor product) Ch. 7 - 56 Step 1 CH3 H3C C CH3 CH CH3 O CH3 H3C H CH3 CH CH3 OH2 H + H C O H protonated alcohol + H O H Ch. 7 - 57 Step 2 CH3 CH3 H3C C H3C CH OH2 CH3 H3C C CH CH3 CH3 o a 2 carbocation + H O H Ch. 7 - 58 Step 3 CH3 H3C C CH3 δ+ δ C CH + CH CH3 H3C CH3 CH3 CH3 2o carbocation transition state (less stable) 3o carbocation o The less stable 2 carbocation rearranges o to a more stable 3 carbocation. CH3 H3C C CH (more stable) CH3 CH3 Ch. 7 - 59 Step 4 A (a) (b) H H (a) or (b) CH2 C C CH3 CH3 CH3 (a) (b) (major) (minor) CH3 H3C HA + C H3C H H2C C C CH3 less stable alkene H3C C CH3 + HA CH3 more stable alkene Ch. 7 - 60 Other common examples of carbocation rearrangements ● Migration of an allyl group CH3 H3C C CH CH3 CH3 o a 2 carbocation methanide migration CH3 H3C C C CH3 CH3 3o carbocation Ch. 7 - 61 ● Migration of a hydride H H3C C CH CH3 CH3 o a 2 carbocation hydride migration H H3C C C CH3 CH3 3o carbocation Ch. 7 - 62 8B. Rearrangement after Dehydration of a Primary Alcohol R R H H C C C H R H H +H O A C H E2 H C R H R H H R +H C H A protonation H + A H H C C C R R H H C C R H + A H R A O +H R C C + H C C H H C H deprotonation C R C H H +H A Ch. 7 - 63 9. The Acidity of Terminal Alkynes Acetylenic hydrogen sp sp2 H H C C H H C C H pKa = 25 sp3 H H pKa = 44 H H C C H H H pKa = 50 Relative basicity of the conjugate base CH3CH2 > CH2 CH > CH CH Ch. 7 - 64 H Comparison of acidity and basicity of 1st row elements of the Periodic Table ● Relative acidity OH > H pKa 15.7 OR > H CR > H C 16-17 NH2 > H 25 CH 38 CH2 > H 44 CH2CH3 50 ● Relative basicity OH < OR < C CR < NH2 < CH CH2 < CH2CH3 Ch. 7 - 65 10. Synthesis of Alkynes by Elimination Reactions Synthesis of Alkynes by Dehydrohalogenation of Vicinal Dihalides H H C C Br Br NaNH2 heat C C Ch. 7 - 66 Mechanism R H H C C Br Br R NH2 H R E2 R Br NH2 R R Ch. 7 - 67 Examples Br H NaNH2 (1) H (2) Br Ph Ph heat Br2 CCl4 Br Ph (78%) H Ph H NaNH2 heat Br Ph Ph Ch. 7 - 68 Synthesis of Alkynes by Dehydrohalogenation of Geminal Dihalides O R Cl PCl5 CH3 0oC R Cl CH3 gem-dichloride 1. NaNH2 (3 equiv.), heat 2. HA Ph H Ch. 7 - 69 11. Replacement of the Acetylenic Hydrogen Atom of Terminal Alkynes R The acetylide anion can be prepared by H NaNH2 liq. NH3 R Na + NH3 Ch. 7 - 70 R Acetylide anions are useful intermediates for the synthesis of other alkynes R' X R R' + X ∵ 2nd step is an SN2 reaction, usually o only good for 1 R’ o o 2 and 3 R’ usually undergo E2 elimination Ch. 7 - 71 Ph Examples NaNH2 liq. NH3 Ph CH3 H I Na I H SN2 Ph CH3 + E2 Ph H + NaI + I Ch. 7 - 72 13. Hydrogenation of Alkenes C C C C H2 Pt, Pd or Ni solvent heat and pressure H H C C H2 Pt, Pd or Ni H H solvent heat and pressure C C H H Hydrogenation is an example of addition reaction Ch. 7 - 73 Examples H H2 H Rh(PPh3)3Cl H2 Pd/C H H Ch. 7 - 74 14. Hydrogenation: The Function of the Catalyst Hydrogenation of an alkene is an exothermic reaction ● ∆H° ≃ -120 kJ/mol R CH CH + H2 R hydrogenation R CH2 CH2 R + heat Ch. 7 - 75 Ch. 7 - 76 14A. Syn and Anti Additions An addition that places the parts of the reagent on the same side (or face) of the reactant is called syn addition C + C X C Y C X C C + H H syn addition Y Pt C H C H Catalytic hydrogenation is a syn addition. Ch. 7 - 77 An anti addition places parts of the adding reagent on opposite faces of the reactant Y C C + X C Y X C anti addition Ch. 7 - 78 15. Hydrogenation of Alkynes