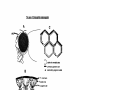
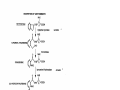
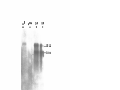
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Minimal genome wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Ridge (biology) wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Skewed X-inactivation wikipedia , lookup
Genetic drift wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Oncogenomics wikipedia , lookup
Genetic engineering wikipedia , lookup
Gene therapy wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Public health genomics wikipedia , lookup
Saethre–Chotzen syndrome wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Gene nomenclature wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
History of genetic engineering wikipedia , lookup
Population genetics wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Genome evolution wikipedia , lookup
Gene desert wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
X-inactivation wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Point mutation wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Dominance (genetics) wikipedia , lookup
Gene expression programming wikipedia , lookup
Gene expression profiling wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Genome (book) wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Designer baby wikipedia , lookup
GENETIC AND MOLECULAR ANALYSIS OF THE garnet EYE COLOUR GENE OF Drosophila melanogaster by VETT LLOYD BSc., The University of British Columbia MSc., The University of Geneva A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY in THE FACULTY OF GRADUATE STUDIES Genetics Programme We accept this thesis as conforming to the required standard THE UNIVERSITY OF BRITISH COLUMBIA July 1995 © Vett Lloyd, 1995 In presenting this thesis in partial fulfilment of the requirements for an advanced degree at the University of British Columbia, I agree that the Library shall make it freely available for reference and study. I further agree that permission for extensive copying of this thesis for scholarly purposes may be granted by the head of my department or by his or her representatives. it is understood that copying or publication of this thesis for financial gain shall not be allowed without my written permission. (Signature) Department of cO6 Y The University of British Columbia Vancouver, Canada Date DE-6 (2/88) 4. /2 Abstract. The garnet eye colour gene of Drosophila melanogaster is one the group of genes called the transport group of eye colour genes. The garnet gene resembles other members of the transport group of eye colour genes in its phenotype and shows extensive genetic interactions with them. The most significant interaction is between garnet and a cryptic allele of the white gene, first identified as a mutation called enhancer of garnet (we(g)). The phenotype of garnet mutations and the extreme sensitivity to decreased levels of the white+ gene product suggest that garnet, as well as other members of the transport group of eye colour genes, act as positive regulators of the white gene. This interaction may occur at the protein level. A simple model for the physical interactions between the gene products of garnet, white and other members of the transport group is proposed. A critical test of this model requires molecular cloning and analysis of the individual members of the transport group of eye colour genes. Preliminary molecular analysis of the garnet gene is reported in chapter two. The garnet gene is expressed in many different tissues at different stages in development. Two messages are produced from the garnet gene in wild type embryos. Conceptual translation of a 4kb c-DNA reveals a novel protein. In the final chapter I describe the use of the garnet gene to study an example of epigenetic gene regulation. I have examined a mini-chromosome which variegates for the garnet gene. The variegation of this mini-chromosome is extremely unusual in that it depends on the sex of the fly transmitting the mini chromosome. In this way it conforms to the conventional definition of parental genomic imprinting. I examined a number of possible mechanisms which might be responsible for the parental imprinting of the mini-chromosome The results suggest that heterochromatin formation is responsible for the somatic expression of the genomic imprint, but a different system may operate to establish the imprint. III TABLE OF CONTENTS Abstract ii Table of contents iv List of Tables Vi List of Figures x Acknowledgments xv Dedication xvi General introduction 1 Materials and Methods 39 Chapter One Interactions between garnet and other eye colour genes. Introduction 45 Results 47 Discussion 102 Chapter Two Analysis of the garnet gene Introduction 116 Results 118 iv Discussion Chapter Three 187 Imprinting of a mini-chromosome in Drosophila melanogaster. Introduction 205 Results 220 Discussion 269 Bibliography 287 Appendix 1 Determination of eye pigment levels 306 Appendix 2 Cloning of the garnet gene 312 V Index of Tables: page Table 1. A list of genes which affect eye colour in Drosophila melanogaster. 3 Table 2. Genes proposed as members of the transport group of eye colour genes. 32 Table 3. Eye colour genes with known or proposed functions. 36 Table 4. The Effect of 3C deficiencies on white and enhancer of garnet. 54 Table 5. Complementation between e(g) and different white alleles. 60 Table 6. Rescue of the enhancer of garnet effect by white+ transgenes. 64 Table 7. Sensitivity of the enhancer of garnet effect to garnet dosage. 68 Table 8. Effect of we(g) dosage on the enhancer of garnet 71 effect. Table 9. The effect of the enhancer of garnet mutation on other eye colour mutants. 78 vi Table 10. Interactions between garnet and other eye colour genes. 82 Table 11. Summary of interactions between other eye colour genes and enhancer of garnet and garnet. 84 Table 12. Phenotypes of garnet double mutants with different white alleles. 89 Table 13. Epistatic interactions between the wa3 and the alleles. 2 g 91 Table 14. Effect of zeste on garnet. 96 Table 15. Effect of zeste modifiers on the zeste-garnet genotype. 100 Table 16. Pigment levels of various garnet alleles. 123 Table 17. Quantitative assessment of pteridine pigments after chromatographic separation of pigments. 128 Table 18. Effect of various garnet alleles on colour of 131 malpighian tubules. Table 19. Effect of various garnet alleles on testes sheath 134 colour. vii Table 20. The phenotype of various garnet alleles in combination with a deficiency. 137 Table 21. Summary of lesions in different garnet alleles. 158 Table 22. List of genes with sequence similarity to the garnet gene. 179 Table 23. List of published garent alleles. 189 Table 24. List of other names given to imprinting phenomenon. 208 Table 25. Imprinting and human medical conditions. 212 Table 26. Parental effects in Drosophila melanogaster. 215 Table 27. The effect of different garnet alleles on the imprint. 242 Table 28. Maternal effect of the garnet gene. 245 Table 29. Variegation of the Dp(1;f)LJ9 mini-chromosome: The effect of developmental temperature. 255 Table 30. Variegation of the Dp(1;f)LJ9 mini-chromosome: 257 The effect of sodium butryrate. viii Table 31. Variegation of the Dp(1;f)LJ9 mini-chromosome: The effect of Y chromosome dosage: 259 Table 32. Imprinting of the Dp(1;f)LJ9 mini-chromosome: The effect of developmental temperature. 262 Table 33. Imprinting of the Dp(1;f)LJ9 mini-chromosome: The effect of sodium butryrate. 264 Table 34. Imprinting of the Dp(1;f)LJ9 mini-chromosome: The effect of Y chromosome dosage. 267 Table 35. The effect of attatched versus free sex chromosomes on imprinting of Dp(1;f)LJ9. 284 ix Index of Figures. page Figure 1. Diagram of the eye, structure and organization of different cell types in the ommatidia and organization of the primary and secondary pigment cells of Drosophila melanogaster. 11 Figure 2. Diagram of the biosynthetic pathway of xanthommatin production in Drosophila melanogaster. 16 Figure 3. A possible pathway of pteridine pigment biosynthesis in Drosophila melanogaster. 18 Figure 4. Three different models for pteridine biosynthesis in Drosophila melanogaster. 20 Figure 5. Phenotypes of three severe garnet alleles in conjunction with the enhancer of garnet mutation. 49 Figure 6. Cytological localization of the enhancer of garnet 52 mutation. Figure 7. Diagram of the structure of the white gene of 57 Drosophila melanogaster. Figure 8. Comparison between the severity of different white alleles and their effect on garnet. 73 x Figure 9. Analysis of garnet transcription by in situ hybridization to rosy null tissues. 86 Figure 10. The effect of zeste mutant alleles on garnet phenotype. 94 Figure 11. The phenotype of zeste-garnet combinations in females and males. 98 Figure 12. Nolte’s model for the interaction between eye colour genes. 110 Figure 13. Model of the physical interactions between the products of white, brown, scarlet and the transport group of eye colour genes, including garnet. 113 Figure 14. Spectrum of eye colour phenotypes of different alleles of the garnet gene. 120 Figure 15. Chromatographic analysis of pteridine pigments of garnet alleles. 126 Figure 16. Life span of wild type and various garnet mutants. 140 Figure 17. Diagram of the mutation rate to garnet upon the 144 S6-1 strain. xi Figure 18. Diagram of the mutation rate and phenotypes of derivative garnet mutations derived from the original gP mutation. 146 Figure 19. Southern transfer and hybridization analysis of the gP allele and gP derivative mutations. 150 Figure 20. Southern transfer and hybridization analysis of the wild type garnet region and the g 1 allele. 153 Figure 21. Southern transfer and hybridization analysis of garnet mutants. 155 Figure 22. Northern transfer and hybridization analysis of wild type embryos and garnet mutant adults. 160 Figure 23. Restriction fragment length analysis of lambda phage clones containing garnet and flanking sequences. 163 Figure 24. Restriction fragment map of garnet and surrounding region. 165 Figure 25. Strategy used to sequence the imaginal c-DNA 168 clone of the garnet gene. Figure 26. Sequence and conceptual translation of the imaginal disc c-DNA clone of the garnet gene. XII 170 Figure 27. Analysis of garnet transcription by in situ hybridization to various tissues. 183 Figure 28. Southern analysis of regions of sequence similarity to the garnet gene in Drosophila melanogaster. 186 Figure 29. Map of the garnet gene generated by intragenic recombination. 193 Figure 30. Diagram of the structure and origin of the Dp(1;f)LJ9 mini-chromosome. 222 Figure 31. Meiotic stability of the Dp(1;f)LJ9 mini-chromosome. 224 Figure 32. The garnet phenotype in flies with maternally and paternally derived Dp(1;f)LJ9 mini-chromosome. 228 Figure 33. The garnet phenotype in malpighian tubules of flies with maternally and paternally derived Dp(1;f)LJ9 mini-chromosome. 231 Figure 34. Phenotype of narrow abdomen and tiny in flies bearing maternally and paternally derived Dp(1;f)LJ9 mini-chromosomes. 234 Figure 35. Variegation of narrow abdomen and tiny in genotypically identical flies bearing either maternally or paternally derived 236 Dp(1;f)LJ9 mini-chromosome. XIII Figure 36. The Y chromosome does not cause the imprint. 240 Figure 37. Test of the physiological compensation model. 249 Figure 38. Under-representation of the garnet gene in the Dp(1;f)LJ9 mini-chromosome. 252 Figure 39. Model of parent-dependent spread of heterochromatin responsible for the imprint of the Dp(1;f)LJ9 mini-chromosome. xiv 273 Acknowledgments: I would like to thank those who have helped me. It is not possible to list here all those who have given me support and inspiration, emotional, intellectual and financial, over the many years that this work was in progress. Nevertheless, those who have helped me know who they are. They have my gratitude, and I hope, will allow me to opportunity to reciprocate. Although any work of science depends on the prior work of others, the specific contributions of some must be mentioned. The garnet gene was cloned by Dr. D. Sinclair, working in the laboratory of Dr. G. Tener, biochemistry U.B.C. Dr. Sinclair initiated the molecular analysis of the garnet gene, brought the parental effect of Dp(1;f)LJ9 to my attention and provided me with advice and encouragement at every step. The gP allele, which facilitated cloning of the garnet gene was isolated by R. Wennberg, in the laboratory of Dr. T. Grigliatti. The program used to analyze the pigment data was written by Dr. J. Berger. As with any genetic work, I am indebted to the fly stock centres, at Bloomington, Indiana, Bowling Green and Umea, Sweden. A number of undergraduate students also contributed to this work in the course of doing a directed studies project. D. Dyment and K Swanson contributed data to the pigment assay controls in appendix one. M. Maharaj made some of the stocks used to test the effect of the e(g) mutation on other eye colour genes. G. Mahon mobilized the CaSpeR element construct and characterized the new insertion strains that were used to test the rescue of the enhancer of garnet effect. A. Rivers assisted in the sequencing of garnet genomic clones. B. Lee contributed to the test of the enhancer of garnet effect of different white alleles. L. Harvey tested the effect of some garnet alleles on longevity. The details of the experiments are given in the appropriate section. In addition to these students, I also had the opportunity to supervise a number of other undergraduate students, from whom I learned at least as much as they from me. Although their work is unpublished they are: Melanie Klenk, Keyvan Hyunda, Cara Warrington, Carol Lee, Gurdip Lalli, Lynn Ma, Chaucer Wong, and Mahnaz Kermati. Finally, I would like to thank my supervisor Dr. T. Grigliatti for allowing me the rare privilege of complete freedom of research. xv Quotation: There is no answer. There has never been an answer, there will never be an answer. That’s the answer. -Gertrude Stein. xvi General Introduction The garnet gene. 1 General introduction. The problem: The eye colour genes of Drosophila melanogaster have attracted the attention of biologists for as long as the organism has been used as an experimental system for genetics. Aside from their intrinsic beauty, eye colour genes have played a role in the genesis of many important genetic concepts: sex linkage, position effects, pleiotropy, the chromosome theory of inheritance, autonomous and non-autonomous gene action, intra allelic complementation, taxonomy, the action of genes through hormones and developmental regulatory cascades. One of the features of Drosophila eye colour mutants which first appealed to biologists was the number of mutations which have an effect on eye colour. While an invaluable genetic resource, the remarkable number of eye colour genes also posed practical and theoretical problems. The overriding problem posed by eye colour genes is the sheer number of them. Breme and Demerec (1942) list 45 eye colour genes. In 1950, 38 eye colour mutants were extant (Nolte 1950). By 1976, 51 eye colour genes were known (Phillips and Forrest 1976). The list shown in Table 1 is derived from the list of all mutants described in Drosophila melanogaster up to 1992 (Lindsley and Zimm 1992). It lists 110 genes; even if pattern and secondary effects are excluded, there are still 85 genes whose primary effect is on eye colour. Nor are these 110 genes likely to be all the genes which affect eye colour. Amongst eight eye colour mutations isolated from a cellar and a vineyard in Italy, in a study of naturally occurring eye colour variation, three defined novel eye colour genes (Calatayud, Jacobson and Ferré, 1989). A system allotted considerable genetic resources would be expected to be essential or at least important for the 2 Table 1. Genes which affect eye colour in Drosophila melanogaster. The first two columns list the name and gene designation. The eye colour of the mutant is indicated in the third column. Where there is an allelic series the colour of the first allele is given. This is only an approximate indication of the eye colour which varies with allele, age and often sex. The final column gives the pigment group which is affected. Frequently the pigment or pigment group affected is not known and is only inferred from the eye colour. This instance is indicated by a question mark. In some cases the effect on eye pigment is a secondary effect of patterning defects which change cell fate specification as a result of which pigment cells fail to differential. These instances are indicated by “pattern”. In other instances alteration in eye colour or pigmentation is a secondary result of other alterations such as decreased body size or increased melanization. These instances are indicated as “secondary”. This list was derived from Lindsley and Zimm (1992). 3 Table 1. Genes which affect eye colour in Drosophila melanoqaster mutant amy bis bo bos bre bri buo bur bw ca car cast cd cho cm ci cm cmd cml cn cop cr-3 dcm diI--3 dk dke Dke-2 dn dor Dr drb dyb E(z) g gi Hn je kar kpn/awd lix It ltd Ixd amethyst bistre bordeaux bordosteril bright-eye bright burnt-orange burgundy brown claret carnation cast cardinal chocolate cinnamon clot carmine carminoid caramel cinnabar copper cream-3 dark carmine dilute-3 dark dark-eye darkened eye doughnut deep orange Drop dark red brown dusty body enhancer of zeste garnet glass Henna jelly karmoisin killer of prune little isoxanthopterin light Iightoid low xanthopterin dehydrogenase 4 colour pigment affected purple brown purple brown orange orange orange brown brown both brown brown bright brown brown dark orange orangish brown orange brown pale brown paler darker brown darker pattern pale orange darker darker brown pale paler darker dark brown pink orange wild type wild type pale pale wild type pteridines? pteridines? both? pteridines? ommochromes? ommochromes? ommochromes? ommochromes pteridines both both? pteridines? both pteridines? ommochromes pteridines both both? pteridines? ommochromes pteridines? both? pteridines? both? both pattern pattern? pattern both pattern both? secondary? both both pattern? pattern? both? pteridines pteridines pteridines both both pteridines ma mah ma! man Me me! mk mot-28 mot-K mot-321 mot-36c msd(gl) mtb mud! mur nrs ocr or osh p pd Pdr Pec pers pn po port port-b pr Pu pur pw pw-c pwn pym/ade2 ral ras rb rdb red rl rm rs rud rv rwi ly Sa sb maroon mahogany maroon-like mandarin Moire melanized murky mottled 28 mottled of K mottled 321 mottled 36 modifier of sexual dimorphism of gi matt brown mudlike murrey narrow scoop ochracea orange outshifted pink purpleold purpleolder Pupilla ecentrica persimmon prune pale occelli port port-b purple Punch purplish pink wing pink wing c pawn polymorph raisin raspberry ruby reddish-brown red malpighian tubules rolled rimy rose ruddle raven red wine rosy Salmon soft brown 5 brown brown brown orange both darker darker mottled mottled mottled mottled darker brown brown purple darker orange orange brown pink dark pink rosy-like pattern orange brown brighter pale browny browny pale ruby pink lighter brown browny brown browny paler brown wild type darker brown pink browny dark browny browny browny browny pteridines pteridines? pteridines ommochromes? pattern secondary secondary pattern pattern pattern pattern secondary secondary? both? both? secondary ommochromes both secondary both both both? pattern ommoch romes? pteridines pattern? both? pteridines? pteridines pteridines pteridines secondary? secondary? secondary pteridines pteridines? pteridines both? both? both? pattern pteridines? pteridines? pteridines? secondary pteridines? both? both? pteridines? se st sf-3 she som St swy syn te trl It U ups v yin w We z very dark brown brown brown dull brown orange darker brown dark purple lighter mottled dull, rough orange browny white paler lighter sepia safranin safranin-3 sherry sombre scarlet swarthy syndrome tenerchaetae translucent tilt Upturned upright vermilion yin white Washed eye zeste 6 pteridines pteridines? pteridines? pteridines? secondary ommochromes secondary secondary secondary pattern secondary pattern pattern ommochromes pteridines? both pattern? both viability of the organism. Yet few eye colour genes affect the viability of the fly. The clearest example is that of the white gene. Flies with no pigments due to null mutations in this gene are completely viable and fertile. In fact, the first mutation isolated in Drosophila melanogaster was a such a complete loss of function mutation for the white gene (Morgan in 1910 cited in Lindsley and Zimm 1992). That it was isolated from a wild population suggests no great loss of biological vitality. The sheer number of mutations which alter eye colour was seen as a problem by early authors (e.g. Nolte 1 952b) and has not yet been adequately resolved. Even a cursory inspection of the two pigment biosynthetic pathways (Figure 2 and Figure 3) reveals that there are approximately 15 enzymatic steps required to produce the pigments found in the wild type eye. Thus, even allowing for co-factors, there remains a considerable excess of genes involved in the production of the pigments of the wild type eye. In addition to enzymes and co-factors, it is reasonable to suppose that some of this apparent “excess” of eye colour genes are concerned with transport, sequestration and control of deposition of the eye colour pigments. The study of any eye colour gene must ultimately address the question of pleiotropy and redundancy of these genes. Both the number of genes and the dispensability of many of them suggest a diverse range of functions, many of which may be shared by other genes. Despite their historical importance, the disposition and biogenesis of the pigments in the eye, the chemical structure of the pigments, the biosynthetic pathways responsible for the pigments, the genes involved and the complex physiological, developmental and tissue interactions required to produce wild type eye colour remain obscure and still subject to debate. In addressing these 7 questions, the physical context of pigments, the pigment cells of the eye, and the complex developmental regulation of pigment biosynthesis, as revealed by the intersection of genetic and biochemical studies on the pigments, must be briefly summarized. The eye. The eye is the most thoroughly studied of the four pigmented structures in Drosophila melanogaster (excluding melanized structures). The structural and developmental complexity of the eye is underscored by studies which show that approximately two thirds of randomly selected lethals have defects in the eye (of those two thirds, one third is responsible for general cell viability functions, the other third is eye specific, Thaker and Kankel 1992). The development and structure of the compound eye have been the focus of extensive investigation. I will briefly summarize this work as it relates to pigment deposition. Both the signals leading to specification of cell fate in the development of the eye, and the nervous connections between the eye and the brain are subjects of intense research. These subjects have been thoroughly and often reviewed (Meyerowitz and Kanker 1978, Renfranz and Benzer 1989, Pak and Grabowski 1980, Zipursky et al 1984, Tomlinson 1985, Venkatesh, Zipursky and Benzer 1985, Ready 1989, Zipursky 1989, Campus-Ortega 1988, Ranganathang, Harris and Zuker 1991) and will not be treated thoroughly here. Development of the eye. The compound eye of Drosophila melanogaster, like that of many insects, is composed of hundreds of reiterated units, the ommatidia (Figure 1). The eye analage arises from approximately 20 cells which invaginate from the embryonic ectoderm and which eventually form the eye imaginal disc (reviewed by Venkatesh, Zipursky and Benzer 1985, Zipursky 8 1989, Ranganathang, Harris and Zuker 1991). During embryogenesis, first and second instar stages, the eye imaginal disc cells proliferate but remain undifferentiated. During the proliferation stage, the eye disc is attached to the brain by the optic stalk. Later elaboration of the nervous system results in precise spatial correspondence between individual ommatidia and their connections in the brain. During the third instar a dramatic wave of morphogenetic activity sweeps over the disc. This morphogenic furrow is associated with differentiation of the various cell types of the eye. The pigment and cone cells are among the last to differentiate and are recruited from the undifferentiated epithelium by underlying photoreceptor cells. As with other cell types in the eye, cell fate is not clonally determined but is determined by position-dependent induction. Finally, during pupation the eye imaginal disc evaginates to form the adult compound eye. Structure of the eye. The compound eye of the adult Drosophila is composed of 700-800 ommatidia. The structure of three adjacent ommatidia is shown in Figure lB. The distal end of each ommatidium consists of the corneal lens and pseudo cone which functions to gather and focus light. Proximal to the dioptic apparatus are the 8 photoreceptor cells with central rhabdomeres which transduce light to nervous impulses. Each rhabdomere has different wavelength specificity and connects to the brain via a complex network of neural connections. Surrounding each ommatidium is a sleeve of pigment cells (arranged as shown in Figure 1C). The pigment cells act to regulate light exposure and to optically isolate the ommatidia. The pigment granules within the pigment cells are not static. In bright light they move towards the rhabdomere thus reducing the amount of light reaching the rhabdomeres 9 Figure 1. Diagram of the eye, structure and organization of different cell types in the ommatidia and organization of the primary and secondary pigment cells of Drosophila melanogaster. A. Schematic diagram of the head of Drosophila melanogaster. B. Diagram of a longitudinal section through the eye of Drosophila melanogaster showing three adjacent ommatidia. The various cell types which compose the ommatidia are shown. Redrawn from Nolte 1950, figure 12. C. Schematic diagram of the organization of primary and secondary pigment cells and the ommatidial lens. Redrawn from Nolte 1950, figure 11. 10 The eye of Drosophila melanogaster A C outline of ommatidia lens primary pigment cell — B secondary pigment cell e cell o cell nucleus • pigment granule membrane o p /0 4—post retinal cells —layer of monopolar cells . external optic glomerulus external chiasma 11 whereas in low light they migrate to the periphery of the ommadium increasing the light exposure but decreasing visual acuity. Pigment cells There are two types of pigment cells, the primary and secondary pigment cells. The primary pigment cells lie more distally in the eye directly surrounding the pseudo cone, while the secondary pigment cells lie more proximally, principally surrounding the photoreceptor cells. These two types of pigment cells cooperate to completely encase each individual ommatidium (although each pigment cell is shared by adjacent ommatidia). The pigment cells have an abundance of pigment granules in their cytoplasm, however, they are not the only cells with pigment granules. The photoreceptor cells also have pigment granules in their lateral cytoplasm, although fewer than the pigment cells. The post retinal and basal cells may also have some pigment granules (Mainz 1938, Nolte 1950, Reaume, Knecht and Chovnick 1991), although this has been the source of some dispute. Pigment granules. Early light and electron microscopy work defined differences in pigment granule morphology (Shultz 1935, Nolte 1950, Shoup 1966). There are two types of (normal) pigment granules, named type one and type two. Both types of granules are ribosome-sized, multi-subunit (Hearl, Dorsett and Jacobson 1983, Hearl and Jacobson 1984) membrane-bound organelles which originate in close proximity to the golgi apparatus and are likely derived from it (Shoup 1966), although alternative origins have been proposed (Reaume, Knecht and Chovnick 1991). Type one pigment granules first appear approximately two days before eclosion, which corresponds to the first time that the ommochrome pigment can be detected (Schultz 1935, Nolte 1950, 1954a). They are electron dense, grow in size after eclosion and contain only 12 ommochromes. This type is found in primary pigment cells and most probably in the photoreceptor cells. Type two granules are complex membrane bound, fenestrated structures found only in the secondary pigment cells. They are the site of pteridine pigment deposition. Development of these granules and the first detectable appearance of the pteridine pigments coincide at approximately one day before eclosion. These granules do not change size in development but do become denser after eclosion. Change in both the size and morphology of type one and two pigment granules continues for a few days after eclosion of the pharate adult, concomitant with increase in the amount of ommochrome and pteridine pigments. Eye colour mutants often show a variety of complex changes in the colour and morphology of these pigment granules. For example, histological analysis of the g 3 allele, the only garnet allele for which histological information has been recorded (Nolte 1950), shows normal numbers, distribution, development and morphology of pigment granules. The only change from wild type was alteration in the intensity of colour of both the type one and type two granules. This change presumably corresponds to deficiency for both the pteridine and ommochrome pigments. The discovery that pigments were associated with proteins (Schultz 1935) led to the realization that the pigment granules were largely proteinatious. Coordinate appearance of the pigment granules and pigments in development (Schultz 1935, Nolte 1950, 1954a), in conjunction with the proteinatious nature of the pigment granules led to speculation that pigment granules are not just passive sites of pigment deposition but are complexes composed of the biosynthetic pigment enzymes as well as the pigments themselves (Phillips, Forrest and Kulkarni, 1973). This hypothesis has been contested (Sullivan, Grillo and Kitos 1974) based on the finding that the enzymes in the xanthommatin pathway are 13 found in different subcellular compartments whereas others are free in the cytosol. In addition, there is evidence that suggests that the pleiotropic effect of different eye colour mutants on the various enzymes, on which the enzyme complex model was based, is a result of failure to differentiate enzymatic and non-enzymatic conversion of pigment intermediates (Wiley and Forrest 1981). Nevertheless, more recent work suggests that pigment granules contain at least some of the enzymes involved in pigment biosynthesis (Dorsett, Yim and Jacobson 1978, Hearl, Dorsett and Jacobson 1983, Hearl and Jacobson 1984). Biosynthesis of the eye colour pigments: The eye colour of the wild-type Drosophila eye is derived from the deposition of two biochemically distinct types of pigment, the ommochromes and the pteridines. Both types of compounds are widely present in nature in both the plant and animal kingdom. The ommochromes are fairly simple compounds derived from the amino acid tryptophane. The pteridine compounds, however, are remarkably complex. The pteridines were first isolated by Hopkins (1889) from the wings of an English brimstone butterfly but chemical identification of the red pigment in Drosophila as a pteridine was not made until 1940 (Lehderer 1940). The structure of some of the pteridines is still subject to dispute. While the fundamental steps of the biosynthetic pathway for their production appears conserved from the prokaryote, Escheria coil through Drosophila to humans, the details are still under intense investigation (Phillips and Forrest 1976, Pfleiderer 1993). Figures 2 and 3 show the biosynthetic pathways for the production of these two compounds. While the biosynthetic pathway for the ommochrome pigment, xanthommatin, has been determined, based in large part on Drosophila genetics, the pathway for the pteridine pigments remains 14 Figure 2. Biosynthetic pathway of xanthommatin production in Drosophila melanogaster. The structure of the various intermediates in xanthommatin biosynthesis and the enzymes responsible for their production are shown. Adapted from Phillips and Forrest 1976. 15 BIOSYNTHESIS OF XANTHOMMATIN NH2 TRYPTOPHAN •CH2/\COOH tryptophan pyrrolase vermilion + N-FORMYL KYNURENINE NH2 formamidase KYNURENINE cinnabar ÷ 3-HYDROXYKYNURENINE COOH HCI4H2 phenoxazinone synthetase XANTHOMMATIN DIHYDROXANTHOMMATIN 16 Figure 3. A possible pathway for pteridine pigment biosynthesis in Drosophila melanogaster. The structure of the various intermediates involved in pteridine biosynthesis and some of the enzymes and genes responsible for their production are shown. Adapted from Brown et al. 1978, Brown 1989, Calatayud, Jacobson and Ferré 1990 and Pfleiderer 1993. 17 A possible pathway for pteridine pigment biosynthesis i n Drosophila melanogaster 0 N NJXj> 2 H rj,-LP guanosine triphosphate + OHOHHP HA’d-dLcL-P GTP cyclohydrolase Pu + cm, ma-I, JN) 1 H2NI Ii dihydroneopterin-P3 sepiapterin synthase A pr sepiapterin synthase B -N F-12N H4 pterin * 6-acetyl-homopterin aka-pyrimidodiazepine 0+ H2N H’ N1X)OH 2 H H2 pterin dihydrobiopterin 0 HH H 0* HH H HA()H H2N’ &HI isoxanthopterin xanthopterin H4 biopterin drosopterin 18 biopterin Figure 4. Three models proposed for pteridine biosynthesis in Drosophila melanogaster. Figure 4 shows three pathways proposed for pteridine biosynthesis. The structures of the pigments and intermediates are as shown in Figure 3. A. The pathway proposed by Calatayud, Jacobson and Ferré 1989. This is the pathway shown in Figure 3. B. The pathway proposed by Brown 1989. C. The pathway proposed by Ferré et al. 1983. 19 Three pathways proposed for pterindie biosynthesis I n Drosophila melanogaster A. pathway proposed by Calatayud, Jacobson and Ferre, 1989. GTP H2 neoperinP3 pyruvoyl -144 pterin Iactoyl-H4-pterin H4-ptenn I H2-rin y-H2Lmopterin pterin isoxanttpterin Htbiopterin ‘terins droso H4-biopterin B. Pathway proposed by Brown, 1989 spiapterin biopterin GTP H2 hydroneopterin H2-Pterin PyruvoyI-t4Pterin Lactoyl-H4 Pterin H4 Biopterin Isoxanthopterin osopterin H2Bitterin S epiapterin C. Pathway proposed by Ferre et al., 1983. GTP 4 4 H2-6-(V, 2’-dioxopropyl)-pterin? H2-Neopterin H2-Biotteri n Pterin I soxanthopterin p” Biopterin “Drosoprins” 20 H4-Biopterin somewhat speculative. Figure 4 shows three recent models of the pteridine pigment biosynthetic pathway. The first major advance in understanding the formation of pigments was the realization that the wild type pigmentation was composed of two pigment types; a red-orange pigment and a yellow-brown pigment. In 1924 Johannsen noted that there were two types of pigment granules, one red and the other yellow (Johannsen 1924). This observation was related to two biochemically distinct pigment groups by Casteel (1929), Schultz (1935) and Mon (1937). Studies of combinations of eye colour mutants provided complementary evidence of two independent genetic systems contributing to the wild type eye colour of Drosophila. In 1931, Wright and his genetics class crossed a variety of eye colour mutants. They observed that white-eyed flies arose from a cross between brown and scarlet and between brown and vermilion. (Wright 1931). Based on these observations of synthetic white mutants he proposed that the wild type pigment was a compound of two independent genetic pathways. These, he also related to the two types of pigment granules reported by Johannsen. A similar observation led Glass to the same conclusion (Glass 1934), as did the breakdown of a white-eyed mutant into two component eye colours (Nolte 1943 and 1944). Mainz (1938) generated extensive combinations of eye colour mutants and examined their effects on the histology of the eye. His results generally supported the idea of two independent pigment pathways. Nolte, however, could not reproduce these results (Nolte 1950, 1 952a, 1 959a) and disputed this conclusion as well as some of the morphological observations. Although the pathways leading to the red and brown pigments may be biochemically, and genetically distinct, both pigments are associated with the 21 pigment granules of the pigment cells of the eye and thus might be expected to interact at least at a physiological level. This association may be manifest by altered pigment granule morphology in mutants with simple enzymatic lesions which should alter only one of the two pathways (Nolte 1950, 1952, Reaume, Knecht and Chovnick, 1991). The independence, or lack thereof, of the eye pigmentation pathways is still a subject of debate (Schwinck, 1975 1978, Ferré etal. 1983). Finally, the biosynthesis of eye pigments should be discussed in context of the complex inter-relation of the different tissues and developmental stages involved (reviewed by Tearle 1991). For example, production of the one ommochrome pigment, xanthommatin involves four organs and two developmental stages. During the larval stages tryptophan is absorbed by the fat body and malpighian tubules and the excess converted into 3hydroxykynurenine and kynurenine, respectively. During metamorphosis these organs release these stored compounds which, in addition to tryptophan derived from protein catabolism during metamorphosis, is taken up by the eye and occelli. The interplay between the various organs involved in pteridine biosynthesis and metabolism is likely equally complex, but is unfortunately largely unknown. Analysis of the biosynthesis of the pigments is further complicated by the fact that not all of the pigmented organs have all the necessary biosynthetic enzymes. For example, all four of the enzymes necessary for production of xanthommatin are found in the eye pigment cells whereas the occelli has only the last two and nether organ synthesizes xanthine dyhydrogenase, the product of the rosy locus, which does nevertheless affect the pigmentation of these organs. 22 Much of the impetus for investigation into these compounds stems from their role in nucleic-acid metabolism. For example tetrahydrobiopterin is an essential cofactor in amino-acid metabolism, neu rotransmitter biosynthesis and molybdoenyzymes in general (Bel and Ferré 1986 and McLean, Boswell and O’Donnell 1990). Since these compounds are derived from GTP, alterations in their metabolism is of relatively dire consequence in humans. In this regard Drosophila has been an immensely useful research tool. Not only have certain eye colour mutations (Henna and Punch) been proposed as models for metabolic defects such as phenylketonuria (probably more as a conceptual than physiologically accurate model) but the pigments provide an abundant, readily isolated source of an otherwise scarce and highly labile material. These have proved to be both difficult to isolate from mammals, as would be expected for a cofactor and metabolic regulator, and to synthesize chemically (Brown and Fan 1975). The central role of these compounds in cellular metabolism may provide the biological impetus for sequestering them in the specialize organelles, the pigment granules. This function also makes the complex tissue and developmental regulation less surprising. History of the garnet gene Eye colour genes of Drosophila hold an prominent place in the history of genetic analysis. The first mutant isolated in Drosophila was an allele of the white gene (Morgan in 1910 cited in Lindsley and Zimm 1992). The first mutant allele of garnet was recorded not long after by Bridges in his pioneering work on the chromosomal theory of inheritance (Bridges 1916). Since then it has featured in innumerable genetic studies. As a result of its long history a literature review of genetic studies involving the garnet gene is essentially 23 tantamount to a survey of the history of genetic analysis in Drosophila melanogaster. Consequently the following survey of genetic analysis involving the garnet eye color gene will necessarily be somewhat cursory. The first mutant allele of garnet was isolated on February 19, 1915 as a spontaneous mutation in a sable mutant stock (Bridges 1916). This mutation ) was established as an allele of a sex linked gene. As a sex linked marker it 1 (g was an invaluable tool in a number of studies. It was instrumental in showing that the behavior of genetic elements paralleled exactly the movements of chromosomes in both regular and irregular meiosis. Thus garnet played a pivotal role in the demonstration of the chromosome theory of inheritance. In the same study garnet was used to demonstrate that recombination occurs at the four strand stage, that non-homologous chromosomes (the X and Y) will pair regularly and, by defining the mechanism of non-disjunction, emphasized the regularities of meioses. Finally, interestingly in context of chapter 3, in this same study the garnet gene was used to demonstrate the general rule of equivalence of maternal and paternal genomes. The eye colour genes next resurfaced in the literature in context of the profound problem of how the actions of genes can determine the final phenotype of an organism. Investigators seized upon the eye colour mutations of Drosophila as an amenable model system in “higher” eukaryotes for the one-gene-one enzyme theory. It was initially expected that the relationship between the many genes which affected eye colour could be defined genetically whilst their products were studied biochemically. These studies were not as immediately fruitful as was hoped. Genetic studies revealed only that there were two largely independent pathways (Schultz 1935, Mainz 1938) and biochemical analysis 24 was hindered by the complex, colourless, highly labile and light sensitive nature of the many intermediate products of these biosynthetic pathways. In addition, the sheer number of genes affecting eye colour posed tactical as well as conceptual problems (Nolte 1952b, Lucchesi 1968). Spurred by Sturtevant’s pioneering work on the cellular autonomy of gene action in somatic mosaics (Sturtevant 1932), between 1935 and 1937 Ephrussi and Beadle produced a flurry of reports (Ephrussi and Beadle 1 935a, Ephrussi and Beadle 1 935b Ephrussi and Beadle 1 935c, Beadle and Ephrussi 1 935a, Beadle and Ephrussi 1935b, Beadle and Ephrussi 1936, Beadle and Ephrussi 1937, Ephrussi and Beadle 1 937a, Ephrussi and Beadle 1 937b, Beadle 1937) which focused on the relationship between gene action and development. They attempted to dissect genetic and biochemical interactions between eye colour genes using a series of tissue transplants between donors and hosts of different genotypes. Their results showed that most of the eye colour genes, including garnet, behaved autonomously upon transplantation. These studies, among others, were instrumental in nucleating concepts of diffusable versus cell autonomous substances, the former providing a working model for the action of hormones, tissue specific interactions (Beadle 1937), fluctuating levels of substances in development (Hamly and Ephrussi 1937), conservation of biological functions across species (Howlan, Glancy and Sonnenbilick 1937) and pleiotropy (Hadorn and Mitchell 1951, Hadorn 1962). Although important in emphasizing many concepts in development, a contemporary review of these studies stated “Because of the rather heavy injection of new theoretical considerations and laboratory symbolism, the uninitiated person is to be warned that he may find himself bewildered in trying to follow these investigators to their final conclusions or to comprehend what their conclusions 25 actually are.” (Blakeslee 1938), suggesting that this profusion of studies may not have been met with unabated enthusiasm. While these studies were vital to the development of our current understanding of genetics, the role of the gamet gene was somewhat incidental. It was simply a convenient marker for the X chromosome. The garnet gene also featured prominently in a debate between Chovnick and Hexter on the complexity of genetic loci. In contrast to the previous studies, an intrinsic physical property of the garnet gene, its relatively large size, was the basis of its role in the imbroglio. This debate centered around the question of whether or not garnet was a complex locus. The debate arose from the concept that genes were, by definition, indivisible by recombination. Genetic complexity was defined functionally; if recombination occurred between various alleles, defined as such by similar position, phenotype and failure to complement, the alleles were termed pseudo-alleles (a term coined by McClintock 1944, for another purpose entirely). A gene with pseudo-alleles was then termed a complex gene and was proposed by Lewis (1951) to consist of a series of duplicated genes. This point was raised as an issue in assessing the universality of genetic material. It had been established that alleles of prokaryotic genes could recombine (Benzer 1955). If this was also true of a “typical” eukaryote, Drosophila melanogaster, an important property of genetic material would be conserved between prokaryotes and eukaryotes. The core of the conflict revolved around the inability to recover double mutant chromosomes from intragenic recombination (Hexter 1956, 1 958a, 1 958b and 26 Chovnick 1956, 1957, 1 958a, 1 958b and 1961, reviewed rather more soberly by Carlson 1959). The situation was further complicated by gene conversion events, a phenomenon which at that time, was not documented in Drosophila melanogaster. Although impossible to determine exactly the source of this discrepancy, it might be posited that the double mutant chromosome arose from g5 allele was converted to g+, as suggested d a conversion event in which the 3 by Hexter (1958). The four other garnet alleles used in these studies were g ’ 1 ,g 2 g 3 and g . Data presented in chapter 2 (Table 21) may explain why it was 4 d 5 3 allele that might have converted. The g only the g 3 and g 4 alleles are insertions and thus would be expected to convert at a much lower frequency than point mutants (Chovnick 1964). The failure to recover recombinants or convertants between g 1 and g 3 is compatible with molecular data which indicate that these insertions are highly similar and possibly identical (Figure 20 and 21). In retrospect, both intragenic recombination, as established in prokaryotes (Benzer 1955) and conversion, shown in Drosophila virills (Demerec 1928) probably occurred at the garnet locus. Although these issues were finally resolved by his studies of another eye colour gene, the rosy gene (Chovnick 1964), the debate concerning the complexity of the garnet gene is notable for being the smallest pseudo allelic locus studied and for furnishing the first evidence of gene conversion in Drosophila melanogaster. The “transport” group of mutants: The garnet gene alters the levels of both the pteridine and ommochrome groups of pigments. The garnet gene is not alone in this property. A number of investigators have suggested that the eye colour mutants which alter both types of pigments are functionally related. Based primarily on this phenotypic 27 criterion, Nolte (1955) grouped the mutants carmine, carnation, claret, garnet, light, maroon, pink, prune, purple, purploid, rosy and ruby together as the arbitrarily named ruby group. On the basis of genetic and histological analysis of these genes he further subdivided the groups (and their proposed function) into the sub-groups shown in Table 2. The difficulty with grouping these mutants based on fine gradations of eye colour is that the groupings become a rather arbitrary reflection of the allele used. The eye colour of the garnet alleles, for example, ranges from a pale orange which Nolte would likely have classed as a member of the light group, to weak alleles that would be classed as members of the red or dark groups. The hazard of this approach is illustrated by results reported for interactions between white and carnation. These gave very different results depending on the alleles used. Inspection of the data presented in Nolte’s 1955 paper allows a slightly different grouping, which, while also dependent on phenotype, is based on an effect on one versus both pigment systems and histology (Table 2). This grouping should be less dependent on the choice of allele. Encouragingly, I show extensive genetic interactions within the first group identified by this criteria. The temptation to categorize eye colour mutations lured others. Schwinck (1975) again classed eye colour mutations phenotypically. Her criterion was based on relative concentrations of the drosopterin pigments. Her less colourfully named grouping, the group two mutations, is shown in the third row of Table 2. These mutations were unique in that they were the only mutants to respond to implants of phenylalanine by increasing drosopterin production. This might indicate a common metabolic defect. Tearle (1991) also proposed a functional grouping based on phenotype, this one based which organs were 28 Table 2. Genes proposed as members of the transport group of eye colour genes. The first and second column shows the name given to the group and the reference. The third column shows the names of the genes proposed as members of that particular group. The ruby group proposed by Nolte (1956) was latter subdivided in to smaller groups (indicated here by parenthesis) for which he proposed slightly different functions. The first group contains, rosy, pinkand purploid and is defined by normal pigment granule morphology and a brown eye colour. The second group contains claret and maroon, has normal pigment granule morphology and a light brown eye colour. The third group consists of carnation, carmine, garnet, ruby and light and is defined by normal pigment granule morphology and decreased levels of both red and brown pigments. The fourth group consists solely of purple and is defined by normal pigment granule morphology, less red pigment and elevated levels of brown pigment. The final group consists solely of prune and has abnormal pigment granule morphology. I have regrouped the genes carnation, carmine, claret, garnet, light, pink and possibly maroon and purploid as one subgroup defined as having normal pigment granule morphology and less of both classes of pigments. A second subgroup consists of rosy and possibly maroon and purploid with normal pigment granule morphology, less red pigments and normal levels of brown pigments. The last two groups are identical to Nolte’s last subgroups. 29 The group II mutants proposed by Schwink (1975) is defined by their increased drosopterin production in response to implants of phenylalanine. The transport group proposed by Sullivan and Sullivan (1975) consists of white, scarlet, claret and lightoid for which defects in keynurine transport were demonstrated, and carnation, garnet, light, maroon and pink for which transport defects were proposed. The group defined by their unusual response to transplantation of eye discs consists of carnation, carmine, claret, garnet, pink, ruby, maroon-like and rosy. The grouping proposed by Tearle (1991) is based on the organs affected by mutations in these genes. Those he listed as group 1A affect pigmentation of all organs examined. Finally the group defined as participating in synthetic lethal interactions rosy ry-SOD, 3 Hennar , includes: purple-Purploider, prune-Killer of prune, 6 light-carnation, deep orange-carnation, deep orange-rosy, deep orange purploid, deep orange-cinnabar-brown, deep orange-fused. Some of these combinations may reflect special situations. prune is lethal when combined with Killer of prune (Sturtevant 1956). The latter gene is a dominant allele of the awd (abnormal wing disc) gene and is a nucleotide diphosphate kinase. The prune gene product has been identified as a ras GTPase activating protein. The dominant Kpn allele may cause excessive stimulation of ras-like proteins (Teng, Engele and Venkatesh 1991). It would not be surprising if such an interaction 3 and iy 6 has also been touted as a synthetic was lethal. The combination Hnr 6 lethal combination (Taira 1960), however this interaction is specific to the ry allele; it does not occur with other rosy alleles (Goldberg, Schalet and Chovnick 1962). The gene fused disrupts pteridine metabolism but is not normally considered an eye colour gene. The synthetic lethality of deep-orange with the 30 cinnabar brown double mutant chromosome is specific to this chromosome and is not seen with any combination of double mutants. 31 32 Sullivan and Sullivan (1975) Beadle and Eph russi (1936) Schwink (1975) Green (1955) Reedy and Cavalier (1971) Bridges (1922, purple-Purploider cited in Bridges and Breme 1944) Lucchesi (1968) deep orange-carnation, deeporange-rosy, deeporange-fused deeporange-purploid, deep orange-cinnibar, brown r 3 Hennar 6 osy Tiara (1960), Goldberg, Schalet and Chovnick (1962) Sturtevant (1956) prune-Killer of prune Nash (1971) light-carnation Nickla, et al (1980) Hilliker et al. (1992) rosy-SOD Tearle (1991) claret, carmine, deep orange, garnet, light, lightoid, orange, pink, ruby, scarlet white The transport group Aberrant disc transplantation epistatic interactions Synthetic lethals Pigmentation of all organs Schwink (1975) Group II (carnation, garnet, light, maroon, pink) , ruby and garnet 3 whitea bright, brown, clot, lightoid, cardinal, claret, mahogany, rosy, scarlet,sepia carnation, carmine, claret, garnet, pink, ruby, maroon-like, rosy (white, scarlet, claret lightoid) carnation, garnet, maroon-like, orange, pink, rosy, ruby (carnation, carmine, claret, garnet, light, pink, ruby, maroon? purploid?) (rosy, maroon?, purploid?) (purple) (prune) this work The ruby group revisited (prune) Nolte (1956, 1959) (rosy, pink, purploid) (claret, maroon) (carnation, carmine, garnet, ruby, light) (purple) The ruby group Membership in the “transport” group of eye colour mutations. name reference members of group affected by mutations. His group 1A affects pigmentation of all organs examined. At approximately the same time, Sullivan and Sullivan (1975) documented defects in metabolite transport in white and scarlet mutants. They also discovered similar defects in claret and lightoid mutants. Although not examined in this study, they proposed that the genes carnation, garnet light, maroon and pink were, in addition to claret and light, involved in metabolite transport. Other suggestions of functional similarly between these genes have arisen from a number of studies. Beadle and Ephrussi (1936) noted that while most mutant imaginal eye disc transplants were cell autonomous, transplants between carmine, carnation, claret, garnet, pink and ruby and either vermilion or cinnabar (the latter non-cell autonomous due to diffusion of the “hormone”- the metabolic ommochrome intermediate keynurenine) produced exceptional results. Eye discs from vermilion mutants transplanted into hosts mutant for most eye colour genes, become wild type. Presumably, the mutant discs are able to scavenge enough kynurenine to generate wild type levels of pigment. Different results were seen with members of the “transport” group. Discs from vermilion donors transplanted into carnation and garnet hosts developed an intermediate phenotype, whereas when transplanted into carmine, claret, pink and ruby hosts the vermilion discs develop a mutant phenotype. Minor differences between mutant and intermediate phenotypes were likely due to the hypomorphic nature of the mutant used. For example the g 2 allele used in these experiments is a moderate hypomorphic allele. These results suggest that the transport group of mutations are physiologically distinct from other eye colour mutants. The authors implied that this difference could be due to differences in 33 levels of the “hormone”, although this is not consistent with their data. Schwink (1975) suggest that similar intermediate phenotypes seen in wild type eye discs implanted into maroon-like or rosy hosts is due to negative feedback of the biosynthetic pathways by excess products. However, the transport defect identified by Sullivan and Sullivan (1975) seems a more appealing cause for these aberrant results. Nevertheless, these results suggest a physiological equivalence between many of the members of the transport group. Finally, there have been a few other hints of functional similarity between various members of the transport group. Green (1955) found that some alleles of garnet and ruby were unique in showing no additive interactions in combination with certain white alleles. Certain members of these groups also show highly specific synthetic dominant (Nolte 1 952a) and synthetic lethal interaction (Luccessi 1968, Nash 1971, Nickla 1977). I shall use the term “transport group” genes to refer to members of this group as they show extensive phenotypic similarity and share a number of physiological properties, of which a role in transport is the best defined. A direct role in transport has, however, been shown for only one of these mutants, and this may not be their primary role. Thus this term is adopted more for its descriptive value than as a claim of function. Summary: The study of eye colour genes is now less fashionable. But, many of the problems noted by the early investigators still remain and the biological function 34 Table 3. Eye colour genes with known or proposed functions. The first two columns indicate the designation and name of those genes for which functions are known or proposed. A “+“ in the third column indicates that the gene has been cloned. The function proposed for the gene and the reference is given in the fourth and fifth columns, respectively. For those genes which encode an enzymatic function, the name of the enzyme is given in part B on the next page. 35 36 v w z St ly se rosy sepia scarlet vermillion white zeste brown claret carnation cardinal cinnamon clot cinnabar Drop garnet glass karmoisin killer of prune littleisoxanthopterin light lightoid low xanthine dehydrog. mal maroon like pink p pn prune pr purple Pu Punch pym polymorph ras rasberry ri rolled bw ca car cd cm cI Cn Dr g gl kar kpn lix It ltd lxd + + + + - + + + + + + + + + + - + + + + + + + + - + - - + + function transmembrane channel transport?/nervous systerm? transport?/nervous system? enzyme-ommochrome pathway molybdenum containing cofactor enzyme-pteridine pathway enzyme-ommochrome pathway cell-cell signaling transport?/nervous system? transcription factor enzyme-ommochrome pathway regulation of GTP hydroxylase enzyme-purine synthesis transport?/nervous system? transport?/nervous system? molybdenum containing enzyme cofactor molybdenum containing cofactor transport?/nervous system? regulation of GTP hydroxylase (Pu) enyme-ommochrome pathway enyme-pteridine pathway enzyme-purine pathway enzyme-pteridine synthesis cell-cell signaling-rhabdomere formation enzyme enzyme-pteridine pathway transmembrane channel enyme-ommochrome pathway transmembrane channel transcription factor A. Eye colour genes with known functions gene cloned Keith et al 1987 Wiederrecht and Brown, 1984 Tearle et al. 1989 Nissani 1 975/Searles and Voelker 1986 Dressen et al. 1988 Pirrotta 1988 Warner, Watts and Finnerty, 1986 Jones & Rowls 1988 Teng, Engele and Venkatesh, 1991 Searle & Voelker 1986 Mackay and O’Donnell, 1983 Henikoff et al. 1986 Nash-personal communication Zipursky et al 1993 Dreesen Johnson and Henikoff 1988 Yamamota et al. 1988 Sullivan and Sullivanl975 Sullivan, Grill and Kitos, 1974 Kamdor, Shelton and Finnerty, 1994 Wiederrecht, Paton and Brown 1984 Ghosh and Forrest 1967/Howells in Tearle 1991 Renfranz and Benzer, 1989 this thesis Moses, Ellis and Rubin, 1989 Sullivan, Grille and Kitos, 1974 Teng, Engele and Venkatesh, 1991 Ordono, Silva and Ferre, 1988 Sullivan and Sullivanl975/Devlin et al. 1989 McCarthy and Nicklal 980 Schott, Baldwin and Finnety, 1986 reference 37 v B. gen e ci cn kar kpn lix pr Pu pym ras ry se dihydroneopterin triphosphate pyrimidodizepine kynurenine 3-hydroxylase phenoxazenine synthetase nucleoside diphosphate synthetase dihydropterin oxidase sepiapterin synthase guanosine triphospate cyclohydrolase formyiglycine amide ribotide aminotransferase inosine monophosphate dehydrogenase xanthine dehydrogenease 6-pyrovoyltetrahydropterin 2-amino 4-oxo-b-acetyl-7,8 dihydro-3H ,9Hpyrim ido [4,5,6]-[1 ,4] diazepine syntetase tryptophane pyrrolase enzymes of most remain unknown. Those eye colour genes which have been analyzed in detail have provided insight into a diversity of biological functions (Table 3). Not only does this diversity suggest a potential resolution of the historical paradox of the apparently excessive number of eye colour genes, but it also suggests that the classical system of Drosophila eye pigmentation is a powerful assay for a diverse array of biological functions. The sensitivity of this system is such that it has the potential to allow the genetic detection even of those genes such as the transport group which may have extensive functional redundancy. The crucial, if still undefined, functions of these genes can only be revealed though detailed analysis of the genes involved. This thesis examines the genetic and molecular properties of one of the eye colour genes, the garnet gene. Chapter one deals with the complex genetic interactions between eye colour genes, specifically those of the transport group, which are likely involved in some aspect of inter or intracellular transport of metabolites. Membership of garnet gene in this group is indicated by extensive genetic interactions between the garnet gene and other members of this group. Molecular analysis of the garnet gene is presented in chapter two. In the final chapter the garnet gene is used as a genetic and molecular marker to examine the question of epigenetic gene regulation. Historically, the garnet gene has been an invaluable tool for those studying a variety of biological and genetic processes. Further analysis of the garnet gene and other members of the transport group should lead to insights into the ubiquitous and essential role of cellular communication in development. 38 MATERIALS AND METHODS All crosses were performed at 220 C unless otherwise stated. Culture medium was standard cornmeal/sucrose media supplemented with antibiotics and 0.04% tegosept as a mold inhibitor. Crosses were generally carried out in 8 dram shell vials with groups of 3-5 virgin females crossed to an equal number of males. For each experiment 3-6 replicates were made. Crosses were subcultured twice at 4-5 day intervals before the parents were discarded. Each set of crosses was scored independently. The data from replicate crosses within a group were subsequently pooled. Mutant strains and chromosomes: The mutations and rearranged variegating chromosomes used in this study are described in Lindsey and Zimm (1992). The g 62 and gS3 alleles were generously provided by Dr. A Schalet via Dr. D. Sinclair. The Dp(1;f)LJ9 mini-chromosome is carried balanced against an attached XX and attached xy chromosome. As the minichromosome carries a region which is diplo-lethal in males the attached XY stock carries the deficiency g-I on the X chromosome. The mini-chromosome was generously proved by Dr. G. Waring via Dr. D. Sinclair. Crosses: Details of the crosses will be described in the appropriate figure and table legends. Assays to quantitate eye pigment. Red pigments, five heads: 39 The amount of pigment deposited in the eye was measured separately for 25 females and 25 males. Flies three to seven days post-eclosion were decapitated by vigorously banging the frozen flies. Flies were held in the dark at 700C for 4 hours-six months before decapitation. No appreciable loss of pigment occurred during this time (data not shown). The heads were placed in wells of a microtitre plate and 30 uL of 0.25M 13-mercaptoethanol in 1% aqueous NH4OH per well was added. The eye pigment was released by sonication for three seconds and a five uL aliquot was removed from each well and applied to a piece of Whattman No. 3 filter paper. The amount of pigment in the dried spot was determined fluorometrically using a MPS-1 Zeiss microscope. The software program for processing the data from the photomultiplier attached to the microscope was written by Dr. J. Berger. Five groups with five heads per group were measured for each genotype and sex. In each case, the amount of pigment in each of the five spots was averaged and then expressed relative to wild type. Red pigments, single heads: The amount of pigment in single fly heads was quantified as above except as follows: only one head was used per microtitre well; 2OuL of solution was removed after sonication and spotted onto filter paper; a minimum of 10 individuals per genotype were measured. Red pigments, spectrophotometric assay: The spectrophotometric assay was adapted from the procedure of Real, Ferré and Mensua (1985). Five samples of five heads each, per genotype and sex, were placed in an eppendorf tube with 150 uL of 30% ethanol, acidified to pH 2 and shaken on an orbital shaker for 24 ± 4 hours. The absorbance of the 40 pigment was then read at 480 nm. Brown pigments, spectrophotometric assay: This assay was also adapted from the procedure of Real, Ferré and Mensua (1985) and Euphrussi and Harold 1944. Three samples of twenty heads per genotype and sex where placed in an eppendorf tube with 450 uL 2M HCI and 0.66% sodium metabisulfite (wlv) and sonicated. 