H2 H H Pt or Pd H2 H H H H Using the reaction conditions, alkynes are usually converted to alkanes and are difficult to stop at the alkene stage Ch. 7 - 79 15A. Syn Addition of Hydrogen: Synthesis of cis -Alkenes Semi-hydrogenation of alkynes to alkenes can be achieved using either the Ni2B (P-2) catalyst or the Lindlar’s catalyst ● Nickel boride compound (P-2 catalyst) Ni O O NaBH4 CH3 EtOH 2 ● Lindlar’s catalyst Pd/CaCO3, quinoline Ni2B (P-2) Ch. 7 - 80 Semi-hydrogenation of alkynes using Ni2B (P-2) or Lindlar’s catalyst causes syn addition of hydrogen ● Examples H2 H Ni2B (P-2) Ph CH3 H (97%) (cis) H2 H Pd/CaCO3 quinoline Ph H CH3 (86%) Ch. 7 - 81 15B. Anti Addition of Hydrogen: Synthesis of trans -Alkenes Alkynes can be converted to transalkenes by dissolving metal reduction Anti addition of dihydrogen to the alkyne H o R R' 1. Li, liq. NH3, -78 C 2. aqueous work up R' R H Ch. 7 - 82 Example o 1. Li, liq. EtNH2, -78 C 2. NH4Cl H H anti addition Ch. 7 - 83 Mechanism radical anion R R C C R C vinyl radical H C NHEt R H C C R R Li Li H R C H C R trans alkene EtHN H R H C C R vinyl anion Ch. 7 - 84 16. An Introduction to Organic Synthesis 16A. Why Do Organic Synthesis? To make naturally occurring compounds which are biologically active but difficult (or impossible) to obtain AcO Ph O O OH BzN H OH TAXOL HO H OH O OAc Anti-tumor, anti-cancer agent Ch. 7 - 85 TAXOL Isolated from Pacific Yew tree Leaves Cones and Fruit seed pollen cones usually appear on separate male and female trees Ch. 7 - 86 TAXOL Approved by the U.S. Food & Drug Administration in 1992 for treatment of several types of cancer, including breast cancer, lung cancer, and melanoma An estimation: a 100-year old yew tree must be sacrificed in order to obtain 300 mg of Taxol, just enough for one single dose for a cancer patient Obviously, synthetic organic chemistry methods that would lead to the synthesis of Taxol would be extremely useful Ch. 7 - 87 16B. Retrosynthetic Analysis target molecule 1st precursor 2nd precursor starting compound Ch. 7 - 88 When doing retrosynthetic analysis, it is necessary to generate as many possible precursors, hence different synthetic routes, as possible 1st precursor A target molecule 1st precursor B 1st precursor C 2nd precursor a 2nd precursor b 2nd precursor c 2nd precursor d 2nd precursor e 2nd precursor f Ch. 7 - 89 16C. Identifying Precursors Synthesis of C C (target molecule) Ch. 7 - 90 Retrosynthetic Analysis o SN2 on 1 alkyl halide: good C disconnection 1 C C + δ− X δ+ C disconnection 2 δ+ X δ− + o SN2 on 2 alkyl halide: poor ⇒ will get E2 as major pathway Ch. 7 - 91 Synthesis C C H NaNH2 C liq. NH3 (SN2) NaI + C Na I C C Ch. 7 - 92 16D. Raison d’Etre Summary of Methods for the Preparation of Alkenes C H C X (Dehydrohalogenation of alkyl halides) H+ base, heat C C H OH heat (Dehydration H2, Ni2B (P-2) or Lindlar's catalyst (give (Z)-alkenes) C C C (Semihydrogenation of alkynes) C of alcohols) Li, liq. NH3 (give (E)-alkenes) C (Dissolving metal reduction of alkynes) C Ch. 7 - 93 Summary of Methods for the Preparation of Alkynes X (Dehydrohalogenation Cl of geminal dihalide) R' H R H H NaNH2 heat NaNH2 heat (Dehydrohalogenation of vicinal dihalide) R' R H X Cl R C (Deprotonation of terminal alkynes and SN2 reaction of the acetylide anion) R C R' 1. NaNH2, liq. NH3 2. R'-X (R' = 1o alkyl group) C C H Ch. 7 - 94 END OF CHAPTER 7 Ch. 7 - 95