0.9 mL of n-butanol (equilibrated with H20 and 0.66% sodium metabisulfite) was then added and the extract was shaken for 30 minutes on an orbital shaker. After centrifugation (5 mm. at full speed in a microcentrifuge) the aqueous layer was removed and 500 uL of H20 and 0.66% sodium metabisulfite was added. The mixture was again shaken for 30 minutes, centrifuged and the aqueous layer removed. This wash was repeated twice. The absorbance of the organic layer was measured at 492 nm. Quantification of separated red pigments by thin layer chromatography: The red pigments were separated and then quantified by a modification of the method described in Hadorn and Mitchell (1951) and Hadorn (1962). Red pigments were extracted as described for the five head red pigment assay above. An aliquot of 5 uL was spotted one inch from the bottom of a cellulose chromatography plate (Kodak) and allowed to dry. Further aliquots of the same sample were placed on the existing spot and allowed to dry, for a total of 6 x 5 uL of solution. The pigments were then separated in a solution of n-butanol and 1% ammonium hydroxide (2:1) for 3-4 hours. The pigments on the dried plates were identified by their distinctive colour under UV light, position and by comparison to the results of Hadorn and Mitchell (1951) and Hadorn (1962). The amount of pigment in each spot was quantitated by scanning the 41 chromatography plates with the fluorescent microscope (wavelength 500 nm). Using this procedure it was possible to identify 9 pigment spots. Assessment of malpighian tubule colour. Malpighian tubule colour was assessed essentially as described by Brehme and Demerec (1942). Malpighian tubules where dissected from healthy wandering third instar larvae in 0.7% NaCl. The isolated malpighian tubules were immediately viewed against a black background. Their colour was assessed in comparison with wild type, carnation, carmine and white mutants. Assessment of testes sheath colour. Testes were dissected in 0.7% NaCl from freshly killed males and immediately examined for colour when placed against a dark blue background. As with malpighian tubule colour, the colour of the testes sheath fades rapidly in 0.7% NaCI, tap water or in frozen whole flies. Molecular analyses: Genomic DNA was extracted by the method of Jowett (1986) with minor modification. Following RNAse digestion the DNA was isolated by spooling from the ethanol-aqueous interface and dissolved in TE. Restriction endonuclease digestion of the DNA was performed overnight, twice, with intervening phenol/chloroform extraction and ethanol precipitation. Isolation of RNA: RNA was isolated from the appropriate stages and genotypes using either the “hot phenol” method of Jowett (1986) or the guanidine 42 thiocyanate method (Fluka). The latter technique was more effective. Isolation of lambda clones: lambda clones and DNA were isolated according to standard procedures (Sambrook, Fritsch and Maniatis 1989). Isolation of plasmid DNA: Small scale plasmid DNA preparations were isolated by the alkaline lysis method (Sambrook, Fritsch and Maniatis 1989) or using “magic mini-preps” (P romega) following manufacturer’s instructions. Large scale plasmid preparations were prepared by PEG precipitation (Sambrook, Fritsch and Maniatis 1989) or using “Qiagen” columns following the manufacturer’s instructions. Southern and Northern transfers and hybridization: Southern and Northern transfers and hybridization were performed according to standard procedures (Sambrook, Fritsch and Maniatis 1989) and following manufacturer’s (Amersham) instructions. Labeling of DNA: DNA was radioactively labeled using 32 P and either the nick translation kit (Amersham)or the random priming kit (Amersham) following the manufacturer’s instructions. For some experiments DNA was labeled with UTP flourescein using the ECL (enhancered chemioluminescence) random prime kit (Amersham) and detected using anti-flourescein antibody conjugated to HRP which catalyzed a light-emitting reaction, following the manufacturer’s instructions. Sequencing: Short overlapping segments of DNA for sequence analysis were generated using the Exo Ill directed deletion kit following manufacturer’s 43 instructions. In some cases primers to previously determined sequences were prepared by the UBC oligosynthesis laboratory and used for sequencing. Sequencing was done using the double stranded dideoxy chain termination method using 35 labeled DNA. Initially the Sequenase (United States Biochemicals) kit was used. Subsequently 17 polymerase (Pharmacia) was substituted for the Sequenase enzyme and all reagents were made following recipes supplied with the T7 enzyme. The DNA fragments for sequence determination were separated on a 6% or 8% Acrylamide:bis acrylamide (40:1) gel or on a 6% “long ranger” (N-methly-acrylamide) (United States Biochemicals) gel in 0.5% TBE buffer. The genebank accession number for the garnet c-DNA is U31351. Sequence similarity search: The search of sequence data bases was carried out using the blastN and blastX algorithms provided by the national center of biotechnology information. This search included all eukaryotic sequences present in the Swiss protein, EMBL and genebank data banks. In situ hybridization: Hybridization to RNA in whole mount embryos and various tissues was done essentially as described by Tautz and Pfeifle (1989) with the exception that hybridization was carried out at 55°C. DIG labeled RNA probes were made following the manufacturer’s instructions (Boeringer Mannheim) and detected with alkaline phosphatase-conjugated anti-DIG antibody and NBT/X phosphate colour reaction. Photography: Flies and fly tissues were photographed using the Wild stereomicroscope adapter at 16 or 40 X magnification on a dark blue background. Kodak Eckatchrome Ti 60 colour slide film was used. 44 Chapter 1 Interactions between garnet and other eye colour genes 45 Introduction-chapter 1. Interactions between garnet and other eye colour genes: The large number of genes which affect eye colour suggests that eye pigmentation is a powerful assay system, able to detect alterations in a wide variety of biological functions (Table 3). Study of these genes, while having a long history is still in its early stages and further work on this group is certain to be rewarding. Of the eye colour genes, those of the transport group are particularly intriguing. Membership in this group has been assigned somewhat haphazardly, based on an arbitrary assessment of phenotype and some suggestive histological and physiological experiments. The garnet gene has been implicated as a member of the transport group but detailed analysis of either the gene itself or of interactions between garnet and other members of this group is lacking. The choice of garnet as a representative member of the transport group, while in part arbitrary, was promoted by the extensive previous work on this gene and the existence of a second site mutant which enhanced the mutant eye colour phenotype of severe garnet alleles. This interaction provides an attractive avenue for genetic analysis of the garnet gene. This chapter presents data on the nature of this second site mutation, the enhancer of garnet mutation (e(g)), its interaction with garnet and with other members of the transport group of eye colour mutants, and finally, interactions between this latter group and garnet. This analysis supports the inclusion of garnet as a member of the transport group of eye colour genes and suggests a simple model for the biological role of this class of genes. The molecular analysis presented in chapter two lays the ground work for testing of this model. 46 Results-Interactions between garnet and other eye colour genes. The enhancer of garnet mutation: Rather fortuitously, the P-element bearing strain (S6-1) that generated the garnet allele, gP, which permitted the cloning of the garnet gene, also contained a second site mutation which made the phenotypes of severe garnet alleles appear more extreme. This mutation was called enhancer of garnet (e(g)). The enhancer of garnet mutation has no independent phenotype. Individuals bearing only the enhancer of garnet mutation have no alteration in eye colour, viability or fertility (data not shown). However, in combination with severe alleles of the garnet gene the enhancer of garnet mutation reduces the amount of eye pigmentation by approximately half (Figure 5a, b and c and Table 16). This sex linked gene mapped, by recombination, near the site of a previously isolated enhancer of garnet mutation (Payne and Denny, 1921) and is presumably allelic to this mutation. Direct test by complementation is not possible as the original enhancer of garnet mutation is no longer extant. Other than its phenotype in conjunction with garnet, little information is available on this mutation. Cytological position of enhancer of garnet: As a first step in identifying the nature of the enhancer of garnet lesion, the cytological position of the enhancer of garnet mutation was determined. The enhancer of garnet mutation had been mapped by recombination to approximately map position 4 (Wennberg 1988). This position corresponds roughly to division 3 on the cytological map. Three deficiencies and one duplication encompassing most of division 3 were obtained from the stock center (Bowling Green) and tested for complementation with the enhancer of garnet mutation. None of the heterozygotes between these deficiencies and the double mutant e(g) g 2 chromosome showed an eye colour 47 Figure 5. Phenotypes of three severe garnet alleles and these garnet alleles in conjunction with the enhancer of garnet mutation. d, 5 3 g O e and g The garnet alleles g5 2 are shown on the left of each panel from top to bottom, respectively. On the right are shown these same alleles in conjunction with the second site enhancer of garnet mutation. 48 . e(g) g53d e(g) g5Oe e(g) g2 49 phenotype (data not shown), consistent with the absence of any detectable phenotype for enhancer of garnet homozygote. In contrast, when the g 2 allele was recombined onto these deficiencies and retested against a e(g) g 2 chromosome, all the deficiencies showed a strong enhancer of garnet phenotype. The deficiency/enhancer of garnet (Df e(g) g /e(g) g 2 ) phenotype is 2 more severe than the homozygote enhancer of garnet (e(g) g /e(g) g 2 ) 2 phenotype indicating that the original enhancer of garnet mutation is hypomorphic. The presence of a duplication for this region rescued the enhancer of garnet mutation on the e(g) g 2 chromosome reducing it to a g 2 phenotype (Figure 6 and Table 4). These results imply that the enhancer of garnet lesion lies within the area of overlap of these four rearrangements. The only area held in common by these deficiencies and duplication is region 3C3, although given the limits of cytological resolution the area of overlap might extend to bands on either side as 3C2 and 3C3 are often difficult to distinguish. These data effectively place enhancer of garnet in cytological position 3C2 - 3C4. The division 3 region has been extensively analyzed at the genetic and cytological level (Lindsley and Zimm 1992). No mutation corresponding to enhancer of garnet has been reported for this region but this is not unexpected as the mutation has only an indirect phenotype. The position of enhancer of garnet is, however, extremely close to that of the white gene which is located at position 3C2. Furthermore, there is complete congruence between removal of the white gene and the occurrence of the enhancer of garnet phenotype or rescue of these phenotypes (Figure 6B). Initial tests of allelism between enhancer of garnet and white indicated complementation; the phenotype of w g+/e(g) 2 g flies is completely wild type. Nevertheless the proximity of the white 50 Figure 6. Cytological localization of the enhancer of garnet mutation. A. A diagram of the first part of polytene chromosome division 3 is shown (adapted from Lindsley and Zimm 1992). Above is indicated the position of the zeste and white genes and the location determined for the enhancer of garnet lesion. Below is shown the cytological limits of three deficiencies; Df(1) DF(1)N8, Df(1) wrJl and Df(1) wrJ3, and one duplication; Dp(1;3)N 238 used to map the enhancer of garnet mutation. The black bars indicate deficiencies, the striped bar the duplication and the clear bars areas of uncertainty in the cytological determination of the extent of the deficiencies. B. The phenotype of the deficiency with respect to white and enhancer or garnet is summarized. A gene, a “-“ “+“ indicates presence of wild type function for that indicates absence of that gene function. See Table 4 for data and crosses. 51 postion Cytological A. of e(g) white 3C2 zeste e ) 3C2-3 s1E_ 19\T1Th • I I 4 I I . I — I ’ 1 r Iii Iii I I I I I b41 1 ‘4 •3D I 11 I Df(1)N8 1 I’, I I Df(1) 3C2-3 ‘3E3-4 I ii I I t 3A I I I I - ni Df(i)w Df(1) 3A1-2 - 3C2-3 B. Complementation of white and enhancer of garnet w Df(1)N8 Df(1) rJ1 rJ3 Df(1) w 38 Dp(1,3) e(g) - I -h I I I I I rJ3 I Df(i)w DfcI) 3C3-3C1 2 238 Dp(i;3)N Dp(1;3) 3B2-3;3D6-7;8ODF + + 52 0 Table 4. The Effect of 3C deficiencies on white and enhancer of garnet. The first column lists the three 3C deficiencies used to cytologically localize the e(g) lesion. The second and third column list the visual phenotype and red pigment values (as percent wild type) for the appropriate deficiency over a white null (w ). The fourlh and fifth columns list the phenotype and red pigment 1 values (as percent wild type) of the appropriate deficiency combined with the g 2 allele heterozygous with the e(g) g 2 chromosome. The last row lists the equivalent values derived using the e(g) g 2 chromosome for comparison. CROSSES: 1. To generate the deficiency garnet chromosomes, P Df(1)*/In(1)dI49 ®e(g)g /Yo’ 2 Fl Df(1)*/e(g) g 2 F2 Df(1)*g / 2 !n(1)d149, g4 selected by garnet phenotype. pair matings with e(g) g /YcJ sibs to isolate deficiency bearing 2 chromosome (no e(g) g cf progeny). 2 0 In(1)d149/Yo’(sibs) 2. To determine phenotypes: A. With respect to the white gene: Df(1)*®w/YQ P Fl Df(1)*/w4 phenotype determined. B. With respect to enhancer of garnet phenotype. P Df(1)* g /In(1)d149 2 Fl Df(1) g /e(g) g 2 2 phenotype determined. 1 /YcJ 2 0 e(g) g * 53 Effect of 3C deficiencies on white and e(g) Deficiency Df/white pigment Df(1)N8 0 Df(1)WrJ1 phenotype /e(a) g 2 Lg 2 pigment phenotype completely white 11 pale yellow 0 completely white ND yellow Df(1)WrJ3 0 completely white ND pale yellow e(g) g 2 88±9 wild type 54 21 + + 1 3 orange gene to the enhancer of garnet mutation led me to test for allelism in a less direct manner. enhancer of garnet is a subliminal allele of white. Two types of tests of allelism between white and enhancer of garnet were made. In the first series of tests, g O e and the 3 g5 mutations were recombined onto d the enhanceable g , 5 2 chromosomes bearing 11 different alleles of the white gene. The white alleles represent a variety of different types of lesions in the white gene (Figure 7). These recombinant chromosomes were then tested in trans with the appropriate e(g) g * mutation to see if the white alleles would complement the enhancer of garnet allele. Table 5 presents measurements of the levels of red eye pigments of these genotypes. In general, all the white alleles acted as extreme enhancers of the three different enhanceable garnet mutations. The one g O e allele. These individuals were exception was wsat in combination with the 5 g O e homozygotes. This visually, and by pigment assay, indistinguishable from 5 result was repeatable (data not shown). This suggests some allele specificity in the interactions between white and garnet, which is borne out by the lack of obvious correlation between the severity of the white allele and its effect on garnet (Figure 8 and see below). Specifically, although the Sat allele is a more severe allele of white than the e(g) allele (column 3 of Table 5) it has virtually no effect on garnet expression. Also interesting, is the result with wa. The wa allele is a moderate hypomorphic allele of white, yet it has a very strong enhancer of garnet effect on all three garnet alleles. This effect is more 1 and 1 w 1 18). pronounced than that of the white null alleles (w As a second type of test of allelism between white and enhancer of garnet the white+ transgene was tested for its ability to rescue the enhancer of garnet 55 Figure 7. Diagram of the structure of the white gene of Drosophila melanogaster. The heavy line represents the molecular map of the white gene (from Levis and Bingham 1985). The size, position and direction of transcription of this gene is shown above the line. The position and identity of inserts in the various white alleles used to complement the enhancer of garnet mutation are shown above and below the line (adapted from Lindsley and Zimm 1992). The molecular lesions associated with sat and w are unknown. The two dashed lines below the heavy line represent the white DNA included in the P-element constructs P[(w,ry)]A4-3 (from Hazelrigg, Levis and Ruben 1984) and CaSpeR (from Pirrota et al 1985). 56 Structure of the white gene w+ transcript _A_A __A__ 3’ 44 5’ h ch ____ 8 will () w RH -5 R K H E +10 w P[(w,ry)]A4-3 Sst ___WIJ( CaSpeR R=EcoRi H = Hind III K = Kpn I S = Sail 57 Table 5. Complementation between e(g) and different white alleles. The left half of the table shows the 11 alleles of white tested for complementation against e(g), their visible phenotype and total red pigment levels (expressed as a percent of wild type pigment levels). The right side of the table gives pigment levels for the appropriate combinations of the genotype w g*/e(g) g*, where * indicates the given allele. The last two rows list the results obtained with the e(g) allele and wild type allele for white for comparison. As an incidental note, these results show that when the amount of pigment in the w g+ / e(g) g 2 strain is assayed the conventionally recessive white alleles are slightly dominant. In contrast the garnet mutations are truly recessive. These data agree with previously reported pigment levels of the white and garnet genes. Nolte (1 959c) in a theoretical treatment of the significance of dominance, found w/w had slightly less pigment than wild type (w+/wj. In the same study, Nolte found garnet to be “truly” recessive. This has caused some difficulties in the interpretation and significance of dominance of alleles but these problems pose conceptual rather than functional problems. CROSS: 1. To generate recombinant w g* chromosomes: g*/g*® w*/YQ Fl F2 + g*/ w’ + 1 ® g*/y’f (sibs) w*g*/Y0 58 (select by phenotype of test cross to g*/g* and w*/w* , balanced to make stock) 2. To perform the complementation cross: P e(g)g*/e(g)g* ® w*g*/Yo Fl e(g) g*/w* g* assayed for pigment levels. Note: This complementation test was performed three times, once by Barney Lee as part of his directed studies course. The values of the individual tests have been averaged as they did not differ greatly. 59 Complementation between e(g) and different white alleles white allele 2 g w*/w* 5Oe w* g*/e(a) 53d g* 1118 w <1 completely white 11±1 1 w <1 completely white 13±1 wa 3± .5 dull orange 5±1 wbf 2±.5 faint yellow 10±1 7±1 3±1 9±1 3±1 3±1 wBXW.7±.1 dull red 6±1 2±1 ND ND 5±1 2±1 wch >5 ± .1 pink-orange 12±2 8±1 ND w’ ND 12±1 8±1 4±1 we 4±.5 pinky 11±1 7±1 ND 1 w .6±.1 faint pink 9±1 3±1 ND 10±1 22±1 4±1 6±1 8±1 ND e(g)g* 32±3 14±1 11±1 w÷g* 38±2 26±1 13±2 sat 7± 1 w dull red browny orange .6±.1 faint pink 60 effect on garnet. Two constructs were chosen to provide the white+ transgene. The first transgene construct contained the full white + gene coding region and approximately 3kb 5’ and 3’ white regulatory sequence (along with rosy+) inserted within a P-element located at position 1 OOF on 3R (Hazelrigg, Levis and Ruben 1984). This particular construct shows an unusual pattern of white + gene expression. The anterior portion of the eye is normally pigmented (red) while a crescent at the posterior margin, approximately one quarter of the eye, remains unpigmented. This construct showed partial rescue of the enhancer of garnet effect (Table 6). The pattern of rescue was similar to the pattern of white+ expression. The posterior portion of the eye in which white+ was not expressed appeared more lightly coloured (enhanced) than the anterior. The second white+ construct used was the CaSpeR P-element transformation vector. This construct contains all the coding region of the white gene but has a deletion of most of the large first intron and all but 300 bp of the 5’ regulatory region and 630 bp of the 3’ region (Pirrotta, Stellar and Buzzetti 1985). As the white gene in this construct lacks most of its 5’ regulatory sequences, the expression of white tends to be weak and highly dependent on the site of insertion of the construct. Initial tests to determine if this construct would rescue the enhancer of garnet phenotype were inconclusive (Table 6-top line). The original insert had marginally more pigment than its e(g) g ; TM3 siblings (26 ± 2 2 vs 23 ± 3) but the difference was not significant. This failure to compensate for the enhancer of garnet effect might have been due to the weak expression of the construct. In order to determine if this construct was able to rescue the enhancer of garnet effect when more strongly expressed, the element was mobilized and over 30 lines that showed strong expression of the white+ gene were generated. Results of this experiment, for eight different insert lines, are shown in Table 6. In all eight lines the white construct was capable of 61 Table 6. Rescue of the enhancer of garnet effect by white+ transgenes. The first column gives the designation of the individual white transgene inserts. The first nine are different inserts of the CaSPeR insert, the first of which, 4-1, is the original insert. These inserts also contain the cdc-2 homolog of Drosophila melanogaster. The 4-3 transgene contains the complete white gene and most of its 5’ and 3’ regulatory sequences. This insert was generously provided by Bob Levis. The last two lines provide control values for e(g) g2 and g2 males. The e(g) g2 control is an internal control derived from the average values of the e(g) g2/Y; ÷/TM3 siblings of the experimental crosses. The second column indicates the strength of the white transgene expression by pigment assay (as percent wild type) and the last column indicates the ability to rescue the enhancer of garnet effect in a e(g) g 2 background also determined by pigment assay. CROSSES: 1. To generate the different inserts of the CaSpeR 4-1 transgene; P 67 casper 4-1/casper 4-1 w ; 67 /w Fl (transposase source) /w +/÷ w ; 67 casper/delta 2-3 c? 0 1 w /Y; F2 .1. 0 w-/Y delta 2-3/TM3’ ‘I, 67 casper/+ w ; 1 /w scored for strong white+ expression and balanced as homozygotes stocks or over FM7. 62 To determine the chromosome on which the transgene was inserted; P /Y; casper insertJTM3 or w’/Y; casper insert/-i-; -i-/TM3cJ ® 1 w /wm CyO/Tft wm ; 4 Segregation of the white insert relative to the dominant markers indicates the chromosome into which the transgene is inserted. Of the 27 independent inserts all were located on the third chromosome. No further effort was made to map them. 2. To assess the strength of the white transgene expression; P /Y; insert/TM3cf 0 1 1 w /w +/÷ w ; “ / 1 w w’orY;insert/+ Fl progeny assayed for pigment levels. 3. To assess the ability of the white+ transgene insert to rescue the enhancer of garnet effect: P e(g) g / e(g) g 2 ; +/+ 2 F1 /Y; insert ‘ and e(g) g 2 /Y; TM3/+ 2 e(g) g progeny assayed for pigment levels. ‘I /Y; insertJTM3 ‘ 1 0 w a’ Generation of the different casper transgene inserts and determination of the chromosome into which the transgene had inserted was done by Gwendoyln Mahon as part of an undergraduate directed studies project. 63 Rescue of enhancer of garnet phenotype by white+ transgenes w+ insert effect on white effect on e(g) w-/Y:insert/÷ e(g) a2/Y: insert/+ 4-1 21±2 26±2 1-16 32±6 97±5 1-18 68±6 32±4 1-22 91±4 39±5 1-23 96±3 34±3 2-6 49±2 37±2 2-23 82±6 43±5 3-1 22±2 31±5 3-29 23±3 36±3 4-3 27±2 e(g) g2IY; TM3/+ g2/Y 23±3 30±2 64 rescuing the enhancer of garnet effect and restoring the full garnet phenotype. While all of these new insertions expressed the white + gene more strongly than the original insert, there was again no clear correlation between the strength of white gene expression and rescue of the enhancer of garnet phenotype (Figure 8 and see below). The final note of interest arising from this latter set of experiments is that the interaction between garnet and enhancer of garnet appears to involve the coding region of the white gene. The white gene in the CaSpeR construct possesses little regulatory white+ sequences so it clearly cannot be providing additional white regulatory regions. Yet it is able to rescue the enhancer of garnet phenotype. This suggests that interaction between garnet and enhancer of garnet is restricted to the coding region and is possibly a post-transcriptional process. Examination of white transcription in a garnet mutant background might resolve this question. In summary, the enhancer of garnet mutation cytologically maps to the same position as the white gene. Eleven white alleles, when combined with the enhanceable garnet alleles, show a severe enhancer of garnet phenotype, in conjunction with the enhancer of garnet and garnet mutations. The enhancer of garnet phenotype can be rescued by a white+ transgene containing white+ coding region. Thus the enhancer of garnet mutation appears to be a cryptic allele of the white gene. The nature and dose sensitivity of interaction between enhancer of garnet and garnet: There are a number of systems of genes and specific modifier genes which have been described in Drosophila. Many of these consist of 65 transposable- or retro-element-induced mutations in one gene, the activity of which is modified by a mutation in another gene. There are five such modifier genes; Su(Hw) which modifies mutations produced by insertion of the gypsy element (Modelell, Bender and Meselson 1983), su(f) which also modifies gypsy-generated mutations (Parkhurst and Corces 1985), su(wa) which modifies copia expression (Bingham and Judd 1981), su(pr) and su(s) which modify mutations produced by the 412 element (Searles and Voelker 1986). Initially it seemed possible that the interaction between garnet and enhancer of garnet was of this nature. The first evidence that this was not the case came g O e and g53d are from the observation that the three enhanceable alleles, g , 5 2 also the most extreme alleles (Table 16). In addition, none of these garnet alleles was associated with insertion of any foreign DNA (Figure 21). In order to determine if sensitivity to the enhancer of garnet mutation was dependent on some specific property of these three alleles (other than insertion of a transposable element) or due merely to the dose of functional garnet gene product, the enhancer of garnet mutation was recombined onto chromosomes carrying four of the weaker garnet alleles (g ,g 1 , g 3 4 and gP). Dosage of the garnet gene product was then manipulated by making these chromosomes heterozygous with a deficiency for the garnet gene. Table 7 shows that although these alleles are not discernibly sensitive to the enhancer of garnet mutation as homozygotes, when the dose of the garnet gene is further reduced by a deficiency for the garnet gene, these alleles become dominantly sensitive to the enhancer of garnet mutation. Dominant sensitivity to enhancer of garnet was also observed when garnet gene dosage was reduced with the severe g53d allele instead of a deficiency (Table 7-legend). Thus the enhancer of garnet effect is highly sensitive to the dosage of the garnet gene and does not appear to be allele specific. As the weak garnet alleles used are hypomorphs 66 Table 7. Sensitivity of the enhancer of garnet effect to garnet dosage The effect of the enhancer of garnet mutation on three strong garnet alleles, g ’ 2 Oe and g53d, and two weaker garnet alleles, g 5 g 1 and gP, was determined by pigment assay. The first column lists the garnet allele. The next two columns report the eye pigment values for the garnet allele over a deficiency for that region and the same genotype heterozygous for enhancer of garnet, respectively. The last two columns list the eye pigment values of the appropriate garnet and enhancer of garnet and garnet homozygotes for comparison. The enhancer of garnet effect was also sensitive to garnet dosage when the garnet gene dosage was reduced using the extreme g53d allele. All values are expressed as percent wild type pteridine pigments. d/÷ gP genotype has 24±4 % WT pteridine pigment levels. 3 The e(g) g5 53 gP genotype has 13 ±2 % WT pteridine pigment levels. g The e(g) d/e(g) In comparison with 50 ± 5 for the gP homozygote. CROSS: To generate the deficiency genotypes, P Df(1)HA97/FM7 ® g/Yor e(g) g/Yo’ Fl Df(1)HA97/g or e(g) g ‘I, g*/g* c( and progeny assayed e(g) g*/ e(g) g* homozygotes are taken from stocks. 67 Sensitivity of garnet alleles to the enhancer of garnet effect and garnet dosage garnet allele g/Df(g) e(g) g/+ Df(g) e(g) g/e(g) g 1 g 40±3 19±3 30±4 57±2 2 g 40±3 25±3 32±3 37±2 Oe 5 g 28±2 24±2 14±1 26± 1 53 g d 16±3 9+2 11±1 13+1 qP 38±4 23±2 56±3 50±5 68 (Table 20), the sensitivity is presumably related to the amount of functioning garnet gene product. To further examine the sensitivity of the enhancer of garnet and garnet interaction to the amount of gene product, the expression of the weaker garnet alleles was tested in genotypes where the dose of the enhancer of garnet mutation was reduced using a deficiency for the enhancer of garnet region. Table 8 shows that only a single dose of mutant enhancer of garnet (DI e(g) g / e(g) g), enhances the phenotype of weak garnet alleles. In contrast to the previous results, the interaction is not dominant, the deficiency for the enhancer of garnet region does not enhance the garnet phenotype in the presence of a wild type allele of enhancer of garnet. Thus interaction between garnet and enhancer of garnet is very sensitive to the dose of the garnet gene but rather less sensitive to the dose of enhancer of garnet. The complementation tests performed between different white alleles and the enhancer of garnet mutation provide a reasonable set of data to quantify the dose dependence between garnet and enhancer of garnet. Figure 8 shows a comparison between the strength of different white alleles and their effect on garnet. Two points are immediately evident. There is no correlation between the severity of the white allele and its enhancing effect on garnet. There is however a good correlation between the strength of the enhancing effect and the severity of the garnet allele. Although this pattern might appear fortuitous, since only three garnet alleles were tested as opposed to the eleven white alleles, the results generally support the findings described above. The interaction between 69 Table 8. Effect of we(g) dosage on the enhancer of garnet effect. The effect of reducing the dosage of the enhancer of garnet locus with a deficiency for that region, on different alleles of garnet is shown. The first column gives the garnet allele examined. The g 2 allele is a strong, “enhancable” allele. The other three, g ’g 1 3 and gP are weaker alleles not normally responsive to the enhancer of garnet mutation (as double homozygotes). The next two columns give pigment values (as percent wild type pigment) of these garnet alleles (shown as g*) in conjunction with a deficiency, heterozygous with either a wild type e(g) allele and g* or the e(g) mutation and g*, respectively. The last two columns show values for garnet and enhancer of garnet homozygotes for comparison. CROSSES: 1. To generate Df(1)N8 garnet strains; P Df(1)N8, w-, N-/In(1)d149, Hw g4 1. 0 g/YcJ Fl Df(1)N8+/+ g F2 Df(1)N8 g/In(1)d149 recombinants selected by garnet phenotype and notched wing. 1 0 In(1)dI49/Y’ (sibs) 2. To generate e(g) g stocks; P /ye(g)cvg ye(g)cvg 2 ‘I, Fl ®yacscpn wvg 1 f/YQ’ or ecctvg YcJ 3 ycvvgPf/Y or /y cv v g 2 ®y e(g) cv g y e(g) cv g 1 cf 2 g* recombinants selected by appropriate markers. y e(g) 3. To generate the Df(1)N8 g/ + g or e(g) g individuals, P Df(1)N8 g*/In(1)d149 Fl Df(1)N8 g*/e(g) g* or “ 0 g/Yor e(g) 70 g*ty Effect of e(g) dosage on the enhancer of garnet effect Dfe(g)g* + g* Dfe(ci)g* g* e(g) 28±5 g* e(cx)g* e(g)g* 7±1 37±2 32+3 13±2 5±1 57±2 ND 3 g 24±2 14±2 45±1 ND gP 16±1 8±1 50±5 ND 2 g 71 Figure 8. Comparison between the severity of different white alleles and their effect on garnet. A. The effect of different white alleles (combined with three different garnet g O e and g alleles, g53d 5 (e.g. w*g*/e(g) g’ as heterozygotes with the 2 enhancer of garnet mutation and the same garnet allele (relevant crosses and genotypes given the legend to Table 5). The percent wild type pigment of the different white alleles are shown on the X axis. The percent wild type pigment of the white garnet / e(g) garnet heterozygote are shown for the three garnet alleles, on the Y axis. B. The effect of different white+ transgenes in rescuing the enhancer of garnet effect. The crosses and relevant genotypes given in the legend to Table 6. The /Y; [white+]/+. The percent wild type pigment of 2 generic genotype is y e(g) cv g the white+ transgene is shown on the X axis. The pigment level of the “rescued” genotype is shown on the Y axis. 72 Comparison between the strength of different white alleles and their complementation of enhancer of garnet. J- — - *g2 g50e 20- — 15- +g53d — 10- — — r I I 5.; I__ — — — — — — I — I n_ — 0 — 23456789 10 pigment of white allele Comparison between strength of white transgene and rescue of enhancer of garnet effect. ‘+z,- lb 40-————— C + — I I C.) •1 -C 30-— 25---20 0 - - - - + + 102030405060708090100 pigment of white transgene 73 enhancer of garnet and garnet is dramatically sensitive to the dose of the garnet gene but the effect of the white gene is somewhat allele specific. Interactions between enhancer of garnet and other eye colour genes: The genetic interaction between enhancer of garnet and garnet points to some previously undetected interaction between their gene products. The white gene product has been proposed, based on both molecular and genetic studies, to function as transmembrane channels in conjunction with the products of the brown and scarlet genes (Dressden, Johnson and Henikoff 1988). Thus the white gene product appears to interact with at least two other eye colour gene products. As the enhancer of garnet allele of white was an invaluable tool in examining interactions between white and garnet, I decided to test other eye colour genes for their sensitivity to the enhancer of garnet allele. The enhancer of garnet mutation was combined with 26 other eye colour genes. Since it was practical to test only one allele of each gene, and it is generally not known which, if any, alleles of the different eye colour genes are amorphic, there is a possibility that some interactions would not be observed. In order to increase the sensitivity of this test, the enhancer of garnet mutation was placed in trans to a deficiency for enhancer of garnet region. Table 9 shows the results of this survey. Of the 26 eye colour genes examined, the mutant phenotype of nine, possibly ten, is enhanced by the enhancer of garnet mutation. These genes share with garnet the property of altering levels of both brown and red pigments. Significantly, brown and scarlet fail to show an interaction with the enhancer of garnet mutation. Thus the interaction between garnet and enhancer of garnet appears to define a different type of interaction than the structural role proposed for brown and scarlet. Interestingly, raspberry 74 Table 9. The effect of the enhancer of garnet mutation on other eye colour mutants. The e(g) mutation was tested in combination with 25 eye colour genes. The first two columns list the symbol and name of the eye colour gene respectively. The next two columns list the amount of total pteridine pigments measured for the given eye colour mutant as a homozygote and for the same homozygous mutation in combination with the a hemizygous e(g) mutation, respectively. All values are expressed as percent wild type pteridine pigment. The generic genotypes are mutant/mutant vs. e(g) mutant! Df e(g) mutant or e(g)/Df e(g); mutant/mutant respectively. The values for the homozygous mutant genotype were derived from internal controls for mutations on the second and third chromosome and external controls (mutant/mutant flies grown concurrently with the test crosses) for those on the first chromosome. The next column gives the percentage difference of the two measurements as hemizygouse(g) with mutant I mutant alone. The last column indicates whether or not this difference constitutes responsiveness to the enhancer of garnet mutation. The e(g) mutant phenotypes are heterozygous for the DF(1)N8 deficiency. This deficiency has two effects on pigment levels unrelated to hemizygosity for the white locus. Flies bearing the N8 deficiency are generally smaller and have a transient, slightly brown eye phenotype shortly after eclosion. These effects have been corrected for using DF(1)N8/ e(g); mutant/Balancer siblings where possible or an arbitrary cut off point of 75% of the control, mutant alone, value. In general there was little ambiguity. 75 CROSSES: 1. For X-linked eye colour genes: A. P Df(1)N8/In(1)d149 Fl Df(1)N8 +/ + mutant F2 Df(1)N8 mutant?/In(1)d149 0 mutant/Yo’ (test cross individual virgin females to select double mutants) 1 ® mutant/Ycy ® In(1)d149/Yo’ (sibs) ‘I B. P /y e(g) cv g 2 y e(g) cv g 2 Fl ye(g)cvg2÷/÷÷+÷mutant®ye(g)cvg2/Yf(sibs) F2 y e(g)? cv+ g+ mutant/Ye’ selected. Stocks were made by crossing to In(1)d149 balancer and the presence of garnet and the eye colour mutant were confirmed by the appropriate test cross. e(g), 0 mutant/Yo’ ‘I, The Df e(g) mutant chromosomes and the first two generations of the e(g) mutant crosses were made by Mitra Maharaj. C. The experimental cross: P Df(1)N8 mutant/In(1)d149 Fl Df e(g) mutant’ e(g) mutant test genotype. “ 0 y e(g) mutant/Yo’ 2. For second chromosome mutants: A. P Df(1)N8/In(1)d149; GIa/ + 0 +/Y; mutant/mutant B. P ; Tft/CyO 2 y e(g) cv g /y e(g) cv g 2 a’ 0 +/Y, mutant/mutanto’ 76 C. /Y; mutant/CyOcf 2 ®y e(g) cv g Fl Df(1)N8/÷; Gla/mutant F2 ; mutant/mutant 2 Df(1)N8/y e(g) cv g ; mutant/CyO 2 Df(1)N8/y e(g) cv g ; mutant/mutant 2 + /y e(g) cv g ; mutant/CyQ 2 +/y e(g) cv g ‘I, (experimental genotype) (NB effect control) (mutant control) (wild type control) 3. For third chromosomes mutants: A. P B. P C. Fl ® +/Y mutant/mutanto’ Df(1)N8/In(1)d149; H/TM3 ; H/TM3 2 /y e(g) cv g 2 y e(g) cv g Df(1)N8/+; TM3/mutant 0 ® ÷/Y mutant/mutantc? /Y; mutant/TM3cI 2 y e(g) cv g ; mutant/mutant 2 Df(1)N8/y e(g) cv g ; mutantiTM3 2 Df(1)N8/y e(g) cv g ; mutant/mutant 2 + /y e(g) cv g ; mutant/TM3 2 +/y e(g) cv g 77 (experimental genotype) (NB effect control) (mutant control) (wild type control) The effect of enhancer of garnet on other eye colour mutants gene bordeaux bo brown bw claret ca carnation car cardinal Cd carmine cm cinnabar cn dke-c dark eye dor deep orange light It lightoid ltd ma! maroon-like Moire Me mel melanized Mot-K Mottled of K mw mottler of w pink p prune pn purple pr raspberry ras ruby rb ry rosy sepia se scarlet St vermilion v white wo occelli phenotype of e(g) mutant mutant 74±5 1 .1±.2 36±4 39±4 97±4 59±10 103±3 74±4 18±2 37± 1 90± 1 50±5 80±3 50±5 100 ±2 1.3± .3 32±2 49±3 37±3 29±2 38±2 60±2 111 ±3 76±5 106 ±4 92 ±7 108±2 1.5± .4 24±4 28±2 113 ±5 10±1 96±2 91 ± 16 5± 1 9± 1 17± 1 105±2 85±6 105±2 79 ±7 1.6 ±.5 14±3 49±2 6±1 41±2 7± 1 38±3 132±2 71±4 103 ±3 99±4 78 difference effect 144% 104% 66% 72 106 13 93 123 29 25 19 208 106 208 79 127 44 100 16 142 19 63 119 93 97 106 NO NO YES YES? NO YES NO NO YES YES YES NO NO NO NO NO YES NO YES NO YES YES NO NO NO NO and maroon-like, lesions in the pteridine pathway, appear suppressed by the enhancer of garnet mutation. The significance of this suppression is not clear. Thus, enhancement of mutant eye phenotype by the enhancer of garnet mutation appears to be a property shared by many eye colour genes in the “transport” class. Interactions between garnet and other eye colour genes: As the garnet gene is not alone in its interaction with the enhancer of garnet allele, and as most of these belong to the transport group of eye colour mutants, it was of immediate interest to test the garnet gene for interactions with other eye colour genes. Nolte (1 952b) found that while alleles of the genes garnet, carnation, carmine and ruby are all normally recessive, in pairwise combinations they show synthetic dominance. I extended these analysis to include 14 other eye colour genes (Table 10). Interactions between pairwise combinations of garnet and the other eye colour genes were assessed by looking for synthetic dominance of garnet (garnet/÷, mutant/mutant< +/+; mutant/mutant), synthetic dominance of the other gene (garnet/garnet or/Y; mutant/-i- <garnet/garnet or garnet/Y; +1+), synergistic or additive, versus epistatic effects on eye pigmentation of the double mutants, and effects on viability and fertility of double homozygotes. Of those mutations which interacted with enhancer of garnet (claret, carmine, deep orange, light, lightoid, pink, purple, ruby, rosy and probably carnation) all but purple also interacted with garnet. In no case was an interaction with garnet observed with a gene that failed to interact with enhancer of garnet. Taken together with the data of Nolte (1 952b) there is considerable congruence between those genes which interact with enhancer of garnet and those which interact with garnet (Table 11). Finally it is interesting to note that, while the partial sterility seen between garnet and other genes probably reflects a 79 Table 10. Interactions between garnet and other eye colour genes. Interactions between garnet and other eye colour genes was monitored in three ways: 1. Epistatic versus additive or synergistic interactions (as monitored both visually and by pigment assay). 2. Synthetic dominance. 3. Synthetic lethality or sterility of the double homozygotes. The first two columns indicate the symbol and name of the eye colour gene tested with in combination with garnet. The next four columns show pteridine pigment values (expressed as percent wild type levels) for; females and males homozygous or hemizygous for the other eye colour mutation and female and male double mutant individuals. To determine if either garnet or the other eye colour mutant acted in a dominant fashion in a mutant background of the other gene, the following genotypes were generated; ÷/g; mutant/mutant to determine if garnet acted in a dominant manner in a mutant background and g/g or g/Y; mutant/÷ to determine if the mutant became dominant in a garnet background. The next three columns show pteridine pigment values (expressed as percent wild type levels) for; individuals homozygous for the other eye colour mutation and heterozygous for garnet, and individuals homozygous or hemizygous for garnet and heterozygous for the other eye colour mutant. The next two columns indicate the fertility of the double homozygotes and notes on their phenotypes. The last column is a summary of the various models of interaction. A “+“ indicates interaction, either of epistatic/synergistic, pigment interactions, synthetic dominance in either direction, synthetic sterility or extensive 80 secondary phenotypes generally indicative of cell death. One “+“ is given for each type of interaction observed. CROSSES: 1. To generate double homozygotes: For eye colour mutants on the X chromosome; P g mutant/In(1)d149 0 g mutant/YJ 2 (double mutant stocks generated in cross 1 B described in legend to Table 8. For second and third chromosome mutants; 53 za g53d; E(var) 18. l2Sp/CyO g y za d/y or E(var)303 ru h eg pP K1ffM3 0 In(1)IN(1)BM1/Y; mutant/mutantcf 53 mutant/CyO or TM3 g Fl In(l)!N(1)BM1/y za d,. y za g53d/y,. mutant/CyO or TM3’ P 2. Genotypes used to examine synthetic dominance; For X-linked eye colour genes, p P g53d/g53d 0 2 g mutant/Yo’ and mutant/mutant 0 2 g mutant/Yo’ For second and third chromosome eye colour mutants the genotype g53d/g53d d; mutant/Balancer were 53 or g53diy; mutant/Balancer and In(1)!N(1)BM1/g generated as siblings of the crosses described above to make double homozygotes. Cross 2 and 3 described in the legend of Table 9 generate equivalent phenotypes with the g 2 allele and second and third chromosome mutations. In all cases the conclusions derived from these crosses were the g5 allele. d same as with the 3 81 82 Interactions between garnet and other eye colour genes gene pigment synthetic single mutant double mutant dominance female male female male ÷7q;m/m j/q;+/m bw brown 1 + .2 0 1±.1 0 2±1 5±.5 ca claret 36±2 43±2 9±1 6±1 35±3 8±1 cd cardinal 101±4 ND ND 5±1 101±3 95±3 cn cinnabar 103 ± 3 ND 108±2 ND ND 106±1 It light 42±5 2±1 1±.5 20±1 7±1 37± 1 ltd lightoid 0 78±10 68±6 0 50±2 8±1 Me Moire ND ND ND 82±3 80±3 79±5 MotK Mottled 100±2 101±3 ND ND ND 95±1 pink p ND ND 34±2 5±1 25±2 45±3 pr purple ND 30± 10 40±10 4±1 7±1 37±4 iy rosy ND 63±3 6±1 64±10 ND 54±3 se sepia ND ND 111±3 ND 112±5 113±4 St scarlet 87± 10 81±10 10±2 7±2 69±5 13±2 wo white ND ND ND ND 92±7 94±4 occelli q/Y÷/m 3±.5 6±1 ND ND 7±1 7±1 ND ND ND ND 16±2 ND 16±2 ND f&m fertile male semi-sterile male fert. f. ND ND f&m fertile male semi-sterile ND ND male semi-sterile f&m fertile f. sterile ND f&m fertile ND fertility +++ ++ ++ + -9 SUMMARY Table 11. Summary of interactions between other eye colour mutations and both garnet and enhancer of garnet. The first two columns give the symbol and the name of the eye colour gene. The third list the pigment pathway effected or the general nature of the defect (from Table 1). The final two columns list whether there was an interaction with either enhancer of garnet or garnet respectively. A gene was considered to interact with garnet if it showed any one or more of epistatic/synergistic interactions, synthetic dominance, sterility or unusual phenotypes of double hete rozygotes. 83 Summary of eye colour genes interacting with e(g) and garnet. mutant pigment interaction interaction affected with e(g) with garnet claret both ca YES YES carnation both car YES ND cd cardinal both NO NO carmine both cm YES NO dor deep orange both YES ND garnet both YES NA g light both It YES YES ltd lightoid both YES YES pink both YES YES p pd purpleoid both ND NO? rb ruby both? YES ND rosy both? YES YES iy YES* w white both NA cn cinnabar ommochrome NO NO scarlet ommochrome NO NO? St vermilion ommochrome NO? v ND bordeaux ND bo pteridines? NO NO bw brown pteridines NO pn prune pteridines NO? ND pr purple pteridines YES NO ras raspberry pteridines NO ND sepia pteridines NO NO se maroon-like ND mal pteridines NO NO ND dke dark-eye pattern Me Moire pattern NO NO Mottled of K pattern NO NO Mot-K rolled NO? ND rI pattern mel melanized secondary NO ND NO ND pw-c pink wing c secondary? YES* z zeste regulatory ? 84 Figure 9. Analysis of garnet transcription by in situ hybridization in a rosy null genetic background. Panel A shows garnet message in brain (b), salivary gland (sg), eye antennal discs (e), leg discs (I), wing discs (w), fat body (f) and trachea, of ry /ry 506 third instar larvae. Panel B shows garnet message in leg discs (I), a wing disc (w), and possibly a haltere disc (h?) of ry /ty third instar larvae at higher magnification. 506 /ry third instar larvae hybridized with 506 Panel C shows two leg discs of ty sense probe as controls. 85 ....• 3 8 I 6s r 4 q 4 • t V cc secondary effect of the small, weak, poorly viable males that died a few days after eclosion, only the rosy gene, when combined with garnet, shows both complete sterility and female sterility. The interaction between garnet and rosy was investigated in more detail by examining garnet transcription in imaginal discs, brain and other tissues derived from rosy null (ry ) third instar larvae. 506 Figure 9 shows these results. Although this is not a quantitative assay, it would seem that garnet transcription is not noticeably altered in the imaginal discs of rosy null larvae. Interestingly, however, there appears to be no garnet transcription in the brain of rosy null third instar larvae, a tissue which shows garnet expression in wild type larvae (Figure 27). Interaction between garnet and white: Two of the garnet interactions deserve additional comments. The interactions between garnet and enhancer of garnet have already been detailed at some length. As the enhancer of garnet mutation appears to be an allele of white it is clear that garnet and white show a fairly complex genetic interaction. It is only by virtue of being a subliminal allele of white that it was possible to investigate these interactions. The conventional alleles of white (necessarily) effect eye pigmentation so that the spectrum of interactions seen with other eye colour mutations is quite limited. Nolte (1 952b) noted that g 3 interacted additively with the white alleles, wCh, 00 This analysis was extended to include 7 additional w , wa, wsat, wbl and . 0 w 1118 completely white), one weak 1 and w white alleles, two extreme (w (wBVlfX 1 and which produces a dull red colour), and four moderate alleles (wbf, we, w g O e and wt, pinky-yellow eyes) in combination with three garnet alleles, g ,5 2 53 (Table 12). The results are consistent with those reported by Nolte. The g d white and garnet alleles act additively to reduce eye pigmentation. One 87 Table 12. Phenotypes of garnet double mutants with different white alleles. Oe 5 The phenotype of 11 different white alleles alone and combined with g ’ g 2 and g53d is shown. The first column shows the white allele, the next column lists its visual phenotype and below, the value obtained from pigment assay for the white allele alone (as percent wild type pteridine pigment). The last column lists the phenotype and pigment value of the appropriate white allele and the d 5 g 3 allele of the genotype w* g53d/y where *“ indicates the given white allele. There was in general no great difference between the three garnet alleles in combination with the white alleles. 88 Additive interactions between white and garnet alleles white allele phenotype white and garnet double mutant phenotype 1 178 w completely white <1 completely white <1 1 w completely white <1 completely white <1 wa dull orange-red 3±1 completely white <1 wbf faint yellow 1±.5 completely white <1 Bwx dull red 1±.2 pale pink-orange ND wch faint pink/orange completely white <1 00 w dull red 6±1 faint yellow 2±1 we pink/orange 3±.5 completely white <1 1 w faint pink completely white <1 sat browny-purple 8±1 pale orange ND w faint pink .6±1 completely white <1 89 Table 13. Epistatic interaction between g 2 and wa3. The first row gives the genotype of males with either single or double mutant combination of g 2 and wa3. The second row gives the values for pteridine eye pigments (as percent pteridine wild type pigment) for each of these genotypes. 90 Epistatic interaction between g 2 and a3 genotype /Y 2 g wa3/Y a3 g /Y 2 pigment 38±3 25 ±3 28 ±...2 91 exception has been noted to this rule. Green (1959) noted, while attempting to functionally distinguish white alleles, that a2 and wa3 in combination with g 2 can not be distinguished from the single mutant. Table 13 shows pigment determinations for a2, g 2 and the double mutant combination. These results confirm that, in contrast to the situation with all other combinations, there is no additive interaction between these alleles. (Green also claims this effect is also found for another member of the “transport” group of eye colour mutations, ruby and .) 3 vva These results further emphasize the allele specificity of the white- garnet interaction. Further study of this aberrant epistatic interaction might help reveal the nature of the physical nature of the white-garnet interaction. Interaction between garnet and zeste. The zeste gene was originally identified as a modifier of the white gene. In the presence of a mutant zeste gene, paired copies of the white gene function less effectively, decreasing pigmentation. Thus zeste acts as a pairing-dependent positive regulator of white. Molecular analysis of the zeste gene has shown it to be a protein which acts as a transcription factor which enhances the expression of white and many other genes (Pirrota 1988). Mutations in the zeste gene have a dramatic effect on g O e and 3 g5 flies without and with d garnet expression. Figure 10 shows g , 5 2 the z 1 and the za alleles. Table 14 shows the corresponding pigment levels. Clearly mutants in the zeste gene further reduce the amount of pigmentation of mutant garnet individuals. The zeste-garnet interaction appears sensitive to 1 allele does reduce garnet expression in chromosome pairing. While the z males, with a single X chromosome, in females where the two X chromosomes may pair, eye pigmentation is essentially abolished (Figure 11). Thus both the zeste gene and the enhancer of garnet mutation reduce garnet expression. Both the cytological mapping of enhancer of garnet (Figure 6) and by 92 Figure 10. The effect of zeste mutant alleles on the garnet phenotype. The effect of the za and z 1 alleles on the phenotype of flies mutant for the three 53 5 g g O e and g garnet alleles, d, 2 (from top to bottom respectively). Each photograph shows the eye colour of the garnet allele alone, the same garnet Qe 5 alleles when combined with the za allele (and also the z 1 allele with the g garnet allele) and the appropriate e(g)-garnet combination for comparison. 93 e(g) g53d Oe 5 1 g z e(g) Oe 5 za g 2 e(g) g 2 g 94 Table 14. Effect of zeste on garnet. The effect of zeste on garnet expression was determined by visual inspection and by determination of pteridine pigment levels (expressed as percent wild type levels) for two alleles of zeste (z 1 and za) and the three most severe alleles 0 5 0 and g5 d). 3 of garnet (g , g 2 Cross: The zeste and garnet mutations were combined as follows: g*/g* ®yz*/Yc/ Fl yz’ ÷/÷ + g* ® g*fyf(sjs) yz*g/Ycf progeny selected as yellow, hence probably zeste, and garnet individuals. These were balanced over the ln(1)d149 balancer to generate stocks. F2 95 Effect of zeste on garnet garnet allele zeste genotype yzg yzag g 1 yz g53d/g53d 3d/y 5 g 16± 1 19±1 10 + 1 12±2 <1 ± 1 10±1 Oe/g 5 g Q e Oe/y 5 g 29±2 33±2 18±3 19±5 <1 ± 1 10±3 /g g 2 31±2 38±2 14 + 1 17+1 <1 ± 1 21±2 /Y 2 g 96 Figure 11. The phenotype of zeste-garnet combinations in females and males. Oe and g 5 The phenotypes of three garnet alleles, g53d, g 2 (from top to bottom) alone and in combination with the z 1 allele are shown. The phenotypes of homozygous and hem izygous z’- garnet flies differ noticeably. The females (where pairing between the two X chromosomes is possible) have essentially white eyes whereas the males show a less dramatically enhanced garnet mutant phenotype. 97 d/y g 3 1 5 z d g 3 3 5 d/zl 5 zl g 121 e g O e/zl 5 g O 5 e/Y g O 1 5 z 1 g z IY 2 z 1 / 2 1 g z Table 15. Effect of zeste modifiers on the zeste-garnet genotype. The effect of two modifiers of zeste, E(z) 1 and Su(z) 302 was determined on d 5 3 genotypes. theyza g 1g 2 and yz Cross: P yzi g*/In(1)d149; +1+ oryza g*/Jn( 1)d149; ÷/÷ Su(z)302/TM3 a’ where * 0 +/Y E(’z,)1 or indicated either g53d or g . 2 The g 4 allele occurs in the complex inversion In(1)In(1)d149. The progeny of this cross were scored visually and by determination of pteridine pigment levels (values expressed as percent wild type). 99 Effect of modifiers of zeste on garnet garnet genotype E(z)1 Su(z)302 yzlg d/÷;Mod.*/÷ 53 d/+; TM3/+ 3 1 g5 yz 53 Mod./+ g 1 d/Y; yz d/Y;TM3/÷ 53 yzlg 19±1 96±2 1 ± .1 11±1 101±2 101 ± 1 15± 1 15±1 /+; Mod./+ 2 y za g /+;TM3/÷ 2 yzag /Y;Mod./+ 2 yzag /Y; TM3/+ 2 y za g 97±2 94±2 10±3 9±1 103± 1 100±1 25±2 18± 1 /+; Mod.!-,4 yz g y z+ g4/÷; TM3/+ /Y;Mod./÷ 4 yzg /Y;TM3/÷ 4 yz+g 102± 1 97±2 29±3 14±1 104±2 101 ±3 38±4 35±1 * Mod. = Modifier of zeste, either E(z) 1 or Su(z)302. 100 complementation between zeste and enhancer of garnet (data not shown) indicate that the enhancer of garnet mutation is not an allele of zeste. This interaction is sensitive to modifiers of zeste. The enhancer of zeste mutation d 5 3 in males and is, furthermore, capable of 1 g further reduces pigment of z inducing dominant garnet expression in z 1 g53d/÷ + females (Table 15). The d 5 3 allele by z enhancement of the g 1 occurs in males as well as females indicating that in this case, pairing of X chromosomes is not necessary. Interestingly the eye phenotype of these flies is a tine grained mosaic of brown spots on an orange background which becomes more pronounced with age. Genetically the interaction between zeste and white resembles that between garnet and white. At least superficially, both appear to be necessary for full white gene expression. Although the zeste gene has been extensively investigated and is known to modify the transcription of a number of genes, there is no mention in the literature of an interaction between zeste and garnet. This interaction could occur either directly, if zeste acts as a transcriptional enhancer for the garnet gene, or indirectly, if it occurs due to compromising the function of the white+ gene. The latter may be more likely as there are no zeste binding sites at garnet. These possibilities could be distinguished genetically by providing another wild type copy of the white gene as a transgene which would be insensitive to pairing-mediated zeste effects. 101 Discussion-Interactions between garnet and other eye colour genes. The enhancer of garnet mutation: The first clue to the extensive genetic interactions between garnet and other genes was the discovery of the enhancer of garnet mutation. The chance of finding not only a mutant white allele, but a cryptic white allele, in the strain in which the gP mutation was induced is truly remarkable. This mutation was pivotal in unraveling the complex network of interactions between white and many other eye colour mutations. These interactions could not have been discovered with a conventional allele of white where the phenotype of the white allele would mask that of the interaction. The enhancer of garnet mutation is a cryptic allele of white: Three lines of evidence indicate that the enhancer of garnet mutation is an allele of the white gene. Firstly, their cytological positions coincide. Secondly 11 different alleles of white acted as strong enhancer of garnet mutations in combination with the enhancer of garnet mutant and the three alleles of garnet whose phenotypes are sensitive to the enhancer of garnet mutation. Finally white+ transgenes were able to rescue the enhancer of garnet effect. Thus the enhancer of garnet mutation should properly be designated we(g) Dosage studies with deficiencies for the white gene indicate that the original enhancer of garnet mutation was hypomorphic. In conventional complementation tests with different white alleles and white deficiencies, the enhancer of garnet mutation complements white mutations. Mutations in the 102 white gene, however, fail to complement the enhancer of garnet phenotype. Thus the enhancer of garnet mutation is a cryptic allele of the white gene. As a cryptic allele it has no independent phenotype. The we(g) mutation reveals interactions between white and members of the transport group. The we(g) mutation was the key tool in the exploration of the interaction between garnet and white. Combinations between we(g) and mutations in 26 other eye colour genes were examined in the hope that e(g) would reveal similar interactions between the white gene and other eye colour mutations. Of these 26 combinations, nine or possibly ten showed interactions which mimic the interaction seen between garnet and enhancer of garnet. More importantly, only those eye colour genes identified as members of the transport group showed this interaction. Neither brown nor scarlet, likely structural components of the transmembrane pore complex, nor genes encoding enzymes in the ommochrome biosynthetic pathway (vermilion and cinnabar) or pteridine biosynthetic pathway (maroon-like, raspberry and sepia), showed this interaction. These results imply that the white gene interacts not only with garnet but also with claret, carnation, carmine, deep orange, light, lightoid, pink, purple, rosy and ruby. These genes are largely the same as those described as the ruby group by Nolte (1955), group two by Schwink (1975) and the transport group by Sullivan and Sullivan (1975). (Two of the eye colour genes which have been implicated as members of this group, orange and maroon, were not tested because mutant alleles were not available). 103 Conceptually, this interaction could arise from a situation where the white gene product interacted separately with all these genes. Alternatively, the white gene product, and the products of the transport group of eye colour genes might interact as members of a large macromolecular complex. The second model predicts that the transport group of genes would interact with each other as well as with the white gene. This was found to be the case. Pairwise combinations between garnet and many of these genes showed a variety of novel phenotypes, including synthetic sterility, cell death phenotypes, synthetic dominance and synergistic/additive effects on eye pigmentation. Although these genes have been previously linked by phenotypic, physiological and histological analysis, genetic analysis of interactions between these genes has been haphazard. There is remarkable congruence between the genes which interact with enhancer of garnet and those which interact with garnet. This strongly suggests that the white gene interacts in the same fashion with all of these gene products. One member of the transport group deserves additional comment; the rosy gene. The rosy gene is exceptional in this group because it is the only one which is known to encode a defined enzymatic function. The rosy gene encodes xanthine dehydrogenase. Unlike other eye colour genes which encode enzymes, no role for xanthine dehydrogenase has been confirmed in either the ommochrome or pteridine biosynthetic pathways, (Reaume, Knecht and Chovnick, 1991). Interactions between rosy and other members of the transport group might suggest a structural rather than enzymatic role for this gene product. While structural enzymes are hardly unprecedented, enzymatic activity appears necessary for both normal eye pigmentation and correct localization of 104 xanthine dyhydrogenase in the pigment granules (Reaume, Knecht and Chovnick, 1991). It is also possible that rosy has a general function only indirectly related to eye pigmentation (Hilliker et a!. 1992). It may be important to note in this context that the interaction between garnet and rosy was unusual in that it was the only combination that resulted in complete female sterility. Further investigation may well reveal that the rosy gene plays a slightly different, and possibly a key role in the function of this group of mutations. The nature of the interaction between garnet and white: Null mutations in the white gene show no pigment deposition in any tissue. Thus the white gene product is necessary for the correct localization of both the pteridine and ommochrome pigments. Early models advanced the white gene as a common element which interacted with the product of the brown and vermilion, cinnabar and scarlet genes to deposit both red and brown pigments (Nolte 1952b). The role of the white gene in pigment deposition has been reformulated based on its similarity to a number of transmembrane channel complex proteins such as bacterial permeases, mammalian multiple drug resistance genes and the cystic fibrosis gene product (Dressen, Johnson and Henikoff 1988). These proteins function as transmembrane pores or parts thereof. One model for its function in eye pigmentation is based on a physical interaction with the products of the brown and scarlet genes, respectively, to form the principal component of the transmembrane channel complexes which allow transport of pteridines and ommochrome pigments, pigment intermediates and metabolites into the pigment cells. The role of the garnet gene in this process is unknown. 105 The extensive complementation testing between garnet and different white alleles allowed me to examine the sensitivity of the enhancer of garnet effect to the dosage of both the white and the garnet genes. The enhancer of garnet effect appears to be very sensitive to the dosage of the garnet gene. The interaction is sufficiently sensitive that if the dosage of the garnet gene is severely reduced, such as when a mutant garnet allele is heterozygous with a deficiency for the garnet region (e(g) g / + Df(g)), the enhancer of garnet mutation acts in a dominant manner. In contrast, the enhancer of garnet effect appears to be sensitive to the specific allele of white rather than dosage of the white gene. The allele specificity may reflect the mechanism of the interaction. In at least one case, that of the wa allele, the lesion at the white gene is associated with aberrant transcripts. If these altered transcripts are translated, the strong enhancer of garnet effect might indicate a protein-protein interaction, conceivably via titration of active garnet gene product. Characterization of the 3 allele, which shows an epistatic interaction lesion at the white locus in the wa with garnet might be illuminating. A further suggestion that the interaction between white and garnet may occur post-translationally can be inferred from the rescue of the enhancer of garnet effect by white+ transgenes with essentially only white coding region. Regardless of the nature of the interaction, genetic interactions between white and garnet clearly indicate a functional link between these genes. Severe garnet mutations and weak white mutations are very similar in phenotype. In this context, it is interesting to note that the chromatography profile of a garnet allele (g ) is indistinguishable from that of a hypomorph of 2 white (Hadorn and Mitchel 1951). Furthermore, Nolte (1959b) noted that change in red and brown pigments of the wbl allele produced by temperature 106 change parallels the changes in these pigments between different garnet alleles. This similarity in phenotype, in conjunction with the proposed structural role of white in forming the transmembrane channel complex suggests that garnet is a positive regulator of white function. Specifically if garnet is non functional, the white gene functions, but much less efficiently. If there is any further compromise of this system, such as with the we(g) or other white mutant allele, pigment deposition, which in the presence of a functional garnet gene product would be unaffected or only slightly diminished, is essentially eliminated. Models of gene interaction in the eye: The most complete model proposed for the role of the many eye colour genes in the structure and pigmentation of the eye was proposed by Nolte (1952b,1959), based on data obtained from his decade of histological and genetic work on these genes. Nolte proposed that the genes which altered levels of both the red and brown pigments, the “transport group”, were involved in general aspects of protein metabolism, specifically protein catabolism. The massive rise in protein catabolism that accompanies metamorphosis and the inability to excrete toxic by-products could result in the evolution of pathways whereby toxic by-products were converted into pigments. Genes which regulate or participate in these functions might alter the amounts of pigment precursors and thus final pigment levels. Nolte (1 952b) derived a genetic pathway that invoked two partially independent systems, controlled by brown and scarlet, which interacted with the white gene. In this scheme garnet, along with ruby, carmine and carnation operated in separate but parallel pathways to provide appropriate levels of precursors. Later he included the genes claret, maroon, pink, purple, purploid, 107 prune and rosy (Nolte 1955) and light (Nolte 1 954b) as other members with similar functions. The genetic interactions, the synthetic dominance and synergistic interactions, between these genes were the impetus for this model and they are certainly consistent with the proposal of an interrelated network of somewhat redundant pathways where mutation in one would lower the threshold for others. Such a system would also explain why lesions in such a general and important function were not lethal. Figure 12 shows the functions derived from genetic interactions as proposed by Nolte (1952b). Nolte’s later (1959b) variation of the model included available information on tissue specificity, developmental control of pigment formation and multiple feedback loops. Little more can be added conceptually to this model. The analysis of some eye colour genes over the past thirty years has, however, suggested more specific roles. This information, in conjunction with the results of this work, allows me to postulate the model shown in Figure 13. The essential features of this model are: 1. The white, brown and scarlet genes are structural components of a transmembrane pore complex. 2. The white gene product interacts separately with the scarlet and brown gene products. 3. The transport group of eye colour genes also interact with the white gene product. 4. This interaction is different from that of brown and scarlet, likely fulfilling a non-structural role. 5. Interaction between the transport group and the white gene is proposed to occur at the protein level. 108 Figure 12. Nolte’s model for interactions between eye colour genes. Figure 12 shows a simplified models of interactions between eye colour genes adapted from models proposed by Nolte (1952b and 1959). The lower circle indicates biochemical reactions proposed to occur in the body whereas the upper portion of the figure indicates reactions proposed to occur in the eye. Roles of a number of eye colour genes are indicated. The genes garnet, carmine, carnation and ruby are shown at the bottom performing similar, redundant functions involved with metabolism of eye pigment precursors. The white gene is shown interacting with the products of both the brown and scarlet gene to produce the red and brown pigments respectively. The cinnabar and vermilion genes are indicated as enzymatic lesions. 109 Nolte’s model of eye colour gene interactions Red chromoprotein chromoprotein • reactions in eye protein brown pigment rec’ pigment(s) sors 1 precu precursors cn + f reactions in body t precursors prersors v+f tryptophan precursors / g+cmar4rb 110 + 6. The products of the transport group genes are proposed to form a complex. 7. The rosy gene may have a different and possibly key role in this process. 8. The function of this complex is to enhance the function of the transmembrane pore complex. 9. These gene products may associate in various combinations to perform similar functions in other tissues, other locations in the cell, or at other times in development These points are discussed in more detail below. The white, brown and scarlet genes appear to encode transmembrane transport proteins. The phenotypes of mutations in these genes, the interactions between these genes, and their physical properties lead to a simple model in which the white gene product associates with either the brown or the scarlet gene products to form the transmembrane channel complexes responsible for transport of the pteridine and ommochrome pigments or pigment intermediates. The role of the “transport” group of mutants appears somewhat different. The garnet, light (Devlin et al. 1990) and pink (Jones and Rawls 1988) genes have been cloned. The garnet gene does not show a strong similarity to transmembrane proteins, nor has such similarity been published for the light gene, however, A.J. Howells (cited in Tearle 1991) reports that the pink gene may have some sequence similarity to the white gene. Genetically, neither brown nor scarlet interact with the we(g) mutation, suggesting that the transport group gene products participate in a different type of interaction with the white gene product from that hypothesized for the brown and scarlet gene products. Rescue of the enhancer of garnet effect by the coding region of white leads me to postulate that this interaction involves the protein products of these genes, although the data are inferential for the garnet gene and there are no equivalent 111 Figure 13. Model of the physical interactions between the products of the white, brown, scarlet and the transport group of eye colour genes, including garnet. The product of the white gene is proposed to interact with the products of the brown and scarlet genes, separately, to form transmembrane channels in the cell membrane to allow the ingress of the pteridine and ommochrome pigments (or intermediates), respectively. The products of the garnet, carnation, carmine, light, lightoid and pink genes are shown interacting in some ill-defined macromolecular complex, the function of which is to enhance the activity of the white gene product. The product of the rosy gene is shown as participating by some independent mechanism. 112 - C) p 0 0 CD 3 0 0 C) 3 9 0 Z O3. C, a CD D CD CD 0 = 0 C•) -‘ -h 0 CD a 0 D -‘ data concerning other members of the transport group. The effect of e(g) other members of the transport group of eye colour genes parallels that of we(g) on garnet. Furthermore, those members of the transport group of eye colour mutations which are sensitive to we(g) also show genetic interactions with garnet. This suggests that these “transport” group of genes are functionally linked. I have interpreted this functional interaction as a macromolecular complex. Transmembrane permeases are typically large multi-subunit complexes but other possibilities for the physical nature of the interaction exist. The rosy gene shows a slightly different spectrum of interactions with garnet, and is the only member of this group which is known to encode an enzyme. For this reason, as well as its extensive participation in synthetic lethal interactions, I have postulated that the rosy gene performs a different, and possibly controlling, function. This function might conceivably be involved in metabolic feedback. The phenotypes of null and extreme members of all of the transport group of genes resemble moderate mutations of the white gene. This phenotype suggests the function of these genes, or of the complex, is to enhance the activity of the transmembrane pore complex. I have indicated that this involves interactions between the gene products. It is, however, possible that these genes all act as transcriptional enhancers of the white gene. Finally, the action of this proposed complex may not be restricted to the plasma membrane of the eye pigment cells. At least one of the enzymes necessary for ommochrome pigment synthesis is found in the mitochondria (Sullivan, Grub and Kitos, 1974). Hence the resulting pigment precursor must be transported across the mitochondrial membrane. The accumulation of pigment in the mitochondria of rosy mutants also implies the need for transport across this membrane (Bonse 1967). And, as the pigment granules themselves are also membrane bound, transport across this membrane would also be necessary for 114 normal pigmentation. The synthetic lethal interactions between certain members of the transport group, as well as the synthetic sterility or garnet and rosy, suggests that different members of the complex may associate in other tissues or at other times in development to perform slightly different functions. This transport system may in fact have a fairly ubiquitous distribution. The fact that the sole phenotype of mutants in the system is an alteration in eye pigmentation may be the results of functional redundancy within the transport group of eye colour genes and the requirement for massive amounts of pigment and pigment intermediate transports, within a short time, for normal eye pigmentation. Thus while the eye cells may place the greatest demand on this transport system, it does not mean that this system is confined to that tissue. The expression of the white gene is extensively regulated. Pairing dependent regulation of the white gene by the zeste gene product is well documented (Pirrotta 1988). Additional regulators have been described which appear to regulate not only white but also the brown and scarlet genes, also proposed to encode structural components of the transmembrane channel complex (Rabinow et a!. 1991, Birchler et al. 1994). These regulators all appear to act at the transcriptional level. If the interaction between garnet and white involves post-transcriptional regulation this will describe a novel form of regulation of the white gene. Testing of this model will require molecular analysis of the transport group of eye colour genes. As an initial step in this process, chapter 2 presents phenotypic characterization of the different garnet mutants and preliminary molecular characterization of this gene. 115 Chapter 2. Analysis of the garnet gene 116 Introduction-Chapter 2. Analysis of the garnet gene. Any attempt to test a model of the biological role of eye colour genes must rest upon the detailed analysis of individual genes. The garnet gene is a member of a particularly interesting class, the transport group of eye colour genes. These genes are defined by alterations in the levels of both the ommochrome and the pteridine pigments. This phenotype likely stems from lesions in inter- and intra cellular transport and communication. Chapter 1 presented genetic analysis of the interactions between the garnet gene and other eye colour genes, which led to a simple model for the function of these genes. This chapter presents detailed phenotypic analysis of a number of garnet alleles, evidence for the cloning of this gene and preliminary molecular analysis. This work lays the necessary groundwork to determine the function of garnet, to test the model presented in chapter 1, and leads to further studies on the garnet gene presented in chapter three. 117 Results: Characterization of the garnet eye colour gene. Phenotype of garnet mutants: The garnet gene was originally described as an eye colour mutation and this is the most obvious phenotype of the garnet mutants. Figure 14 shows the spectrum of eye colours found in a variety of garnet alleles. Eyes of garnet mutant flies range from the slightly brown colour of weak alleles through the browny-orange of the intermediate alleles to the pale orange of the extreme d allele. This latter allele is unusual that in that it displays a fine grained 3 g5 mottling of red spots on a pale background as the flies age. Although the various garnet alleles have been used for many years, quantitative information of the amounts of red and brown pigments exists for only a few garnet alleles ,g 1 (g ,g 2 3 and g , Nolte 1959). Table 16 gives a quantitative estimate of the 4 levels of pteridine (red) pigments and the ommochrome (brown) pigment xanthommatin for 18 garnet alleles using four different methods (comparisons between these methods is discussed in more detail in Appendix 1). Nolte (1950, 1952b and 1959) gives values for the relative amount of red and brown pigments in the g ,g 1 ,g 2 3 and g 4 alleles as 38, 15, 23 and 57 % (red pigments) and 56, 32, 47 and 23 % (brown pigment) respectively. He used a sequential pigment extraction technique to obtain these values. It should be noted that Ephrussi and Harold (1944), as well as the controls presented in both these papers show that this technique is problematic. Nevertheless these data agree reasonably well with those shown in Table 16. 118 Figure 14. Spectrum of eye colour phenotypes of different alleles of the garnet gene. The top portion of the figure shows the phenotypes of wild-type (Canton S) and two garnet alleles, gP and g53d. The two garnet alleles shown represent two extremes of the spectrum of eye colours due to mutations in the garnet gene. The g53d allele is the most extreme of the garnet alleles and has a pale orange eye. The gP allele is a very weak allele. Flies bearing the gP mutation have slightly browner eyes than wild type upon eclosion which then darken to wild type within three days. The lower portion of the figure shows the spectrum of eye colours seen in the g O e, g garnet alleles, g , g53d, 5 4 , 6 1 gS3 and gP. A wild type male , g 1 , g 3 ,g 2 (Canton S) is shown in the center of the figure for comparison. 119 oI Determination of pigment levels in garnet mutants. Table 16 presents values for total relative absorbance of the pteridine and the ommochrome pigments, all expressed as percent of the appropriate wild type pigment levels. As there is only one ommochrome pigment in Drosophila melanogaster, dihydroxanthommatin, the “brown pigment” column gives an adequate estimate of the amount of xanthommatin in these mutant strains. There are however, at least 28 different pteridine pigments and intermediates (Ferré et al. 1983, 1986). All of the pteridine pigment procedures used to generate the values given in Table 16 measure principally the content of the drosopterin pigment (Appendix 1). In order to assess the effect of these mutants on at least some of the other pteridine pigments, the red pigments were separated by thin layer chromatography and the relative amount of each pigment was determined. Adequate discrimination was obtained for only 9 pigments. These results are shown in Table 17 and Figure 15. The garnet alleles tested generally decrease the amount of all the pigments although the various alleles have variable effects on the individual pigments. Quantitative assessment of pigment levels was meaningful only for the drosopterin pigments (see appendix 1) but visual assessment of pigment levels generally support this conclusion. The g53d allele appears to be the most severely affected. No novel pigments or intermediates are detected in any of the garnet mutations. Ferré et al. (1983, 1986) examined pigments for 52 eye colour genes, the g 1 allele among them. The values they reported for the amount of these nine 121 Table 16. Pigment levels of various garnet alleles. The first column shows the garnet allele and sex of the mutant assayed. The next four columns give quantitative determination of the red and brown pigment levels. The first three values are measurements of total pteridine levels by three different methods. The last column is a measurement of the brown pigment xanthommatin. Pterins (5 heads) refers to a measurement of pteridine levels by the 5-head microflourimeter method. Pterins (1 head) is measurement by the single head microflourimeter method and pterins (spec.) refers to measurement by spectoflourimeter. Details of the experimental method are given in the materials and methods. Comparison between the methods for red pigment determination is given in appendix 1. 122 Pigment levels of garnet alleles allele pterins (5 heads) pterins (1 head) pterins (spec.) ommochromes (xanthommatin) 1 g 57±2 72±4 4±1 9±1 9±1 15±3 25±3 66±1 g 2 2 g 32±2 37±2 2±1 2±1 3±.3 57±2 ND 15±1 45±1 50±3 17±2 21±2 18±3 56±18 70±8 52±5 g 4 4 g 25±1 38±3 7±1 10±1 10±1 12±3 28±5 37±3 g5Oe g5Oe 24±2 33±3 1±1 3±1 3±4 35±4 ND 19±4 g53d g53d 15±2 21±3 12±1 3±1 4±.5 19±4 13±2 7±4 gS3 gS3 43±4 44±3 23±3 26±1 26±3 29±7 41±5 31±3 e(g)g2 cf 2 e(g)g 29±2 32±3 ND 1±1 ND 34±3 15±9 4±3 e(g)g50e 12±1 OecJ 15±1 5 e(g)g 1±1 1±1 2±.3 17±2 13±2 4±2 e(g)g53° 10±1 dcJ 12±1 53 e(g)g 2±1 2±1 3±.3 12±2 6±2 4±2 3 g cca 123 S6-1 S6-1 96±2 97±6 47±3 75±5 43±5 82±18 75±5 77±6 gP[P] 50±5 3±1 10±3 ND* gP-Rev1c 93±4 99±4 60±6 101±11 gPReV2 93±4 78±5 86 ±22 ND gP-Rev3cJ 87±6 ND 88 ±20 78±9 gPReV4 89±5 72±7 72 ±20 98±6 2 gPO 3±1 1±1 2±.5 9±1 gPdcc? 60±3 44±3 54± 10 31 ±2 ’ 2 gPX 60±2 48±3 49±10 62±4 c? 124 Figure 15. Chromatographic analysis of pteridine pigments of garnet alleles. Figure 15 shows some of the separated pteridine pigments. The various garnet mutants from which the pigments were isolated are shown below the chromatograms. The names of the pigments are shown on the left of the figure. In addition to the seven pigments identified, there were three unidentified pigments. Two of these migrate slowly in the solvent and fluoresce blue in ultra violet light, the third has a greater mobility and fluoresces yellow. These products might also be degradation products of the pteridine pigments generated during isolation and chromatography. 125 isoxanthopterin xanthopterin residue drosopterins unknown blue spot 1 unknown blue spot 2 N) 0 - unknown yellow spotM sep iapte ri r hydroxypteridiflel 2-amino-4- b lo pte r CD CD CI)- CD ci 40 ________________________________ cJ-1 isosepiapterin.E jJ CA) D ( — -+oq J — CD )C) c - CD -+0 40 4Q01-+Q(71 CA) 0 CD - CD 0 CD CD I) CD ‘— CD CD F’3Q1 Q Q ‘(D CD CD CDCD — CD CD CD 0- ) CD - I Q Ci) I I I -+0 (C) ( CD CD I CD CD DDtJ I I I CD Ci) DC)) I (C) I... ...•. ____________________________________________________________________ Table 17. Quantitative assessment of pteridine pigments after chromatographic separation of pigments. The first column indicates the genotype of various garnet mutants. The remaining columns show the percent of wild type levels of the indicated pteridine pigment. 127 128 g5Oe g5Oe g53d g53d gS3 gS3f gP,’ S6-1 S6-1 gP-R1 gP-R3 gP-R4 gP-O2 1 gP-dc gP-X2f g3 g3cf g4 g4f allele g1 g1f 5 24 16 26 3 13 0 0 2 0 26 18 18 17 106 200 53 112 0 62 71 8 18 46 33 8 8 0 1 3 1 53 52 9 54 108 92 117 97 1 76 77 0 0 0 0 0 0 14 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 167 0 0 0 0 0 0 0 0 0 0 0 0 0 50 0 0 20 0 0 0 43 0 14 0 0 0 0 20 20 40 0 40 0 0 20 0 0 20 0 0 0 129 0 71 0 0 0 0 0 20 60 0 20 0 0 60 0 0 0 0 0 0 267 133 100 33 0 0 0 50 50 50 0 50 0 0 0 Effect of garnet alleles on pteridine pigments pigment residue droso- mystery mystery isoxanth yellow xantho- sepiapterin spot 1 spot 2 o-pterin spot pterin pterin 33 0 0 0 0 0 167 0 37 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 100 0 0 0 0 0 0 0 0 0 0 0 0 15 23 54 15 0 15 86 57 121 58 8 23 0 73 64 27 38 36 36 145 18 2-NH4- biopterin isosepia pterin 4-OHpteridine pigments generally agree with the results here, although their method may provide for greater accuracy. Malpighian tubule phenotype of garnet alleles. There are four pigmented structures in the adult fly body, the eye, ommatidium, the malpighian tubules and the testes sheath. Beadle (1937a, 1937b) has described the colour of g 2 malpighian tubules. Breme and Demerec (1942) examined the malpighian tubule colour of 25 different eye colour mutations. Their survey included four garnet alleles, g’, g , g 2 3 and g . Table 18 shows 4 the results of a similar survey extended to include an additional 13 garnet alleles. The effect of the two garnet alleles surveyed by Breme and Demerec (1942) on pigmentation agree with my estimate of pigmentation, however, the results from the control mutations used to compare colour suggest that they were somewhat more discriminating than I in detecting very low levels of pigmentation. All of the garnet mutations reduce pigment deposition in malpighian tubules and, in general, the reduction in colour is more extreme than observed in the eye. It is not possible to distinguish the colour of the malpighian tubules of most of the more extreme alleles, as they all appear essentially colourless even though their phenotypes in the eye are readily distinguishable. In contrast, the subliminal allele gX, which is associated with an inversion, shows a weak variegated pigment phenotype in the malpighian tubules although the eye phenotype is visually indistinguishable from wild type (data not shown). It is also interesting that the gP revertants (discussed below) do not appear to be complete revertants based on malpighian tubule phenotype. 129 Table 18. Effect of various garnet alleles on colour of malpighian tubules. The first column shows the garnet allele. The second and third give the colour of the malpighian tubules as determined visually or where reported in the literature (Breme and Demerec 1942). 130 Survey of malpighian tubule colour of garnet alleles mutant colour reported colour CS orange bright yellow car very pale/clear pale yellow cm colourless very pale yellow w colourless colourless 1 g colourless very pale yellow 2 g very pale/clear very pale yellow 3 g colourless very pale/clear e g O 5 very pale/clear g53d colourless 1 g 6 very pale/clear 71 g 2 yellow gEMS pale yellow gS3 colourless gX orange/variegated? 2 e(g)g colourless e g O e(g) 5 colourless d 3 e(g) g5 colourless gP pale yellow 131 gPReV1 yellow gPReV2 yellow/orange gPReV3 yellow gPReV4 yellow S6-1 orange O.R orange 132 Table 19. Effect of various garnet alleles on testes sheath colour. The first column shows the garnet alleles. The second indicates the colour of the testes sheath immediately after dissection. 133 Survey of testes sheath colour of garnet alleles mutant colour CS bright yellow 1 g pale yellow 2 g pale yellow 3 g yellow 4 g yellow Oe 5 g very pale yellow g53d bright yellow 61 g very pale yellow 271 g bright yellow gEMS pale yellow gS3 pale yellow gX bright yellow e(g) g 2 pale yellow Qe 5 e(g) g very pale yellow e(g) g53d yellow gP yellow gPReV1 bright yellow gPReV2 bright yellow gPReV3 bright yellow gPReV4 bright yellow S6-1 bright yellow OR bright yellow 134 Testes sheath phenotype of garnet alleles. In general, all the mutants decreased the pigmentation of the testes sheath (Table 19). The effect of the different alleles on testes sheath colour was generally more severe than the effect on the eye pigmentation but less severe d 5 3 allele provides the one than the effect on malpighian tubule colour. The g exception. Three different strains carrying the g53d mutation all showed essentially wild-type levels of testes sheath pigmentation. This suggests a tissue specific pattern of expression in this allele. The search for a garnet null allele. g5 allele confers the least pigmentation of all the d As observed above, the 3 d 5 3 or any other allele might garnet alleles. In order to determine whether the g be a null allele of the garnet gene, I quantitated the effect on eye pigmentation for different garnet alleles when heterozygous with a deficiency. Table 20 shows the results of red pigment levels of 10 garnet alleles when combined with a deficiency that includes the garnet locus. The phenotype of the g53d allele is the most severe. Comparison between the pigment level of a homozygous g53d strain (Table 16) and the hemizygous g53d condition (Table 20) shows no pronounced difference. Thus, by the criterion of Muller (1936), the g53d allele behaves as an amorphic allele with regard to eye pigmentation (this conclusion 3d allele 5 is also supported by Northern analysis of the g - Figure 22). Comparison of the pigment levels of other garnet alleles as homozygotes or hemizygotes suggest that these alleles are all hypomorphic lesions. 135 Table 20. The phenotype of various garnet alleles in combination with a deficiency. The first column shows the garnet allele. The next two columns show the pteridine pigment values of the appropriate hemizygous garnet allele, as determined by microflourimetric assay (5 heads) and by spectrophotimetric assay respectively. CROSS: P Df(1)HA97/FM7 ® g*/jFF “ Fl Df(1)HA97/g* progeny assayed. where g* indicates the given allele of garnet. 136 Pigment levels of various garnet alleles in combination with a deficiency allele percent wild type red pigment microflourimeter spectrophotometer 1 g 40±3 8±4 2 g 33±3 4±1 3 g 40±3 9±3 4 g 32±4 6±1 Oe 5 g 28±2 5±2 d 3 g5 16±3 3±1 61 g 34±3 7±3 gP 38±4 8±3 2 gS 34±4 8±1 3 gS 38±3 10±2 137 Aging and garnet. Shephard et a!. (1989) have reported that a null mutation for the rosy gene has a decreased adulte life span. Furthermore, Hilliker et al. (1992) have reported that mutants of the rosy and maroon-like genes show hypersensitivity to oxygen stress in addition to a decreased life span and Humphreys, Duyf, Hilliker and Phillips have isolated an allele of the pink gene, another member of the transport group of eye colour mutations, in a screen for mutations that are hypersensitive to the free radical generating chemical paraquat (J. Humphreys, personal communication). Since, longevity should be a sensitive assay for biochemically uncharacterized lesions that effect cell viability, I tested the longevity of three of the most severe garnet alleles, with and without the e(g) mutation, at two temperatures. Figure 16 A and B shows the longevity (% survival) of wild type and different garnet genotypes at 22° and 29°. Some of the garnet mutant strains do die earlier than wild type. But reduced vigor is not particularly surprising in a highly inbred population of flies. More significantly, the relative longevity of the various garnet mutant alleles differs at 22° and 29° and the presence of the e(g) mutation, which profoundly reduces the level of pigmentation, has no appreciable effect on the time of death or shape of the death curve. Finally flies mutant for the garnet gene lived longer than the wild d 5 3 outlived type strains in some instances (at 29° g53d and at 22°, all but g the wild type flies of the appropriate sex). Thus there is no evidence that garnet affects longevity. 138 Figure 16. Adult life span of wild type and various garnet mutants. A. Longevity of wild type (Canton S) and g2, g5Oe and g53d females and males is shown at 22° . Minimum population size is 90 for each genotype. B. Longevity of wild type (Canton S) and g2, e(g) g2, g5Oe e(g) g5Oe, g53d and e(g) g53d females and males is shown for at 29°. Minimum population size is 30 for each genotype. The key for the different genotypes is shown on the graph. The 29° aging experiment and the first portion of the 22° experiment was done by Layne Harvey as part of a directed studies project. 139 C I 0. a) C G) U) C > 1 day Lifespan of garnet mutants 22 ° -- -H g53d male —6- g2 male g2female g5Oe male g5oe female g53d female CS male CS female -e —.- C) 0 > 29° day Lifespan of garnet mutants CS fern. g5Oem g5Oef g53d m g53d f. e(g) g53d f —.*— e(g)g2m —+-- e(g)g2f —+— e(g) g5Oe m —+-- e(g) g5Oe f —0— e(g)g53dm —v— —- g2m —K— g2f —rne-— —a-—e— -R- CSrn. —•— Cloning of the garnet gene: Isolation of unstable garnet mutants from dysgenic crosses. The original gP allele was isolated from crosses involving a natural P-element bearing strain, S6-1 (Wennberg 1988). This strain has two P-elements in section 12 of the polytene chromosome, the cytological location of the garnet gene (Wennberg 1988). Weak garnet mutations arise frequently in this strain (Figure 17). The relatively high frequency of garnet mutations arising from this strain is likely due to the proximity of P-elements to the garnet gene (Towers et a!. 1993). These weak P-element induced mutations, as well as the original gP allele, remain active and will further mutate to a variety of garnet phenotypes upon out crossing to non-P bearing strains. Figure 18 shows the frequency with which new, secondary and tertiary garnet mutations occur. The secondary garnet mutations are also subject to further mutation upon outcrossing to non-P bearing strains, although at a much reduced rate. Figure 18B summarizes extensive lineage and out crossing experiments with these strains. Only the original gP mutation, four wild-type and four secondary mutations which were derived from the original gP allele, have been analyzed at the molecular level. Cloning of the garnet gene: Cloning of the garnet gene was facilitated by the original gP mutation isolated by R. Wennberg (Wennberg 1988). The garnet gene was cloned by Dr. D. Sinclair. Details of the cloning are provided in Appendix 2. Briefly, P-element containing clones were isolated from a size fractionated library made from the gP mutation. One such clone hybridized to position 1 2C, the cytological position 142 Figure 17. Diagram of the mutation rate to garnet upon outcrossing the S6-1 strain. A. The top part of this figure shows the lineage of garnet mutants generated by outcrossing the P-element containing strain S6-1. The number and phenotypes of the progeny are shown for five generation of outcrossing. The phenotypes are given relative to the garnet phenotype, g indicates wild type eye colour, gP indicates a weak garnet mutation similar to the original gP allele and gM or gMOd indicates a moderate garnet eye phenotype. B. The rate of conversion and interconversion to the various garnet phenotypes is summarized. 143 Generation of garnet mutants in the S6-1 background 24 “+“ 18 “+“ sterile A. terile / terile _..-31 •‘+ 44 A(437 •• 31 / (3.1—aP( (3.219?) __43 I / /\.(4a94 1/42 I, / “‘ / 4 , / p./ all Fl -i-’ V..V24 +“ ‘Zç38 “+“ — “÷. P 9 \‘1J2S S... \ \ ‘, \ \ \ \ all—30g+ (3.2.2+,4l_409+ (gP) ll.-40 gP (gP) afl.-4OgP all.-4OgP ‘all—4OgP /,‘all.-40 gP .p sterile g+,1 gModl-4OgP 2÷Y3lgP terile .afl-4O gP 66 gP (g.Ste p 9 112g÷ gMod/8i Og allgP 69 gP ‘i_’all_gMod 1 g÷ sterile \\all..gMod aIl.-gMod all—gMod _Øi.-lgMod lg÷/—4OgP Øll.-40 gP Z*sterile 0 +/—4OgP allgP ÷ -i- - 3 gP .+ gP ‘ 30 ÷ 40 +‘• 39 ÷ 33+. 37 (437 44 +, 48 6 p .p24gP S6-1 C(1)DX [MI stock •4( (4 gP g÷/—40 gP g+/.40 gP +“ 26 + 26 ‘+ 33 22 c 45+ sterile ÷“ 49 .,+ 48+ 33+ 6+ gX 35 cl/ 48÷ 43+ sterite 4 26 ÷ \c sterile —0—48 ÷ _p-84 + 42 +‘ 50 “+ sterile c sterile —‘.sterile (8.1=gP) (4 B Summary of garnet mutation rate in S6-1 background. 4P g+ 0.6-2% S6-1 F5 F4 g”P gM 144 weak garnet mutation gP = gM = moderate garnet mutation “+ garnet revertant Figure 18. Diagram of the mutation rate and phenotypes of garnet mutations derived from the original gP allele. A. The top part of this figure shows the lineage of garnet mutants generated by outcrossing the gP mutation. The number and phenotypes of the progeny are shown for six generation of outcrossing. The phenotypes are again operationally defined relative to the garnet phenotype, g indicates wild type eye colour, gP indicates a weak garnet mutation similar to the original gP allele, gM indicates a moderate garnet eye phenotype and gX indicates an extreme or strong garnet phenotype. B. The rate of conversion and interconversion to the various garnet phenotypes is summarized. 145 2gX/ sterile Generation of new garnet mutants from the original gP allele F3 Øall 43X 1 gXI io—P 2gXL_L...iE. A. F2 I gP 43 p 9 32 gP J sterile I27 , I / I som4 Fl P 9 c(x 0 C(1)DX [P1 (stock) F4 M] / I 24gP ,1 69 P / 45 gP 49 gP / 54 gP gP P\ 9 0 most gP g+ some gX (discarded) / , , I ‘- — ,/19X lgMod/ 21 3 gf’ .r2gX,—2OgP,--2Og+ 2gX/40+gP all 40-fgX .all 40+gX 1 gL3 sterile steril*terilalI 40+gP 40 -all 38gP 40÷g÷ c 2gMod/ 34j4.g sterile 10+9+! 32 gI-’ 10+9+! _240+g+ 40+aM 37 gP — ‘4OgP’’ 3g+48g 33gf\\ 9 ‘ I’ all40+gX _,all 40+ P 9 2g 36 gP ‘ M/ 10+ “ ‘ sterile ,.- steri e all 40+gX sterile 40+9+ all 40+gM 309 ,io+gif 10+g+.10+gMI stenre 1X1g+/40+gP I 4o+gP all lgXI4 —20g+,-.2OgP ..20g+....2OgP gp ‘: steriL ,all 40+gM X140+ciM M 9 40+ all 4oi-gM 38 yP all 40+gM Il 40+gP 19X/40+9M 9ll4O+aP \ 36 gP all 40+gM p 9 lr4o+ 9.2 (+ 3g+/ 39 gIll 40+gP all 40+gM 9.3 (+) gX!40+çP ,1gXJ40+gP 59 gP 47 lg+,-.2OgX,-.2OgP 2aXI 35 g B. all 40+gP 40+gP Summary of interconversion rates 33 g1 gX,2gP/40+g+1 g+!40+gP of garnet mutants 45sterile .-20g+,-2OgP all 40+gP 3 gP 1 X3g+/40 1 g+ sterile a? 4b+gP 20a+ -.2OgP all 40’-i-gP 0 30/0 all 40+gP gP = weak garnet allele 1% \all4O+gP 3 2% gP gM all 40+gP gM= moderate garent allele all 40+gP sterile gX = extreme garent allele F2 ! ‘ . 9+ gX 146 = garnet revertant _______ ___ __ _ ____ ____ __ F3 ‘ F5 F4 F6 •_ Jp ral I 40+g X all 43X sterl 40+gM i5ja. 2gX 1 all 40+9X sterile all 40+gX grn3all 2g!,gM terile II 40+ P 0 27 igX 1g+/40+OP X/40+OP 9 10 ,4 II 40+g 43 X 0 4 4o P 9 sterile II 40÷ —p- steri le 2gX -.20 P -20 + ig —P —i- — .. — ______ ______ ____ c -b 40+q* ..40+(JX sliall 40+gP 2gX1g40+gX \38 gP 2gMod/ P sterile 1 0-i-g all 50+ gX lgM/50+gX all 50+ gX /50 iX 9 all 50+ gX all 50+ gX all 50+ gX c::: _zE + 1140+9 40+i+ _ all 50+ gX O+gX +/ 1O-i-g-i4 P 37 0 39 g __ 2:gP 36 g a terile all 40 gX,10g40+g all 40+0 P ic X/40+9P 0+g+/ -_..10irJ+ 0 10+ M / P c-*.41 9 38 gP 1140 4AP 36 pP 39 g P 9 UP ll40 cSO 2Xl P 0 40+ II 40-i-gP X/40+c 0 X 140+9P 40+ M all 9 X/40+g M 0 all 40+ all 40-i-gM 1 140÷ X 0 gM2g+/40+gX 40÷ Ox zi+gP P 22 9 all 40+gP X,2gP/40+ + 0 33 OP 1 lg+/40+ P 9 sterile -. P 9 i-,— 20 20 all 40+gP 4xall I gxg÷/ all all 40-i-gP all 40+9 P all 9 40-iP all 40+gP sterile __ all all all all 50+ 50+ 50+ 50+ Ox gM gM M 9 all 50+ gP all 50÷ 0P 2g+/50+gP qX/40+yM all 50+ çjX 40+rJMMul40 .Ig I 40+rjM tVi40+gMM cjP +pP 2gP,9gX/50+OM all 50+gX all 50+gM P all 50+ 9 ,ll 40-i gX 40+ - 1 gM/50+clX all.,0÷gX M/50+9X 0 all 50+ Ox - a 1l40+gP all 50-i-gM a 63 iOgP/50+g+ 147 of the garnet gene. Unique sequence DNA flanking the P element was used to isolate lambda clones containing inserts of wild type genomic DNA. These inserts shared a 6.6 Eco RI fragment. This fragment was subcloned and used to isolate c-DNA clones from an imaginal disc library. Four lines of evidence indicate that this 6.6 Eco RI fragment identifies at least part of the garnet gene: The spontaneous gP mutant has an insertion into this fragment, the size of which is altered in revertants. The P-element in the gP mutant is inserted into a region corresponding to a 3’ intron of a transcript from this region. Nine, out of fourteen garnet mutants examined, have alterations, detectable by Southern analysis, in this fragment. Finally, of the two garnet mutants examined by Northern analysis both show alteration in the two transcripts derived from this region. These data are discussed below. Molecular analysis of the gP allele and its derivatives. The gP mutation is associated with a P-element insertion into the garnet gene. The P-element that is inserted in the gP allele is approximately 2Kb in length (Figure 19). Thus it is not a complete P-factor. The four revertants of the gP allele all show alterations in the size of the P element inserted as do the spontaneous garnet P-extreme mutations (Figure 19). Neither the g-revertants nor the gP... extreme alleles are associated with complete loss of the original P-element insert. Thus while the revertants are wild type (based on a visual assessment of eye colour) the P-element insert is not completely removed in any of them. This incomplete molecular reversion is consistent with incomplete phenotypic reversion as assessed by pigmentation of the malpighian tubules. Since the 2kb insert in the gP allele causes only a moderate reduction in the function of 148 Figure 19. Southern analysis of the gP allele and gP derivative mutations. The top portion of this figure shows the results of Southern analysis of the gP mutation and four phenotypically wild type revertants. DNA from wild type (Canton S), the gP allele and four revertants was isolated, restricted with Eco RI, separated on a 0.8% agarose gel, transferred to nylon membrane and probed with the 6.6 Eco RI putative garnet fragment. The wild type DNA shows a band at 6.6 kb as expected, as well as a bands at higher molecular weight likely due to incomplete digestion. The size of the 6.6 kb band is increased to 8.2 kb in the gP mutation. Similar analysis with DNA restricted with Bam HI and Hind III show that this change in mobility is not due to polymorphism for an Eco RI site (data not shown). That this change in mobility is caused by insertion of a P-element is shown by hybridization of this band to P-element DNA (data not shown). The size of the 6.6 kb Eco RI band is also altered in each of the gP revertants. In every case the size of the insert in the gP allele is diminished but in no case is the insert completely removed. The two lower figures show the results of Southern analysis of more spontaneous derivatives of the gP allele. In this case the membrane was probed with the 4 kb imaginal disc c-DNA clone. The gP’X2, gPX3, gPX5 and 2 alleles are moderate or strong garnet alleles. The gPX2dC allele is a gPO subliminal garnet allele (males and homozygous females appear wild type but females heterozygous with a strong garnet allele show a weak mutant 2 strain. With the phenotype) which occurred spontaneously in the gPX possible exception of the gPX2 allele each of these gP derivatives show gPO alleles might 5 and 2 alterations in the size of the gP insert. The gPX represent small deletions in the garnet gene due to imprecise excision of the P element. 149 gP Cs gP—Rev2 gP-Rev3 gPRev4 gPRevl 82kb 66k* Cs gP P-X2 gP-X3gP.X5 gP-dC gP-02 9 PX2-dc 8 65kb — 150 65- the garnet gene (based on the weak mutant phenotype of this allele) it is possible that internal deletions of the P-element might relieve whatever impediment this original insertion produced. Sequence extending outwards from the termini of the P-element in the cloned garnet region of the gP mutation identified the position of the insert as corresponding to the most 3’ intron at position 3234 of the imaginal disc c-DNA from the garnet region (see below). Southern analysis of other garnet alleles. Southern analysis was performed on wild type and fourteen garnet mutants to determine the presence and nature of lesions in the 6.6 kb Eco RI putative garnet fragment. Genomic DNA from wild type (C.S) and g 1 mutants was digested with Eco RI, Barn HI and Hind Ill, size fractionated and probed with the 6.6kb Eco RI fragment (Figure 20) and the 4kb imaginal disc c-DNA (data not shown). Southern blots of wild type DNA digested with Eco RI and probed with the 6.6 kb Eco RI putative garnet fragment yield the expected 6.6kb fragment. Barn HI digestion of wild type DNA gives two fragments one roughly 8kb and the other approximately 10 kb. Only the smaller of these is detected by the c-DNA probe (data not shown) indicating that the c-DNA is encoded exclusively by the sequences to the left of the Barn HI site of the 6.6kb Eco RI fragment. Hind III digestion of wild type DNA gives two fragments detected by the 6.6kb Eco RI sequence as a probe, one of 4kb and one of 2.5 kb. Only this 2.5 kb fragment is detected using the c-DNA as a probe (data not shown). Alterations in the size of restriction fragments indicates that the g 1 mutation is due to an insertion of approximately 2kb into this smaller 2.5kb Hind III fragment. Less detailed 151 Figure 20. Southern analysis of the wild type garnet region and the g’ allele Wild type (Canton S) DNA from the garnet region was restricted with Eco RI, Barn Hi and Hind lii and probed with a portion of the 6.6 Eco RI fragment. Similar analysis was performed on the g 1 allele. In the g 1 allele mobility of a band is diminished in each of the digests indicating the presence of an insertion in this mutation. Of note, the mobility of the smaller Hind III fragment, which corresponds to the 3’ end of the transcribed region, is altered in the g’ allele. A schematic diagram of the restriction map of the garnet gene and segment used as a probe is shown in below. 152 1 g Cs HE B H E B .412 48 ‘p .47 .44 .43 1 (&gq g B H L1 RR\H HAB ii\Aii 5’ 1 kb probe 153 RH I I B Figure 21. Southern analysis of garnet mutants. 53 g The top portion of this figure shows Southern analysis of gl, g2, g3, g4, d, Oe and 3 5 g gS The lower portion of the figure shows Southern analysis of , . 61 g gEMS, gim, gS2, gXand T(1;Y)B166. DNA from these flies was isolated, restricted with Eco RI, separated on a 1% agarose gel, transferred to nylon membrane and probed with the 6.6 Eco RI garnet fragment. The g 1 and g 3 alleles appear to have identical insertions into this fragment. The g ,, 4 61 g gEMS and gim also have insertions into the 6.6 Eco RI fragment. The g 53 allele 53 g g Oe, 5 appear to have a small deletion in the 6.6 Eco RI fragment. The d, 2 and gX alleles show no change in the mobility of the 6.6 Eco RI fragment. gS The g 2 allele shows altered mobility of the Eco RI fragment. That this is due to a restriction fragment length polymorphism of the 3’ Eco RI site, is shown by restriction analysis of the g 2 allele with other restriction enzymes, such as Bam HI, shown at the lower right. Finally, the translocation B 166, shown as a g O e, is broken within the 6.6 Eco RI fragment. heterozygote with 5 154 4 41 a) Co T V ci Co ‘Co V 14 I — C’, Co U, 0. U’ Co co—i’ p analysis was pertormed to identify the nature of 12 other garnet alleles (Figure 21). Oe, g53d, , 5 Genomic DNA from g ,g 2 ,g 3 ,g 4 61 gEMS, gim, gS2, gS3 and g T(1;y)B 166 mutants was restricted with Eco RI, separated by electrophoresis, Southern blotted and probed with the 6.6 kb Eco RI fragment. Of the 13 garnet alleles examined in this way, six show insertions into this fragment, one has a deletion, one a translocation break and five show no structural alterations detectable by Southern analysis. Interestingly, Eco RI digest of the g’ and g 3 alleles generated apparently identically sized fragments. This implies that these alleles possess identically sized insertions into the same region of the garnet g O e and 3 g5 alleles which d gene sequence. It is also interesting that the g ,5 2 are the most extreme alleles and also those sensitive to the enhancer of garnet mutation, showed no structural alterations in garnet. These data are summarized in Table 21. Analysis of transcripts arising from the 6.6kb Eco RI putative garnet region. This 6.6Kb. Eco RI fragment was used as a probe to investigate the size and abundance of messages transcribed from this region. Figure 22 shows messages detected by this probe from wild type (Oregon R) embryos and g 3 and g53d newly eclosed adults. Two messages are detected with this probe in wild type embryos, one approximately 3.5 and the other 4 kb in length. Both of these messages are absent from adults of the severe allele, g53d, and only one, possibly slightly larger than the 4kb transcript is present, at reduced levels in the g 3 adults. The absence of both messages from the g53d allele suggests that both messages are derived from the garnet gene and that no other m-RNAs are transcribed from this region in embryos or adults. 156 Table 21. Summary of lesions in the garnet locus in different garnet alleles. The first column indicates the garnet allele. The second and third columns show, respectively, the type of lesion and where relevant, the approximate size. (NA = not applicable.) 157 Summary of lesions at the garnet locus in different garnet alleles allele type of lesion size 1 g insertion 4kb 2 g point mutation? NA 3 g insertion 4kb insertion 1 kb Oe 5 g point mutation? NA g53d point mutation? NA 61 g insertion 3kb gEMS insertion 2kb gim insertion 1 kb gP insertion 2kb gPReV1 insertion .5kb gPReV2 insertion 1.5kb gPReV3 insertion 1 kb gPReV4 insertion 1.5kb gS2 point mutation? NA gS3 deletion 0.5kb gX point mutation?/position effect? T(1;Y)B 166 translocation breakpoint in garnet 158 Figure 22. Northern analysis of wild type embryos and garnet mutant adults. g5 individuals and wild type (Oregon R) d RNA from adult (0-3 days) g 3 and 3 embryos was isolated, electrophoretically separated, transferred to nylon membrane and probed with the garnet imaginal disc c-DNA. There are two messages in wild type embryos. One message, perhaps slightly greater than the wild type 4 kb message is seen in g 3 adults. No messages are seen in g53d adults. The lower figure shows the same four lanes probed with the RP 49 gene as a control for equal loading and RNA integrity. e=em bryo a=adult 159 I A D A 0• cD (DIP 0. Co In summary, the presence of a P-element insert in the 6.6 Eco RI fragment in the gP mutation, which is altered in garnet-P revertants, the number of garnet mutants with alterations occurring in this same fragment and the disruption of the transcripts from this region in garnet mutants all support the contention that this fragment encodes at least part of the garnet gene. Molecular analysis of the garnet gene. Restriction map of garnet and the genomic region encompassing garnet. The unique sequence DNA flanking the insertion site of the P-element of the gP mutant was used to isolate lambda phage containing inserts of the homologous genomic regions of wild type Drosophila melanogaster. Five different lambda phage were isolated and subjected to restriction analysis using Barn HI, EcoRl, Hind Ill and Sal I to generate a crude map of the region surrounding the garnet gene (Figure 23). A summary of these restriction maps is shown in Figure 24. The map includes approximately 30 kb 5’ and 15 kb 3’ to the garnet gene. The phage all held a 6.6 Kb Eco RI fragment in common. This fragment was subcloned and subjected to more detailed restriction and sequence analysis. Detailed restriction map of the garnet region: A restriction map of the 6.6Kb Eco RI fragment using the enzymes, Eco RI, Barn HI, Sal I, Hind III and Kpn I is shown in Figure 24. Below this figure is shown a restriction map of the imaginal disc c-DNA clone isolated using this 6.6Kb Eco RI fragment as a probe. The restriction analysis of the c-DNA also include the restriction enzymes Sst I, Pst I, Sph I, and CIa I. 161 Figure 23. Restriction fragment analysis of lambda phage clones containing the garnet gene and flanking sequences. Restriction analysis of five lambda phages which were isolated from a genomic library screened with single copy sequences adjacent to the P-element responsible for the gP mutation. The five lambda clones, g212, g?13, g?14, g22O and gA21 were digested with the restriction enzymes, Barn HI, Eco RI, Hind Ill and Sal I, single and in pairwise combinations. The DNA fragments were electrophoretically separated, transferred to nylon membrane and probed with the same g?. DNA. The information derived from these restriction digests as well as from probing these blots with portions of the garnet gene (data not shown) was used to generate the restriction map shown in Figure 24. 162 41 9 0 I r%3 (0 •4 48 I I * • I. I I o1 *1 •11 ., • II • . (0 I • •fl * I S — • ,.J• I jISf .. Figure 24. Restriction map of garnet and surrounding region. A. The top of this figure shows the extent of the five lambda phage clones which encompasses the garnet region. Below is a crude restriction map of this region. The 6.6 Eco RI fragment which includes the garnet transcript is indicated by a heavier bar. B. The 6.6 Eco RI fragment which identifies the garnet gene is shown in greater detail. The site of the P-element insertion in the gP mutant, as well as the location of three introns, the site of the putative hydrophobic domains and of the polyglutamine repeats is shown. The arrow below indicates the location and direction of transcription for the 4 kb c-DNA clone isolated from the imaginal disc library as well as additional restriction enzyme sites. The sequence of the most 3’ intron into which the P-element is inserted in the gP allele is TATGCCGCGATNTTTGNANNATCGAAGAGTATG*TTCCAG where the indicates the insertion point of the P-element. 164 Restriction map of garnet and surrounding region A. gM4 g2O gM3 gx21 — B RB II — gx12 — BRH HBBR I III 9-SRRH liii II II U BSHSR Ffl-IS1BB RHBHSRBHRSRHBSR BBR 11111 III 11111 I I liii liii liii I III 5kb R/ / / / / / / / / / / / / / / // / / / \ \ \ \ \ HB I I I BHO R R Sst SsçxbaH S Sst ClaCla CIa I I II I I II III CIa=Cial H=HindIIl Sst=SstI B=BamHI Ssp=Sspl S=SaIl Xba=XbaI R I • I R=EcoRI \ \ B ELiBIII I = 5’ \ \ HS Inn / / / R 3’ H hydrophobic domains introns polyglutamine region 1kb 165 Sequence analysis of the garnet gene. The DNA sequence was determined for a 1.2 kb DNA fragment isolated from an embryonic library, a 4kb imaginal disc c-DNA, and part of the 6.6Kb EcoRl genomic fragment which encompasses the bulk of the garnet gene. The sequencing strategy used to sequence the imaginal c-DNA and the equivalent section of genomic DNA is presented in Figure 25. Sequence analysis of the approximately 1.2 kb DNA fragment derived from the embryonic c-DNA library showed that, relative to the imaginal disc c-DNA sequence, the 3’ end of this DNA segment is in the intron at position 3234 and the 5’ end, marked by a poly C tail, occurs 3’ to the disc c-DNA, presumably in the genomic region flanking the coding region (data not shown). As this DNA fragment encompasses both intronic and genomic sequences, lacks a substantial open reading frame, and is in the opposite orientation to the imaginal disc-derived c-DNA, it seems likely that this fragment is a genomic contaminant in the c-DNA library. This explanation is consistent with the absence of a 1kb message in embryos (Figure 22). The sequence of the 4kb c-DNA isolated from a 3rd instar imaginal disc library is shown in Figure 26. The sequence is 3825 bp in length. There is an ATG at position 297 which could initiate a potential polypeptide of 1054 amino acids. No poly A tail is present in this c-DNA clone however the size indicates that it might nevertheless be complete. Comparison between the sequence of the genomic and imaginal c-DNA revealed that the c-DNA extends 221 bp 5’ to the 6.6 Eco RI genomic fragment. Thus the genomic fragment does not contain all of the imaginal c-DNA. 166 Figure 25. Strategy used to sequence the imaginal c-DNA clone of the garnet gene. This figure diagrams the segments of the imaginal c-DNA clone that were sequenced. The thin scale bars represent the size, in base pairs, of the imaginal c-DNA sequence. The underlying bars represent independent sequence determination where the location of the bars indicates which section was sequenced, the arrow indicates the strand sequenced and the thickness of the bar represents the number of independent sequence determinations of this segment. The solid bars indicate c-DNA sequence, the stippled bars indicate sequences from the corresponding genomic region. Introns are shown as triangles above the scale bar, in the appropriate location. 167 Sequencing stratagy for the I 0 III liii I ‘‘‘I 400 II ‘‘I I 111111111 I liii 100 200 IJ III liii 500 1111111111 1200 I I I 900 800 garnet gene III 1300 III I II I I 600 I I - I I I I II IF 1 I - I 111111111 I 300 joo I 700 11111 I 1000 11111 I liii 1400 liii I - 11J [ 800 1111111111 1100 1200 4 I liii 1500 II I — I I I 1600 - * 1IIIIIIIIII 1111111111 IIIIIIIII•I 111111111 I 1700 1800 1900 1600 2000 1111111111 2000 1111111111 2400 r ri 3200 I till 3600 I III II I 11111 2200 11111 111111111 2300 I 2400 IIøI E;.* 2800 III 2100 i III 2500 2900 iii i i II I till 3700 1111111 600 I i liii 3000 I III I—I II 111111111 34n0 np - ill 11111 I ‘‘‘ 3800 I I I III — — 168 ‘ I liii 2700 I III 3100 I I ::spo I 3900 — III I I I 11111 III 11111 — genomic c-DNA — lx 2X 3X I 2800 I 3200 I 600 Figure 26. Sequence and conceptual translation of the imaginal disc c-DNA clone of the garnet gene. The sequence of the imaginal c-DNA clone from the garnet gene is shown. The sequence is 3825 bp long. Below the sequence is shown the conceptual translation of the imaginal c-DNA clone. A long open reading frame starts at an ATG at position 297 which could encode a polypeptide of 1054 amino acids. The direction of transcription was determined by tissue in situ hybridization with RNA probes (shown in Figure 27). This putative polypeptide has three indifferent hydrophobic domains and a stretch of polyglutamine residues. A schematic summary of the sequence motifs found in this putative polypeptide is shown in Figure 24. The genebank accession number for this sequence is U31 351. 169 garnet Sequence 10 20 30 40 50 1234567890 1234567890 1234567890 1234567890 1234567890 AAATTCCGCG ACCCCGTCCT CTCGGGAATC CGTCATAGCT GAGTATTCAC 50 CGGAAAACCC GAAATACGCT CAAGTCACCG GCTTTAAATC AATCATCCGG 100 TTTAATGTAG CTGAACGTTT TGCCTAACGC TAATGACAAA GAATCGCAAG 150 CACCAAGCAC CAAAAAAAAG TTAATGATTG GACTCGTGAT TAATATGAGT 200 GTTCTAGGTA TTCTTCCGGA ATTCGTACTG TAAGGAGATT TATAAAAATA 250 CAATTAGCCT TTTTTAATTC TGCGCCAGCT TTCAGCTCGT TGATTACATG M 300 TCCAATTCAA TGTCCAGGCT CGATGCGGAT CATTTGGATC GTATTTGCCA SNSM SRL DAD HLDR ICH 350 TCCTTGTCCT CGCTGTCCGA CATGGAGACA CTTAGGGCAT GTCCAGCGTG PCP RCPT WRH LGH VQRG 400 GTGTTCACAA TGTGCAATGG TTTCGGTTCA CCTTCCGATT CGGAGCGCTG VHN VQW FRFT FRF GAL 450 TTGTATGCCA CTTTATTTTT GGACCTTTTT GCTCTTTTTA CCCTTTTTAT LYAT L FL OLE AL FT LFM 500 GCTTCTTCTT GCCATCCTTG CTGCCCTGCG CCGCCTGCTG TTCCTGCAGG L L L A I L A ALR RLL FLQV 550 TACTTATCCG ACCGTTTGGT GATGCCCACG GCCAGAGCGG CCACACCCTC LIR PFG DAHG QSG HTL 600 170 garnet Sequence 10 20 30 40 50 1234567890 1234567890 1234567890 1234567890 1234567890 CATGTCCAGT GGCAGCTCCG TAATGGGTAT GTCATCAATA TTGTCATACT HVQW QLR NGY VINI VIL 650 GATCCGCATT CGAGGCACCA CGTCCGTGGG GCGTGGACTT GAGGTAGTGA IRI RGTT SVG RGL EVVR 700 GGATTGTTCG ACTGCTCAAT GAGACGAGCC ATCTGGCCGC TCCAGTTGCT IVR LLN ETSH LAA PVA 750 CGGGGCGTGA GCTCCCAGAT GCTGCGCAAC CAGCTGTCCG CGTCGACCGA RGVS SQM LRN QLSA STD 800 TGCCATGGCC ATGGACACGA CGACGGAGGG CGGCATTCCG GTGGCCATTG AMA MDTT TEG GIP VAlE 850 AGATTGTCCA GGAGATGACG CTGCTGTTCA CAGGCGAGCT AATACCGGTG IVQ EMT LLFT GEL IPV 900 GCGCCCAAGG AACCGGCATG CCCCTGCCAG ATGGTCTCGA TCTGGA(i IF APKE PAC PCQ MVSI WTL 950 GAGTGGATTA ATGCACCGCC ACCGGAAGAT GCGGCAACAG AGTTCATCCT SGL MHRH RKM RQQ SSSS 1000 CGGAGCACGA CAAGGACGCA GCTATTCGGT TAGTGCCACC CGAGGCAGGA EHD KDA AIRL VPP EAG 1050 ACGGGGAGCT GCATAGCGCG AAATTACCGC CCGTTTGGTG ATGCCCACCC TGSC IAR NYR PFGD AHP 1100 GCAGACGGCN CACACCCTCC ATGTCCAGTG GCAGCTCCGT AATGGGTATT QTA HTLH VQW QLR NGYF 1150 TCGATCAATA TTGTCTACTT ACTGATCCGC ATTTCGAGGC ACCACGTCGC DQY CLL TDPH FEA PRR 1200 171 garnet Sequence 10 20 30 40 50 1234567890 1234567890 12467R9 1234SE7RqO 1234567890 CGTG GGCTGC GTACTTTGAA GGTAGTGAGG ATTGTTCGAC TGCTCAATGA RGLR TLK VVR IVRL LNE 1250 GACGAGCCAT CGCCGCTCCA GTTGCTCGGG CGTGAGCTCG AGACTCTGTC TSH RRSS CSG VSS RLCR 1300 GCNCTTTTCG CCGCCATCAG CTCCCGTTCC TGGCCTTTCG TGGCACTAAA XFR RHQ LPFL AFR GTK 1350 CGAATAGCTC GTCCTTGGTC GTGCTCCGAG GATCAACTGC TTGCCGCATC RIAR PWS CSE DQLL AAS 1400 TTCCGGTGGC GGTGCATTAA TCCACTCGTC CAGATCCGAG ACCATCTGGA SGG GALl HSS RSE TIWT 1450 CGGGGCACCT TCCGTTGCCT TGGGGCGCCA CCGGTATTAG CTCGCCTGTG GHL PLP WGAT GIS SPy 1500 AACAGCAGCG TCATCTCCTG GACAATCTCA ATGCGCACGC GGAATGCCGC NSSV ISW TIS MRTR NAA 1550 CCTCCGTCGT CGTGTCCATG GCCATGGCAT CGGTCGACGT GGACAGCTGG LRR RVHG HGI GRR GQLV 1600 TTGCGCAGCA TCTCGATCAG CATGCAAGCG GAATTGGCTC GCTCTTGCAC AQH LDQ HASG IGS LLH 1650 CTCAATGTCG CTGGAACCAT TGAAGTGCTG AAGCTTGTCC AGCACATGGT LNVA GTI EVL KLVQ HMV 1700 CGCAGAGCTA CCAATGATAA TGGTAATACT CAGAATACAA TCATTTGAAA AEL PMIM VIL RIQ SFEN 1750 ATACGTATAG AGAGAGTCAC TTACTGTCAC CAAGCCAGGC AGATCCTGAA TYR ESH LLSP SQA DPE 1800 172 garnet Sequence 10 20 30 40 50 1234567890 1234567890 1234567890 123457R90 1234S7Rø GCTCTAAACA CGTGGTGGCC AGGCGAGCGA ACAGCTTCAT CAGCTTCTGC ALNT WWP GER TASS ASA 1850 ACATAGACAC CTGGATGTGA CCGGGCCAGT AGCTTCGGAC GAGCAGATGT HRH LDVT GPV ASD EQML 1900 TAAGCGTCTC TCGCATCCTC CAGTTCGCCA GCAAACTCGC CAACGATCCA SVS RIL QFAS KLA NDP 1950 GGCGGCGGCG TAGAGCACCT CGTACATGGA ATTACTTTGC GCCGAAACGG GGGV EHL VHG ITLR RNG 2000 TGAACGTGTC AGCATGATTG GTCATCTCGT TGACGGCAAA TTGCCGGACC ERV SMIG HLV DGK LPDH 2050 ACAGGCACTG CAATGGCCNT NGTTNTCGCT ATAGGCGCCG TGCGTGGTNG RHC NGX XXRY RRR AWX 2100 AGCTCGACGA CGACAGTTAG ATACCACTCG AAGTTGGTCA CATACAGATA SSTT TVR YHS KLVT YRY 2150 CGAACTCTGC GCGCAATATC TCGATCACCT TGTAAAGCCA ATTCGTCCCG ELC AQYL DHL VKP IRPD 2200 ATAGGCCGAA CCCTCTGACC CGCTGCCATG TGGCCCAGCA ATCGCTTAAC RPN PLT RCHV AQQ SLN 2250 AATCTCCATA AGGTTCTTCT TCGAGACCAT GCCGTAGAGC AGGTCCAGGG NLHK VLL RDH AVEQ VQG 2300 CGCGCAGACG TATCGATTCG TCCTTGTCGT CCAGGCAGGC GAGTATGAGA AQT YRFV LVV QAG EYEI 2350 TCCTTGTGCG CCTGCACACT CTTCGGGTGC GTCTTCAGGA TTTTCGACAT LVR LHT LRVR LQD FRH 2400 173 garnet Sequence 10 20 30 40 50 1234567890 1234567890 1234567890 1234567890 1234567890 GGCCAACAGT CCCAGATATT TCAAGTTCTG GTCCGCGTCC TCGATGAGGA GQQS QIF QVL VRVL DED 2450 TGCGCAGCTT CTGCACGCAG AGCTGAATGG AGGCACTGTG GTTGGGCATG AQL LHAE LNG GTV VGHA 2500 CCGCTGCTAA TGCTGATAAC GACCGCGATG ACCGTGTTGA TGCACTCATA AAN ADN DRDD RVD ALl 2550 CAGAGACTCA TGGCGAGTGC TGTGATAGTT GGGTTTGGTT TTTGGTTCGT QRLM ASA VIV GFGF WFV 2600 I I iGi iii GT1 I ICATTT TTTGTTGGTG TTCGGTTAGT TTTAGTAATT VFIF CWC SVS FSNF 2650 I I I I TTGTTT [Ti [TI F I TT GTGATTTGTA TTAGTTTGAG AAAAAAAAAA 2700 GTAGGAACAT GTATTAAGAG TAAGCAGCGG GTAAAACGCA CAACATTGCA VGTC IKS KQR VKRT TLH 2750 CTACAACAAT AGCAATAGCA AATTAGGCAA TGGCAACAAC AACGGTAGCA YNN SNSK LGN GNN NGSI 2800 TATCGATAGC ATACTATACT ATACATATAC TACTAGCTAC TTGCGGTACG SIA YYT IHIL LAT CGT 2850 AAGGTAACAA TTAGCGATTA TTGCGATAGA CATTGGGGAA AGAGAACATT KVTI SOY COR HWGK RTL 2900 GCAAAGCAAC GCAGCAATGG CAACCAAAAA GAAAAACGTC ACTAAACAAC QSN AAMA TKK KNV TKQQ 2950 AGCAACAACA ACAACAACTG CAACGGCTCG TCATTTGTTT TTCTTTCGCT QQQ QQL QRLV IC.F SFA 3000 FCF FCF FEE VICI 174 SLR KKK garnet Sequence 50 30 40 20 10 1234567890 1234567890 1234567890 1234567890 1234567890 TCATCCGATT TGACTAGTTT AGAACTTTGG ATCTCAATGA GTGTGCTCGA VLD ELW ISMS TSL SSDL 3050 TAAGCAAAAA ATCGATAGGC AAACGATGAA TTATAGAAAC AAAGACAAAC YRN KDKL TMN IDRQ KQK 3100 TTAAGCAGTA TGGCGACAGT CATAAGTTGA GCGAGTGGGA GAGAGAAAGA RER EWE HKLS GDS KQY 3150 GATAGACAGA GAGAGAGAGA GAGAGAGAGT ACGCTAGAGC TAGAGAATTG TLEL ENC RES ERE DRQR 3200 TACAGTAAAT GATATAACGA ATATATCCAG TCACACGACA ATCATCGAGC HTT IIEQ ISS TVN DITN 3250 AGCTTCAATT ATCGATCATT GATATCGACC TTTTAATCGG TCACTTTCGA HFR LIG DIDL LQL 511 3300 TTTGATTTTT CGAATTTTTT CTTTGCTTTC GCCTTGCTTT GTTGCAATCG CNR ALLC FAF NFF FDFS 3350 TTTTCCACAC ATTCTTGGGA AATCGTATCG TATTTTACAT TTTCAGTTCA ILH FQFS SYR FPH ILGK 3400 GTTCAGTTGA TTTGTATTTG TATTTTTGTT TTGTTTTGTT TTGTTTGTTT CLF VLF YFCF LYL SVD 3450 GTTTTGCAAT GATTTTAAGA CTTGCCTATG AATTAGATTT GTGAGTGGTT VLQ. 3500 CTATTAATTT CTTTCCTAGC CGGGGTTCTA AGGGGGTTAA AGCGCCAAAC 3550 TGCAAATGCA AAGAGAAAAA GAAACAAGCA AGAAATTATA AATTACATAC 3600 175 garnet Sequence 10 20 30 40 50 1234567890 1234567890 1234567890 1234567890 1234567890 AATCGAAATC ATCAACGTCC TTATCCACAA CTAAAACTAG AACTAAAACT 3650 AAAGCTAAAA CCGAAAACGA AACTAGAAAA TGAAGTGTTC AAGAAAATGG 3700 TAAACTGGAA CTGGAATACT CTAAGAAGTA ATTTAACTTT CTCTTAAACT 3750 GGTCCTGGTC CTTTTCCATT CGGATCGAAT CTCTTGATGA TATGTCTATA 3800 TATTTTTGTG TATCTCAGGG TGGAA 3825 176 Comparison between the c-DNA sequence and genomic sequence reveals at least four introns, the most 3’ of which is the site of the P-element insert in the gP mutation. Conceptual translation of the garnet c-DNA. The direction of transcription was determined by in situ hybridization of digoxigenin labeled RNA probes to imaginal discs of third instar larvae. The garnet c-DNA from the Hind Ill site at 1720 and the Sst I site at 2143 was subcloned into pBS KS. Sense and antisense RNA was produced by transcription of this garnet fragment by T3 and T7 polymerase respectively. The T7 transcribed probe detected message whereas the T3 transcribed probe did not. Thus the direction of transcription is as shown in Figure 26. There is a reasonable open reading frame in this orientation, starting at position 297 encoding a potential polypeptide of 1054 amino acids. Conceptual translation of the long open reading frame yields the protein shown in Figure 26. This protein has a number of motifs. There is a poly glutamine stretch (8 repeats) at position 3145. There are three reasonable hydrophobic domains. Comparison between this sequence and the EMBL and Swiss protein data banks reveals no informative similarities (Table 22). The first two sequences show essentially exact correspondence with the garnet gene sequence. The slight discrepancies in sequence can be ascribed to sequencing errors. As they are sequence tagged sites from the European genome mapping project this data will assist in aligning the genetic and molecular maps for the X chromosome but is not otherwise informative. The other sequences show short 177 regions of similarity restricted to repeated sequence motifs. Thus it would appear that if garnet -homologous sequences exist in other organisms, (garnet Table 22. Genes with sequence similarity to garnet. The first column gives the name and accession code of the gene. The second and third columns give the region of sequence similarity, relative to the garnet imaginal c-DNA sequence and the other gene, respectively. The next columns give the percent similarity within this region and an indication if the similarity is restricted to a repeated sequence motif. 178 179 7. Rat Na+,K+ ATPase alpha2 subunit gene and 5’ flank seq. 5833-5925 2222-2319 3429-3531 1728-1 808 3745-3775 51988-51933 584-653&45125-46260 33732-33785 20477-20502 910-977 2834-292 1 2839-2911 3092-3179 2575-2670 3094-3190 2572-2654 3395-3425 3059-3186 2558-2669 3383-3435 3770-3798 3094-3161 2600-2687 2565-2637 79% 65% 67% 70% 70% 70% 79% 75% 65% 63% 75% 65% 78% 85% 76% 74% 1188-1253 3717-3765 3721-3768 4262-4300 3096-3175 3395-3446 2600-2670 3534-3572 95% % similarity 97% 1-221 region of similarity in other gene 363-428&428-468 2-68&1 30-171 2352-2514 region of similarity in garnet 4. C. grisous dhfr origin of replication emb X520341 CGDHFRORI 5. Rat MHC Class I Ag gene RT1-u haplotype gb M64795 RATMHRT 1 6. Mouse beta-globin complex bho,bhl,bl,b2,bh3 and bh3 emb X14061 MMBGCXD 1. Drosophila melanogaster STS emb Z31953 DM189B8T 2. Drosophila melanogaster STS emb Z32301 DM7C5T 3. Rat 5.5kb DNA fragment containing repetitive DNA emb Xl 3424 RN55REP gene Genes with sequence similarity to garnet. GAGA repeat T repeat GUT repeat GAGA repeat T repeats GUT repeat AT repeat GAGA repeat T repeats T repeats GAGA repeat T repeats GAGA repeat T repeats T repeats A repeats non-repeat non-repeat motif 180 10. Rat hepatic steroid hydroxylase 3091-3216 hAl gene 2566-2641 gb M33312 RATCYP2A1 3111-3196 687-75 1 3376-3435&2587-2646 180-258 9. Mouse myoglobin exon 1 and flanking regions emb X04405 MMMYOGG1 17713-17834 4891-4966 577-663&6399-6518 9845-9918 3092-31 88 2583-26667 8. Mouse Hox 3.1, 3.2 genes and intergenic region gb M35603 musHOXMAA 8043-8067 492-516 emb D90049 RNATPA25 70% 67% 79% 65% 73% 70% 84% GAGA repeat GTTT repeat GAGA repeat GUT repeat GAGA repeat T repeats GTT repeat is present in sibling species of Drosophila melanogaster (Sturtevant et al. 1925, SturLevant and Novitski 1941)) these have not been entered into the data bank. Tissue distribution of garnet transcripts. The tissue specificity of garnet transcription was investigated by in situ hybridization to embryos and various organs present in third instar larvae and adults. Figure 27 presents these results. The garnet gene is clearly transcribed in the eye-antennal imaginal disc, the anlage of the adult eye. Interestingly it is also present, in other imaginal discs as well as other tissues such as the larval brain and ovarioles. It is also abundantly expressed in embryos. Thus garnet is expressed in a variety of tissues during at least three stages of development. Do sequences similar to garnet exist in Drosophila melanogaster? Genetic evidence suggests that the garnet gene is one of a group of genes that are functionally redundant. Functionally redundant loci might have sequence similarity. To test this possibility, genomic DNA from wild-type flies was probed with the 6.6 Eco RI fragment under conditions of reduced stringency. No extra bands were seen under conditions where sequences sharing approximately 66% identity should be detected (Figure 28). Thus, there do not seem to be any sequences in the Drosophila melanogaster genome which are highly similar to the garnet gene. 181 Figure 27. Analysis of garnet transcription by in situ hybridization to various tissues. A. The first three images (viewed top to bottom, left to right) show garnet message in blastula, gastrula and neurula stages of Drosophila melanogaster embryos. B. The next two pictures show garnet message in the leg and eye-antennal imaginal discs. C. Below is shown garnet transcription in an ovariole. D. The three pictures on the lower right show the same tissues hybridized with the sense probe (T3 probe) after 90 minutes of staining. E. garnet transcription in the brain and the eye-antennal imaginal disc (for comparison) of wild type third instar larvae. The embryos and the larval brain were stained for ten minutes before the staining reaction was stopped. The imaginal discs and ovarioles were stained for 90 minutes. 182 - a a V 3 Figure 28. Southern analysis of regions of sequence similarity of the garnet gene in Drosophila melanogaster. DNA from wild type flies was isolated, restricted with Kpn I and Xho I (which do not cut within the 6.6 Eco RI fragment which contains the garnet gene), separated on a 0.8% agarose gel, transferred to nylon membrane and probed with the 6.6 Eco RI garnet fragment with reduced stringency. Under the conditions used sequences with approximately two thirds similarity should have been detected. With the exception of a high molecular smear evident in the Xho I lane, likely due to incomplete digestion, no additional bands were seen. The gel was overloaded with DNA and then the filter overexposed to allow detection of any weakly hybridizing bands. 185 981 IL ‘I Iudi 19 Ioq Discussion-garnet Phenotype of the garnet alleles. The garnet alleles affect both pteridine and ommochrome pigments. This is not a phenotype that can easily be explained as a simple enzymatic deficiency. As the two pigment biosynthetic pathways are biochemically distinct, an enzymatic lesion should alter only one group of pigments. In addition there is no evidence for novel pigments, or pigment intermediates, that might be expected to accumulate in a blocked pigment biosynthetic pathway. The garnet mutant alleles alter pigmentation of all of the three major pigmented structures in Drosophila melanogaster, the eye, the malpighian tubules and the testes sheath. Pigmentation of the malpighian tubules is generally more severally altered than that of the testes sheath, which in turn is more severely compromised than that of the eye. This difference could reflect the timing of differentiation, the type or amount of pigment in these organs. 53 allele presents an important exception to this pattern of decreased g The d pigment levels. Although this allele is the most severe mutant allele, and genetically behaves as an amorph, based on dosage tests in the eye, it exhibits essentially wild-type levels of pigmentation in the testes sheath. Interestingly, Tearle (1991) reports that this allele does not have a pronounced effect on pigmentation of the occelli either. The obvious conclusion is that this particular allele is not a typical amorph or hypomorph but has an alteration in tissue specific regulation of garnet expression. Many alleles of the garnet gene have been isolated and described (Table 23). Mutant alleles have been isolated as spontaneous mutations, and induced by treatment with X-rays, gamma rays, 187 Table 23. Published alleles of garnet. The first column gives the allele designation. The second column gives the inducing agent, when known. The third and fourth columns gives the name of the investigator responsible for isolating the allele and the reference. This list was adapted from Lindsley and Zimm (1992). 188 Published garnet alleles allele origin discoverer g 1 2 g ” 2 g 3 g 4 g B 7 gl spontaneous spontaneous unknown spontaneous X-rays X-rays X-rays SMS SMS SMS SMS spontaneous spontaneous spontaneous spontaneous spontaneous spontaneous spontaneous spontaneous spontaneous spontaneous X-rays unknown unknown spontaneous spontaneous X-rays EMS spontaneous spontaneous X-rays X-rays X-rays X-rays spontaneous EMS spontaneous Bridges Bridges 1916 Bridges Lindsley and Zimm 1992 unknown Lindsley and Zimm 1992 Bridges Lindsley and Zimm 1992 Glass Lindsley and Zimm 1992 Valencia Valencia 1966 Sobels Lindsley and Zimm 1992 Sobels Lindsley and Zimm 1992 Sobels Lindsley and Zimm 1992 Sobels Lindsley and Zimm 1992 Sobels Lindsley and Zimm 1992 Wallace Lindsley and Zimm 1992 Bridges Lindsley and Zimm 1992 Emerson Lindsley and Zimm 1992 Bridges Lindsley and Zimm 1992 Ives Lindsley and Zimm 1992 Duncan Lindsley and Zimm 1992 Mossige Lindsley and Zimm 1992 Ecken Lindsley and Zimm 1992 Mather Lindsley and Zimm 1992 Bridges Lindsley and Zimm 1992 Green Lindsley and Zimm 1992 King King 1950 unknown Lindsley and Zimm 1992 Hexter Hexter 1958 Lindsley and Zimm 1992 Williams Lindsley and Zimm 1992 Ives Maddorn Hayman, Madden 1967 Schwinck Schwinck, Schwinckl 972 Najera 1985 Najera Demerec Lindsley and Zimm 1992 Demerec Lindsley and Zimm 1992 Hoover Lindsley and Zimm 1992 Hoover Lindsley and Zimm 1992 Gottschewski Lindsley and Zimm 1992 unknown Lindsley and Zimm 1992 Waddle Lindsley and Zimm 1992 ° 2 g 61 2615 g 2641 g 2810 g 284 g ° g29h Od 3 g d 2 g3 33 g J 33 g ’ 34 g e g37f k 7 g3 g38b 42 g a 9h 4 g Oe 5 g g53d g55k b 4 g6 g68d Ok 7 g ’ 7 g 9 2712 g 2716 g 2719 g 2711 g ° ge gEMS gF 189 reference gim g’ gSl 2 gS gS3 gtUh4 gtUh2 gW gX unknown unknown spontaneous spontaneous spontaneous spontaneous spontaneous spontaneous X-rays unknown unknown Schalet Schalet Schalet Kuhn Kuhn Muller Muller 190 Lindsley and Zimm Lindsley and Zimm Chovnick 1961 Schalet 1986 Schalet 1986 Kuhn 1972 Kuhn 1972 Lindsley and Zimm Lindsley and Zimm 1992 1992 1992 1992 chemical mutagenesis (Table 23) and P-elements (Wennberg, 1988 and this work). It is interesting that of the 56 published alleles and the many P-element derived alleles described in this work, none appear to be true null mutations, Of the over 300 mutants described for the white gene, which has a similarly sized transcript, approximately a third 98/344) are phenotypic nulls. This might suggest that a null mutation of the garnet gene has a phenotype other than reduced eye pigmentation, although the paucity of null garnet mutants may not be beyond the bounds of bad luck. Genetic and molecular limits of the garnet gene: Perhaps the most useful issue of the Chovnick/Hexter debate on the complexity of the garnet locus was a recombination fine structure genetic map of the garnet gene (Figure 29). This map was generated by selecting rare wild type intra allelic recombinants between different garnet alleles. Each of these studies involved visually scoring more than a hundred thousand (Hexter 1958 -583,416, Chovnick 1958-762,429 and Chovnick 1961 - 176,526) flies for rare wild type recombinants in a background of brown-red eye mutants, the phenotypes of some of which approach wild type with age. The value of working on a gene where such extensive work has been performed, by others, cannot be overstated. The intra-allelic genetic map shown in Figure 29 should, in principle, provide a basis for correlating the genetic and molecular limits of the garnet gene. Unfortunately, the gSl allele which defines the right-most limit of the gene is no longer extant. The molecular lesions responsible for the three left-most alleles are not known. They are not associated with deletions or insertions. The g 1 and 3 alleles are however associated with seemingly identical insertions which g 191 Figure 29. Map of the garnet gene generated by intragenic recombination. The top line indicates the genetic limits of the garnet gene. Below is shown the order of six garnet alleles, derived from Chovnick 1958, Hexter 1958 and Chovnick 1961. If the g 3 lesions are in the 3’ region of garnet as 1 and g suggested by the results of Figure 21 then the direction of transcription can be oriented relative to the genetic map as shown. 192 RECOMBINATIONAL MAP OF THEgarnet LOCUS d 5 g 3 I I 2 g Oe 5 g I 1 g 5’ I gSl 3• TELOMERE CENTROMERE 193 have occurred at apparently the same point in the garnet gene. These alleles are genetically inseparable. This restriction fragment is near the 3’ end of the gene and is consistent with their recombination position in the rightmost third of the map. It is also interesting that the 3 leftmost, or possibly 5’, alleles are the most extreme. The g53d allele might be a lesion in the 5’ regulatory region. If this were so it would indicate that the orientation of the garnet gene relative to the centromere is with transcription away from the telomere. Evidence that the cloned region corresponds to the garnet gene: The weak P-element induced gP allele was used to clone the garnet gene. There are three lines of evidence that indicate that the cloned region, specifically the 6.6 kb Eco RI fragment does in fact contain the garnet gene. First, a partial P-element interrupts this fragment in the gP allele. This P-element is altered in both revertants and extreme secondary derivatives of the gP mutation. Sequence analysis of wild type garnet DNA, the equivalent region of the gP mutation and a large c-DNA arising from this region places the P element in a 3’ intron. Secondly, garnet mutations have a high frequency of alterations in this fragment. Of the 14 garnet alleles examined by Southern analysis, 9 had either insertions or deletions in this 6.6 Eco RI fragment. Finally, analysis of transcripts from this region identify two transcripts from wild type flies, both of which are absent in the extreme g53d mutant and one of which is absent and the other possibly altered in the hypomorphic g 3 allele. Thus it seems reasonable to propose that the garnet gene is located within the 6.6 Eco RI fragment and that the c-DNA which comprises approximately two thirds of this region, and corresponds to the only major open reading frame within this DNA segment identifies the garnet gene product. Final proof will, however, require rescue of the garnet mutant phenotype by P-element mediated transformation. 194 Expression pattern of the garnet gene: Tissue in situ analysis indicates that the garnet gene is transcribed at many stages in development including embryos, third instar larvae and adults. It is not highly transcribed at any developmental stage. Pigment deposition in the eye and testes sheath starts two to three days before eclosion, about midway through the pupal stage, and darkening continues up to a week after eclosion (Schultz 1935). Pigmentation of the malpighian tubules can be detected from the first instar larvae onwards (Breme and Demerec 1942). The presence of garnet m-RNA in third instar larvae and young adults is consistent with a role in eye pigmentation. However, the ubiquitous, low levels of garnet transcription in embryos is less obviously related to pigmentation. Interestingly, this pattern of expression is also seen for the light gene (Devlin, Bingham and Wakimoto 1990), another member of the transport group. The tissue distribution of the garnet transcript is also somewhat surprising. As expected the garnet gene can be detected in the eye-antennal disc of third instar larvae. It is, however, also present, albeit at somewhat reduced levels, in the leg and wing imaginal discs, larval brain as well as in ovarioles and embryos. In addition, garnet mutations cause diminished pigmentation of the adult eye, malpighian tubules, fat body, occelli, and testes sheath, suggesting that garnet is also expressed in these tissues. Sequence analysis of the garnet gene. Sequence analysis of the garnet gene has not been particularly revealing. Conceptual translation of the 4kb imaginal disc c-DNA yields a putative protein of 1054 amino acid residues. This putative protein has three indifferent 195 hydrophobic domains and a polyglutamine stretch. Polyglutamine repeats have been found in a number of neurogenic genes but are not necessarily diagnostic. They have also been implicated in parental imprinting (Green 1993). In these instances the polyglutamine repeats tend to be long and are not necessarily either translated or transcribed. Polyglutamine repeats have also been implicated in protein-protein interactions. This function might be related to the proposed interactions between the garnet gene product and those of the other eye colour genes. Function of the garnet gene. The considerable genetic resources devoted to the production, developmental and tissue specific regulation of eye pigments raises the question of their function. The function of the garnet gene product has not been explicitly addressed by any author. Nevertheless, there is extensive genetic evidence, detailed in chapter 1, that the product of the garnet gene interacts with not only the white gene, but with other members of the transport group of eye colour genes. Various investigators have proposed functions for this group of genes. The function of pigments in optically isolating the ommatidia and providing for light adaptation seems incontestable, Interestingly, a similar function in both short and long term light adaptation has been proposed for pteridines found in the mammalian retina (Cremer-Bartels 1975), although the evidence for this is not compelling. Nevertheless, many authors have sought additional roles for the eye colour genes. 196 garnet and cell metabolism: The first conceptual approach to studying the function of eye colour genes derived, appropriately, from attempts to resolve the paradox of the apparently excessive number of eye colour genes. The first, and obvious, attempt to deal with this problem was to group the mutations by similar phenotypes. Nolte (1 954a) proposed that there were in fact only 6 groups of eye colour mutations, the vermilion group, the light group, the dark group, the red group, the variegating group and the ruby group, of which garnet is a member. Detailed studies of the red and brown pigments of these groups (Nolte 1 954b, 1955, 1959) led to the realization that while the vermilion group might be united in generally disrupting ommochrome synthesis (Nolte 1954a), the rest of these groups did not represent biochemically or functionally related genes. Nolte (1 954b, 1 959b) then proposed that the genes of the ruby group represent lesions in general aspects of cell physiology. Specifically, he proposed that they are involved with the protein catabolism that generates precursors for pigment production. This hypothesis is mirrored in various forms in most of the ensuing proposals for the function of the transport group of genes. garnet as a transport gene. Sullivan, Grillo and Kitos (1974) and Sullivan and Sullivan (1975) provided data for a specific variation of Nolte’s hypothesis. They proposed that this group of genes encoded products responsible for metabolite transport. Based on the results of a series of experiments where isolated organs, eye discs and malpighian tubules, were cultured in vitro in labeled kynurenine, an intermediate in the ommochrome pathway, they proposed that many of the eye colour mutants were defective in transport of metabolites and pigment intermediates. 197 The data presented by Sullivan and Sullivan on possible transport defects in eye colour mutations did not include the garnet gene. They did however, propose a list of criteria that would identify a gene primarily concerned with pigment transport. These criteria are: cellular autonomy, effects on both pigment pathways, effects on all of the pigmented organs, and diminished pigment levels in conjunction with normal biosynthetic enzyme activity. Cell autonomy would be expected for a membrane based gene product such as a transmembrane channel protein. Alterations in both ommochrome and pteridine pigments suggests that the transport apparatus handles more than one metabolite or compound. This is not unprecedented (Christensen 1973). One example, possibly quite relevant to the transport of pigments in Drosophila, is provided by the mouse pallid locus. Defects in this gene are associated with diminished transport of tryptophane, L-dopamine and Manganese ions (discussed by Wiley and Forrest 1981). Alterations in the pigmentation of different organs would suggest reasonably ubiquitous use of the transport mechanism. The genes white, brown, scarlet, lightoid, claret, carnation, light, maroon and pink fulfill all of these criteria. The garnet gene fulfills the first three of these criteria; the last has not been fully tested (although Glassmann (1956) reported that the g 2 allele has normal levels of the enzyme kynurenine formanidase). Nevertheless, an alteration in the transport of kynurenine or any other compound remains to be shown for garnet. In retrospect, in light of the complex tissue and developmental interactions, the known excretory function of the malpighian tubules and fat body, the cell autonomous nature of many mutations and importance of transport in cell function, a role in metabolite transport for some of the eye colour genes may seem obvious. Nonetheless, these authors were the first to furnish data for the 198 role of transport mechanism in the final production of wild type eye pigmentation. Finally, it should be noted that transport probably involves the mitochondrial membrane, the pigment granule membrane and possibly the golgi body membrane, as well as the plasma membrane. Defects in this transport may have a variety of consequences, such as alterations in intracellular storage or movement of precursors and excretion of waste compounds, as well as diminished accumulation of pigments. garnet in the brain. More recently, McCarthy and Nickla (1980) have proposed that the genes carnation and light, both members of the transport group, are involved in a variety of (unspecified) functions and have an essential role in the development and function of the nervous system. Their studies were based on extensive genetic and histological examination of double mutant light-carnation individuals. Flies homozygous for either one of these mutations survive, whereas, flies homozygous for both die. The lethal phase of the double mutant is protracted and depends on dosage and activity of light (Nickla, 1977). The synthetic lethal focus maps to the ventral blastoderm, site of the presumptive ventral nervous system (Nickla, Lilly and McCarthy, 1980), and double mutant individuals display abnormal brain morphology (McCarthy and Nickla, 1980). These authors propose that in addition to a role in pigmentation, carnation and light perform an essential function in neural development. The garnet transcription seen in the larval brain, as well as the apparent absence of this transcription in rosy null larvae, might suggest a similar function for the garnet gene in this tissue, however further genetic and histological analysis is required. It should be noted that unlike the carnation-light double mutant, the rosy-garnet double homozygotes are viable, and other than female sterility, display no obvious behavioral or physical defects. 199 The suggestion that groups of eye colour genes represent an essential and redundant function is supported by findings that certain pairwise combinations of these genes behaved as synthetic lethals. Synthetic lethal combinations have been known in Drosophila for some time but are not common and offer a powerful tool to identify functional identity between redundant genes. Although synthetic lethal combinations not involving eye colour genes exist, the representation of not only eye colour mutations but specifically of mutants of the transport group of eye colour genes (Table 2) is intriguing. Notwithstanding, a systematic search for synthetic lethal combinations amongst pairwise combinations of eye colour genes has never been done and is not a task to be undertaken casually; the number of eye colour mutations, even discounting the numerous alleles of most, and the need to examine multiple allele combinations would make this an onerous task. A search for interactions amongst the smaller set of the transport group of eye colour genes might, however, prove revealing. The garnet gene does not display lethal interactions with at least prune, light, rosy or deep orange (other members of the transport group have not been tested). It does however display a full spectrum of other interactions with these genes, including female sterility, synthetic dominance and a variable spectrum of cell death phenotypes. These other types of interactions may suggest specialized roles for the garnet gene. garnet and intracellular transport Very recently the g 2 allele has been identified as an enhancer of the quartet mutation (C. Cheney- personal communication). The quartet mutation is a female sterile mutation which seems to be involved in localization of the nanos posterior determinant in eggs. The abnormal phenotype of the quartet mutation is proposed to result from a defect 200 in intracellular transport. The action of the g 2 allele as an enhancer of this defect might implicate the garnet gene as being involved in intra- as well as intercellular transport. Preliminary results indicate that nanos transcription may g5 homozygote (S. Gorski, d be severely reduced in the female sterile e(g) 3 personal communication). While the females sterile phenotype could be due to a general and non-specific physiological effect, examination of nanos localization in the female sterile garnet and rosy-garnet double mutants may prove to be informative and is being pursued by the Cheney laboratory The metabolic role of some of the pigments and intermediates is an area of intense research. In the earliest work on the chemical nature of these compounds, Schultz (1935) speculated that as they were highly susceptible to oxidation-reduction reactions, their function related somehow to this property. More recently Hilliker et a!. (1992) have observed that mutants for the rosy gene have increased sensitivity to oxygen stress and a reduced life span. This suggests an evolutionary impetus for the development of pigments might stem from an alternative use of metabolic waste products. Metamorphosis is a metabolically active portion of the insect life cycle. In holometabolic insects, during this period, excretion of all but gaseous waste products is restricted. Conversion of toxic by-products of amino acid and nucleic acid catabolism into stable, non-toxic molecules which could be deposited in high concentrations in different organs such as the eye, would be useful. If these products served or enhance some other functions in these organs, there should be considerable evolutionary impetus to develop such a system. While this may be true of a subset of eye colour genes this function is unlikely to be restricted to this class and there is no evidence that mutations in the garnet gene alter longevity. 201 While the suggestion of intercellular transport, cell communication and neural function are certainly compatible, these studies have yet to do more than imply an important, but undefined, redundant biological role for this group of eye colour mutants. Resolution of the biological role of garnet gene will require further study. Testing the model of the function of the garnet gene: In summary, the biological function of the “transport” group of eye colour genes, including garnet remains speculative. Their proposed functions in general cellular metabolism, intercellular transport, intracellular transport and a role in neural formation and function are certainly not mutually exclusive. Genetic analysis suggests that this group of genes possess a ubiquitous essential and redundant, if unknown function. Based on the similarity between the phenotypes of mutants in the transport group of genes and white hypomorphic mutants, similar effects on pigment accumulation and hypersensitivity to the cryptic we(g) allele described in chapter 1, I suggest that all of these gene products associate with the product of the white gene. The unusual epistatic interaction found between the a2 and the g 2 allele, as well as the similar interaction reported for wa3 and ruby might provide an avenue to investigate the physical nature of this interaction. The functional redundancy of the transport group of genes, implied by the pleiotropic genetic interactions compared with their rather weak phenotypes as single loci mutations, suggests that they may coordinately perform some essential function(s). In contrast, the phenotype of other combinations, such as light and carnation, suggest that they may perform other more specialized functions. The absence of synthetic lethal combinations involving the garnet gene suggests that this gene may operate only with many other gene products. However, the female sterile phenotype of the double mutant rosy-garnet combination 202 intimates that this pair of genes might have a more unique role in the female germ line. One may envision a complex involved generally in aspects of transmembrane transport, various members of this complex associating in different cell types, in different subcellular compartments, and at different stages of development to perform variations of this function. These proposed functions remain completely speculative and await further investigation. The cloning of the garnet gene should lead to further definition of the biological role of not only the garnet gene but possibly also that of the “transport” group of eye colour mutations. Direct physical proof of the existence of a macromolecular complex which regulates a transmembrane pore as proposed above, must await direct analysis of the garnet gene product. Antibodies to the garnet gene product could be used to examine the subcellular location of the garnet gene product. Co-localization and co-immunoprecipitation with anti-garnet and anti- white antibodies or use of the yeast dihybrid selection system would offer a direct means of ascertaining if the genetic interaction between these genes was mirrored by a structural association. In the interim, the genetic and molecular analysis of the garnet gene makes this gene a useful tool to investigate other phenomenon. In the next chapter, I describe a system where the garnet gene is used to study genomic imprinting in Drosophila melanogaster. Genomic imprinting has recently attracted attention by the association between imprinting and some human genetic syndromes but it has been described in insects and is a well defined phenomenon the sciarids and coccids. In the final chapter I describe a mini-chromosome which is imprinted. The imprinting is manifest as parent dependent expression of the garnet gene. The ease of examining eye colour to monitor imprinting as well as the sophisticated genetics of Drosophila 203 melanogaster have allowed tests of a number of possible mechanisms of imprinting. The garnet gene has a long history of being used as a tool to examine other interesting biological phenomenon. The next chapter continues in this tradition. 204 Chapter 3. Imprinting of a mini-chromosome in Drosophila melanogaster 205 Introduction-imprinting of a mini-chromosome in Drosophila melanogaster 1 encompasses a number of processes The phenomenon of genomic imprinting whereby a gene or a region of a chromosome is reversibly modified so that it retains a “memory” of its genetic history. The term imprinting was originally coined by Crouse (1960) to refer to the complex behavior of the X-chromosome in the dipteran insect Sciara. She defined imprinting as the “differential behavior of the members of a pair of homologous chromosome which is predetermined several to many cell generations before the stage in development at which resulting behavioral differences become obvious.” This definition is fairly broad. As a result, the term imprinting has been applied to a vast number of exceptions to normal Mendelian segregation of traits. Conversely, for historical and traditional reasons, phenomena, that would be defined as imprinting by contemporary criteria, have been given a variety of other names. Table 24 provides a list of these terms, the organism with which they have been used and the probable type of imprinting. A working definition of imprinting has been proposed by Reik (1992) as a process whereby “epigenetic information is introduced into chromosomes and is stabley replicated together with the chromosomes as cells divide”. But even this definition is sufficiently broad that it encompasses a number of biological oddities which are undoubtedly mechanistically distinct. This problem was first addressed by Monk (1990) who proposed subgroups to encompass four general classes of phenomenon which have been collectively called genomic imprinting. These include species-specific imprinting, differentiation, epimutation and parental imprinting. 1 The term “imprinting” properly refers to a phenomenon in behavioral psychology whereby an immature animal learns appropriate behavior from adults. For the purposes of this thesis it will be understood that the term imprinting refers to the phenomenon of genomic imprinting. 206 Table 24. Terms used for genomic imprinting. The first column gives the term used to describe the phenomenon of genomic imprinting. The second column gives the general group of organisms with which this term has been used and the third gives the type of imprinting as defined by Monk (1990). With the exception of the phrase “Non-Mendelian ratios” (Hall 1990) and parental effects (Baker 1963) all these terms are explained in greater detail in Heslop Harrison (1990). 207 Other terms used for genomic imprinting Name of phenomenon organism group Parental effects Insects (Drosophila) type of imprinting parental Block transference of characters plants species Genetic affinity plants species Suppression plants species Selectivity of expression plants species Cryptic structural differentiation plants species Skewed back cross ratios plants species Homeosis plants species Character pseudo-linkage plants species Non-Mendelian ratios humans parental 208 Strain or species specific imprinting The term imprinting has been used to refer to the variable phenotypes of the hybrids resulting from crosses between different subspecies or strains. This type of imprinting has been called species or strain-specific imprinting (Monk 1990) as the hybrid phenotype appears influenced by a “memory” of the parental species. The difference between a mule (horse mother, donkey father) and a hinney (donkey mother, horse father) is the classical example of this type of imprinting. This type of imprinting likely reflects the preferential action of maternally deposited activators (Castro-Sierra and Ohno 1968) or repressors (Schmidtke, KuhI and Engel 1976) on the subtly different regulatory regions of the genes of the two subspecies or strains. As such, speciesspecific imprinting is dependent of different information encoded in the DNA of the two species and is not an epigenetic process. Somatic imprinting or differentiation. The term imprinting has also been used to refer to the processes of determination and differentiation whereby the developmental potential of a mitotic clone is restricted (Paro 1990, KIar 1987, 1990). From both a mechanistic and theoretical point of view the processes involved in somatic versus germ line “memory” are expected to differ. Mechanistically, the packaging of DNA is grossly different between the germ line and soma. More importantly, meiotic products must remain totipotent in order to produce the complete spectrum of cell types present in the next generation. Somatic cells do not face such demands so that loss of totipotence implied by parental imprinting posses no conceptual difficulties. While determination remains a central question in developmental biology, renaming this process “imprinting” adds nothing to our understanding of the processes involved. 209 Permanent imprinting. The term imprinting has also been used to describe parent-specific, permanent changes in gene activity. This phenomenon is manifest as a permanent alteration in gene activity after passage though one parent or genetic background (Hadchouel et al 1987, Reuter 1985, Dorn et al. 1993) and has been called epimutation by Holliday (1987) and allele-specific imprinting by Monk (1990). The mechanism where by a permanent alteration is produced in one parent and then maintained remains somewhat obscure. In the situation described by Hadchouel et al (1987) the gene inactivation was associated with methylation of multiple CpG islands of the hepatitis B surface antigen transgene, but only after passage through the female germ line. These investigators proposed that if methylation was reversed at only a low frequency in males, possibly due to chromatin remodeling in spermatogenesis, then the extensive methylation in females would constitute a virtually permanent alteration. The mechanism responsible for this phenomenon in Drosophila remains unknown although it has been implicitly associated with changes in chromatin structure. Parental imprinting In parental imprinting the activity of the imprinted gene is determined by the sex of the parent transmitting that gene. This type of imprinting has also been termed gamete specific imprinting (Monk 1990). Parental imprinting is distinguished by its transient nature, specifically, the imprint is completely reversed in one generation by passage through the other sex. The result of parental imprinting is the functional nonequivalence of the maternal and paternal genome. In mammals the consequence of this non-equivalence is drastic. Embryos (either parthenogenic, gynogenic or androgenic) or tissues (such as ovarian tumours and 210 Table 25. Human diseases in which imprinting has been implicated. The first column gives the name of the disease or condition in which imprinting has been implicated. The second column gives the reference. 211 Human diseases and conditions in which imprinting has been implicated Condition or syndrome Reference Angelman/Prader Willis syndrome Nicholls et al 1989/Hall 1990 Carmillia De Lange syndrome Clarke 1990/Hall 1990 Duchenne muscular dystrophy Hall 1990 Familial glomus tumours Clarke 1990 Floating harbour syndrome Clarke 1990 Fragile X mental retardation Laird 1987 Huntington’s chorea Laird 1990/Sapienza 1990 Hydatidiform moles Kajii and Ohama 1977 Myotonic dystrophy Clarke 1990/Hall 1990 Narcolepsy Hall 1990 Neurofibromatosis Clarke 1990 Osteogenic sarcoma Toguchida et al. 1989 Ovarian tumours Linder et al 1975 Philadelphia chromosome Haas, Argyriou and Lion 1992 Retinoblastoma Clarke 1990 Rhabdomyosa rcoma Dryja et al 1989 Rubinstein-Taybi syndrome Clarke 1990/Hall 1990 Russel-Silver dwarfism Hall 1990 Sotos syndrome Clarke 1990 Weaver syndrome Clarke 1990 Wiedemann-Beckman syndrome Hall 1990 Wilm’s tumour Clarke 1990 Wolf-Hirschor syndrome Clarke 1990 212 hydratidiform moles) that are derived from two complete maternal or paternal genomes are not viable even though they have the full complement of genetic information. This lethality is thought to be due to the cumulative effect of many imprinted genes, at least some of which are growth regulators acting early in development. Parental imprinting has been most thoroughly described in mammals and is currently being intensely investigated. Much of the impetus for these investigations comes from the implication of imprinting as the underlying cause of a number of human syndromes. Table 25 lists diseases and medical conditions which are associated with either aberrant imprinting or the aberrant transmission of imprinted regions. The evidence for involvement of imprinting in these conditions is well defined in some cases (e.g. fragile X mental retardation) and considerably more inferential in others. Parent-dependent gene expression or parental imprinting is, however, found in many other eukaryotes, including plants (Kermicle and Alleman 1990), C. elegans (Gilchrist and Moerman 1992), and a variety of insects including the Homopteran Coccids, mealy bugs and other amoured scale insects (Chandra and Brown 1975), the Hymenopteran wasp, Nasonia vitripennis (Nur et aL 1988), the Coleopteran coffee berry borer beetle, Hypothenemus hampei (Brun et a!. 1995), and the Dipterans, the fungus gnat Sciara (Crouse 1960) and the genetically well characterized fruit fly Drosophila melanogaster (see below). The consequence of parental imprinting is far less drastic in these groups of organisms. For example, gynogenic Drosophila melanogaster are completely viable and fertile (Fuyama 1984) as are, apparently androgenic flies (Muller 1958). Parental imprinting in Drosophila: Imprinting phenomena have been recognized and studied in Drosophila, albeit under the name of parental effects, for more than 50 years. All of the reported parental 213 Table 26. Imprinting (parental effects) in Drosophila. Six examples of parental effects published for Drosophila are shown. The first column give the direction of the imprint. Following the terminology of Reik (1992) the parent transmitting the inactivated allele or chromosome is indicated. The second and third columns give the name of the rearrangement which shows the parental effect and the reference. The last example occurs in D. hyde!, all the others are found in D. melanogaster. 214 Imprinting (parental effects) in Drosophila Direction of imprint Chromosome Reference Paternal 7 T(1;2)dorvar Demakova and Belyaeva 1988 Paternal Dp(1;4)wm254.58aBaker and Spofford 1959 Spofford 1959 Hesser 1961 Spofford 1961 Baker 1963 Maternal Dp(1;3)wVCQ Maternal Khesin and Bashkirov 1978 Khesin and Bashkirov 1978 Paternal In(1)sc and 8 Dp(1;f)1187 Prokofyeva-Belgovskaya 1947 Karpen and Spradling 1990 Paternal Dp(1;f)LJ9 this work Maternal In(1)wm2 Hess 1970 215 (imprinting) effects (Table 26) involve chromosome rearrangements that exhibit position effect variegation. Position effect variegation is a process whereby a fully functional gene becomes inactivated due to its relocation adjacent to a broken segment of constitutive (Spofford 1976) or facultative (Cattanach 1970) heterochromatin. As the gene inactivation is correlated with the adoption of heterochromatic morphology in the appropriate section of the salivary gland chromosomes (Hartmann-Goldstein 1967), it is thought that the genetic inactivation is due to the spread of heterochromatin which packages and condenses the normally euchromatic region in such a way that necessary transcription factors cannot access the gene. The result is variable genetic inactivity. Imprinting as seen in Drosophila shows many intriguing similarities to the imprinting described in mammals. The key features of genomic imprinting have been defined by Metz (1938), based on his work with the Dipteran insect Sciara, but are true of imprinting phenomenon in all organisms. The primary criterion of imprinting is that the process affects genetically identical DNA, and is thus a strictly epigenetic phenomenon. The second criterion is that the imprint persists for only one generation. Thus the imprint is reversed by passage through meiosis. Genetically this is evidenced by parental effects but no grandparental effects. This latter feature may be a conceptual definition rather than a genuine mechanistic distinction. Long term effects such as seen in epimutation (Darn et al 1993), or medium term effects with diminishing grandparental effect (such as seen with wmVC0; Khesin and Bashkirov 1978) may be merely mechanistic variations on a theme. The third criterion for parental imprinting is that the imprint is mitotically stable. This stability generates functionally distinct clonal regions. These clones are readily apparent in Figure 32 and have been noted as intrinsic features of imprinting in mice (Allen et al. 1988), maize (Kermicle and Aliman 1990) and insects (Nur 1990). The clonal nature of the gene expression results in cell 216 to cell variability within a tissue. This variability suggests a stoichiometric process operates in imprinting and may provide a clue to the mechanism responsible for imprinting. A fourth criterion is the physical continuity and extent of the imprint. The imprinted region frequently affects entire regions of a chromosome and may encompasses more than one gene. Such an effect is self-evident in coccids where the imprint encompasses an entire chromosome (Nur 1970) but is also seen in mammals. For example, the closely linked genes thought to be involved in the pathology of Prader-Willis syndrome, SNRPN, SNF127, PAR-i and PAR-5, are coordinately transcribed, replicated and maternally imprinted (Gunaratne et al. 1995). The neighboring genes H19 and !GF2 (Reik 1992) are also imprinted, although in this case in opposite directions. Cohen (1962) found that the closely linked genes white, split, notchiod, facet and roughest were all maternally imprinted in the wm254.58a rearrangement. Imprinting of contiguous genes is also seen with the mini-chromosome examined here, the garnet gene and the nearby genes narrow abdomen and tiny (Figure 35) are paternally imprinted. It should be noted, however, that most examples of parental imprinting in mammals have been reported for isolated genes. The relative rarity of mammalian imprints which encompass more than one gene may reflect the lower resolution of genetic studies of the imprint in mammals or possibly the greater genome size of mammals as opposed to Drosophila melanogaster which might act to limit a domain to one gene. Finally, the formation of aberrant chromatin structures appears to be the most universal aspect of imprinting. Heterochromatin formation is likely responsible for the altered gene expression seen in position effect variegation which is involved in all of the reported cases of imprinted effects in Drosophila. Likewise imprinting seen in Coccids and Sciara (Chandra and Brown 1975) clearly involve large segments of cytologically visible heterochromatin. Cytological studies of the imprinted heterochromatic chromosomes of coccids have shown that the imprint can spread and can be somewhat variable (Nur 1970). Heterochromatin is also clearly 217 involved in mammalian X-chromosome inactivation (Lyon 1993) which is imprinted in both marsupials and some tissues of eutherian mammals. Paramutation (an epigenetic phenomenon probably closely related to imprinting) seems to be associated with altered chromatin structure in maize (Patterson, Thorpe and Chandler 1993). Aberrant chromatin structure has been proposed as the causal feature leading to mis-expression of the fmrl gene responsible for fragile X mental retardation (Laird 1987) and a number of other recent studies have implicated chromatin binding proteins in the establishment of the imprint in mice (Sasaki et al 1992, Bartolomei et al 1993, Brandeis et al 1993, Ferguson-Smith et al 1993, Stoger et al 1993, Chaillet et al. 1995) and humans (Monk 1988, Barlow 1994, Varmusa and Mann 1994). This chapter describes a mini-chromosome which shows parent specific imprinting. The imprint is manifest as clonally repressed expression of the wild type garnet gene when the mini-chromosome is inherited from the father. The expression of at least two other genes on this mini-chromosome is also imprinted. The immediate cause of the repressed garnet expression is position effect variegation. This variegation is unconventional in that it is strictly dependent on the sex of the transmitting parent. Thus it would appear that chromatin packaging in this mini-chromosome is imprinted. Using the cloned garnet gene sequence to analyze this mini-chromosome I have tested and eliminated a number of factors which might cause the imprint. To assess the role of heterochromatin in the imprinting process, I have tested the stability of the imprint in this region using chemical, environmental and genetic modifiers of position effect variegation which are thought to alter heterochromatic formation. These modifiers of heterochromatin formation and integrity alter the expression of the imprint but not the initial decision of whether or not to imprint. This implies that while heterochromatin is involved in the maintenance and somatic expression of the imprint, 218 it probably does not establish the imprint. Thus the imprinting decision may be under independent genetic control. 219 RESULTS-imprinting The Dp(1;f)LJ9 mini-chromosome. Origin: The Dp(1;f)LJ9 mini-chromosome is derived from the In(1)sc 29 chromosome. The 29 chromosome is an inversion between the tip of the X chromosome (1 B) and In(1)sc the region adjacent to the garnet gene (13A2-5) which places this region near the tip of the X chromosome. The Dp(1;f)LJ9 mini-chromosome was induced by X-ray treatment and is a deletion of most the X chromosome euchromatin from In(1)sc 29 (Hardy et al 1984). Only the euchromatic bands from 12A10 to 13A2 and the distal tip, 1A1 to 1 B remain. This region is appended to the centric heterochromatin of the X chromosome. The general structure and origin of the Dp(1;f)LJ9 mini-chromosome is diagrammed in Figure 30. Although not mentioned by Hardy eta!, (1984) it may be assumed that the heterochromatin was broken by the X-ray treatment as the most proximal euchromatic gene (su(f)) is missing from the mini-chromosome and variegation for a number of genes adjacent to the heterochromatin is observed (see below). Mitotic and meiotic stability. As the centric heterochromatin (or a region within) is involved in the normal segregation of chromosomes, I tested the stability of the mini-chromosome in both meiotic and mitotic cell divisions (Figure 31). The mini-chromosome shows a very low rate of non-disjunction from males (0.1-0.3%, n=1 546). This rate is comparable to that observed for a standard X chromosome (Bridges 1916). The rate of non 220 Figure 30. Diagram of the structure and origin of the Dp(1,i)LJ9 mini-chromosome. The top figure diagrams the wild type chromosome. The middle figure shows the structure of the In(1)sc 29 chromosome and the lowest figure shows the structure of the Dp(1;f)LJ9 mini-chromosome. The relative positions of the narrow abdomen, tiny and garnet genes are shown. 221 Structure and origin of the Dp(1:f)LJ9 mini-chromosome WILD TYPE X CHROMOSOME \F/ 1B In(1)sc 1 B/i 3A2 tAO I 2b ic/i 3B 29 X RAYS n&tyg 1B113A2 Ti 12A10 THE MINI-CHROMSOSOME Dp(1 ;f)LJ9 222 Figure 31. Meiotic stability of the Dp(1;f)LJ9 mini-chromosome. The top portion of the figure diagrams the production of regular and non-disjunction gametes from the attached-X and duplication bearing females (X”X/Dp(1;f)LJ9). The frequency with which the various genotypes arise is shown in the Punnit square. The total number of flies scored for the maternal cross was 745. Cross: X’X/Dp(1;f)LJ9 0 d 3 g5 /y y za d/Dp(1,.f)LJ9 53 g 0 y za & a’ The lower portion of the figure diagrams the production of regular and non-disjunction gametes from the attached XY duplication bearing male (XY/Dp(1;f)LJ9). The frequency with which the various genotypes arise is shown in the Punnit square. The frequency of the y za g53d/Q genotype arising from the paternal cross is given as a range because all of the five individuals of this genotype arose from one vial containing only three females. Thus this genotype may represent a pre-meiotic loss of the mini-chromosome. The total number of flies scored for the paternal cross was 1546. Cross: X’Y/Dp(1;f)LJ9 a’ ® d/y 5 3 za g53d y za g 223 53 g y za d/Dp(1,’f)LJ9c/ DISJUNCTION IN FEMALES regular gametes 53d rn Q37 y Dp 0.47 RIP CROSS: XIDp(1;f)LJ9 non-disjunction gametes 0 Yô(+Op tRIP 0.10 006 RIP y 4 53di y DISJUNCTION IN MALES )t’ 53d 0.56 regular gametes Dp 0.44 CROSS: )&/Dp(1 ;f)LJ9 x 224 non-disjunction gametes fv+Dp 0 0 0.0010.003 diyz yz 53 disjunction from females is 16% (n=745) which presumably reflects the inability of the mini-chromosome to pair and recombine with the full length attached X-chromosomes in the female. Mitotic non-disjunction was monitored by looking for mosaic patches of mutant yellow tissue in a fly bearing a mutant yellow gene on its normal X 53 za g g d/Dp(1;f)L.j9, 5 3 y or y za d/y d/Dp(1;f)LJ9, 5 3 y) as the chromosome (y za g mini-chromosome carries the wild-type allele of yellow. Only 2 instances of mosaicism were seen in over 10,000 flies suggesting that mitotic non-disjunction does not happen at an appreciable rate (interesting but probably not relevant is the fact that these two mosaics were perfect bilateral gynandromorphs and came from the same parent). Thus it would appear that the mini-chromosome is transmitted faithfully through both meiosis and mitosis suggesting that the bulk of the centric heterochromatin, telomere and any other functions necessary for normal disjunction are not compromised. Imprinted variegation of the mini-chromosome. The variegation is dependent on the sex of the transmitting parent: When females carrying the mini-chromosome (XX/Dp(1;f)LJ9) are crossed to yza d/y males, the male progeny (y za d/Dp(1;f)LJ9) 3 g5 53 appeared wild type. This is the g expected phenotype as the mini-chromosome carries the wild type genes for yellow and garnet (the za allele has no independent phenotype and serves only to lighten the garnet mutant phenotype in the background). In contrast, when the males carrying the mini-chromosome were crossed to females of the same y za g53d strain, the wild-type garnet gene on the mini-chromosome showed variegated expression in the d/Dp(1,i)LJ9) 5 3 sons (Figure 32). In most cases the genotypically identical y za g variegation was expressed as no, one, two or three large wild type spots (garnet+) on 225 Figure 32. The garnet phenotype in flies with paternally or maternally inherited Dp (1;f)LJ9 mini-chromosomes. 53 The g A. The top figure shows two male flies of identical genotype; yza d/Dp(1;f)LJ9. fly on the left, with variegated eyes, has a paternally transmitted mini-chromosome whereas the fly on the right bears a maternally transmitted mini-chromosome. g53d/y y za d/Dp(1;f)LJ9 1 53 g ® y za 0 Maternal cross: X”X/Dp(1;f)LJ9 Paternal cross: X’Y/Dp(1;f)LJ9 ‘ 53 za g53d > g ® y za d/y 53 g y za d/Dp(1;f)LJ9f d/y 5 3 za B. The lower figure shows two female flies of identical genotype; y za g 53 The fly on the left, with variegated eyes, has a paternally transmitted g d/Dp(1;f)LJ9. mini-chromosome whereas the fly on the right bears a maternally transmitted minichromosome. 53 za d/Dp(1;f)LJ9 g 53 g Maternal cross: y za d/y a 53 za g5 g dj 3 y za d/y ‘I, d/y 5 3 za d/Dp(1,.f)LJ9 53 g y za g 53 za g5 g d 3 Paternal cross: y za d/y 53 g ® y za d/y/Dp(1;f)LJ9 1 d/y 5 3 za d/Dp(1;f)LJ9 53 g y za g In all cases the genotypes being compared differ only in the parental origin of the mini chromosome. They are otherwise genotypically identical and isogenic. The yellow mutation was used to monitor the presence of the mini-chromosome without bias as to eye phenotype. The zestea allele was used to lighten the background garnet eye 226 d allele. In the absence of wild type garnet expression from the mini3 colour of the g5 d 3 chromosome the background eye colour is a pale orange due to the za and g5 mutations. In cells in which the garnet+ gene on the mini-chromosome is expressed the eye colour is wild type. In most of the following experiments males of yza 53 were used to monitor the imprint as they were the genotype that arose g d/Dp(1;f)LJ9 from both standard maternal and paternal crosses. These phenotypes persist for one generation only. For example, both the females shown in part B will transmit non variegating mini-chromosomes regardless of whether they themselves show variegation for the garnet gene. 227 N the pale orange background colour. These spots appeared to correspond to clonally determined regions of the eye (Jannings 1970). This mosaic expression was probably not due to mitotic loss of the mini-chromosome in the eye since the mini-chromosome is mitotically stable in the rest of the fly. This imprinted expression is seen in both male and female progeny (Figure 32) and persists for only one generation (Table 35). To summarize, genotypically identical progeny, produced by reciprocal crosses and thus differing only in the parental origin of the sex chromosomes and the minichromosome, show very different phenotypes. When the mini-chromosome is derived from the male parent the garnet gene variegates extensively. In contrast, genetically identical offspring resulting from the reciprocal cross in which the mini-chromosome is derived from the female parent, showed no or very weak variegation (Figure 32). Thus the parental origin determines the extent of variegation observed in the genotypically identical progeny. Therefore the expression of the wild type garnet gene is dependent on the sex of the parent transmitting the mini-chromosome. This situation constitutes a classical example of genomic imprinting. The many parallels between this example of imprinting and that seen in coccids and mammals will be discussed below. Tissue specificity of the imprint: This parent-dependent expression is not limited to the eye. Examination of malpighian tubules in individuals bearing maternally versus paternally derived mini-chromosomes demonstrate that the garnet gene expression in this tissue also variegated extensively when the mini-chromosome is transmitted by the father (Figure 33). Malpighian tubules from individuals in which the mini-chromosome is maternally derived show only occasional unpigmented spots. Testes sheaths from individuals of these same genotypes showed a uniform wild type pigmentation as expected since the g53d allele 229 Figure 33. garnet phenotype in malpighian tubules bearing maternally and paternally derived Dp(1;f)LJ9 mini-chromosome. The top figure shows a typical malpighian tubule from a male of the genotype yza 3 where the mini-chromosome was inherited from the father. The d/Dp(1;f)LJgf. 5 g dark areas are individual pigmented cells. The clear areas are regions in which no pigment is produced. The lower figure shows a typical malpighian tubule from a male of the identical 3 In this instance the mini-chromosome was inherited d/Dp(1;f)LJ9. 5 genotype y za g from the mother. Only one unpigmented region is seen. Frequently there are none although up to three unpigmented regions have been found within one tubule from individuals of this genotype. Wild type malpighian tubules are always uniformly pigmented, unless damaged in removal (data not shown). Crosses: g53d/Y & ...). y za d/Dp(1;f)LJ9f 53 g ® y za 0 Maternal X’X/Dp(1;f)LJ9 Paternal: XY/Dp(1;f)LJ9 ‘ d/y 5 3 za g53d —y za d/Dp(1,.f)LJ9f 53 g ® y za g 230 I—. N) () 4- •1 4 I does not affect testes sheath pigmentation. Imprinted expression of narrow abdomen and tiny To determine if the parental effect is peculiar to the garnet gene or is due to some general feature of the mini-chromosome, I tested two other, closely linked, cell autonomous genes narrow abdomen (na) and tiny (ty) to determine if the expression of these genes is also dependent on parental origin. When the mini-chromosome was inherited from the father, the wild type gene on the mini-chromosome showed variable hypomorphic expression (Figure 34 and 35) in males mutant for narrow abdomen or tiny on the standard X chromosome (ty/Dp(1;f)LJ9 or na/Dp(1;f)LJ9). In contrast, when the mini-chromosome was maternally inherited, the genotypically identical progeny were generally wild type. Thus the imprinting effect seems to encompass at least three genes in the small euchromatic portion of the mini-chromosome. The “imprinting” effect for all these genes was paternal. Interestingly, the severity of the imprint, as assessed by the difference between gene expression when either maternally or paternally inherited, decreased with distance from the centromere and the centric heterochromatin. Imprinting and position effect variegation: The clonal phenotype of the garnet variegated expression is distinctly reminiscent of position effect variegation. The variable hypomorphic expression of the genes tiny and narrow abdomen is also indicative of position-effect variegation. As it is probable that the centric heterochromatin was broken when the mini-chromosome was generated I tested several different types of modifiers of position effect variegation to determine if they modified the mosaic expression of the garnet gene. They did, (see below) suggesting that the mosaic expression of the garnet, narrow abdomen and tiny genes 232 Figure 34. Phenotype of narrow abdomen and tiny in flies bearing maternally or paternally derived Dp(1;f)LJ9 mini-chromosomes. The top portion of the figure shows two genetically identical flies bearing a mutation for the narrow abdomen (na) gene on the standard X-chromosome and the wild type allele of narrow abdomen on the mini-chromosome (na/Dp(1;f)LJ9). The individual on the left bears a maternally derived mini-chromosome whereas the genotypically identical fly on the right has a paternally derived mini-chromosome. Arrows point to the abdomen which is considerably more extended (more mutant) in the flies bearing a paternally derived mini-chromosome. Crosses: Maternal X’X/Dp(1;f)LJ9 ® na/Ye’—> na/Dp(1;f)LJ9cI Paternal: XY/Dp(1;f)LJ9 010 na/In(1)d149 —na/Dp(1;f)LJ9o’ The lower figure shows thoracic bristles from flies with a mutation for the tiny (ty) gene on the regular X-chromosome and the wild type tiny gene on the mini-chromosome (ty/Dp(1;f)LJ9). The individual on the left bears a maternally derived mini-chromosome. The genotypically identical fly on the right bears a paternally derived mini chromosome. Arrows point to the thoracic bristles which are smaller and finer (more mutant) in the flies bearing a paternally derived mini-chromosome. Crosses: Maternal X’X/Dp(1;f)LJ9 0 g 2 ty/Yo’—> na/Dp(1;f,)LJ9o’ Paternal: X’Y/Dp(1;f)LJ9 010 g 2 ty/In(1)d149 .-÷na/Dp(1;f)LJ9o’ 233 nalDp(1;f)LJ9 PAT MAT tyiDp(1,f) LJ9 MAT PAT 234 Figure 35. Expression of garnet, narrow abdomen and tiny in genotypically identical flies bearing either paternally or maternally derived Dp(1;f)LJ9 mini-chromosome. This figure shows the level of variegation for three closely linked wild type genes, garnet (position 44.4), narrow abdomen (position 44.5) and tiny (position 45.2), on the Dp(1;f)LJ9 mini-chromosome. The level of expression of these genes was assessed visually as follows: The level of expression of each of these genes was visually estimated. Each fly, or eye in the case of garnet expression, was assigned a score of 0, 1/2 or 1 for extreme mutant, hypomorphic and wild type expression respectively. These values were averaged and this value is presented with the calculated standard error of the mean. A fly was scored as tiny if its bristles appeared severely minute. In practice this meant approximately half the length of regular bristles on the sibs. A fly was scored as narrow abdomen if the abdomen appeared exceptionally long and thin. A fly was scored as a mutant for garnet if its eyes were completely or extensively variegated. As the scoring is somewhat subjective the crosses were scored by using flies grown on the same set of media at the same time and the crosses were scored blind. 235 Parent-dependent expression ofnarrow abdomen. tiny jjsgarnet EXPRESSION OF GENES ON THE MINI-CHROMOSOME mini-chromosome derived from mother mini-chromosome derived from father narrow abdomen (na) tiny garnet (ty) (g) 0.67±0.02 0.52±0.02 0.86±0.02 0.51 ±0.02 0.24±0.02 0.06 ±0.02 236 transmitting parent, the immediate cause of the imprinting of the garnet, narrow abdomen and tiny genes is imprinted position effect variegation. The possible causes of this imprinted variegation are explored below. Possible causes of the imprinting effect. There are a number of possible causes of imprinting. Obviously the mechanism of imprinting must be established on the haploid genome and thus rely on some feature of meiosis or gametogenesis that differs between females and males or on the subsequent fates of the gametes until fusion of the pronuclei. I directly tested the involvement of a number of factors in the establishment and maintenance of the imprint. The following possible causes of the imprinting effect were tested: 1. The Y chromosome 2. Allele specific interactions 3. Maternal effect of the imprinted gene 4. Maternally transmitted modifiers of variegation 5. Unusual euchromatic sequence or DNA structure near the garnet gene 6. Special heterochromatic features Details of experiments designed to test these possibilities are provided below. 1. The Y chromosome. Imprinting is clearly associated with the sex of the parent from which the imprinted chromosome is derived. Thus, imprinting must be a consequence of either physiological or genotypic differences between the parents. The only geneotypic difference between the parents is the presence of a Y chromosome in males in place 237 of one of the X chromosomes. The Y chromosome is a large heterochromatic body, known to affect many processes such as chromosome pairing, expression of heterochromatic genes and position effect variegation. Therefore the parental effect might be due to a direct interaction between the Y chromosome and the minichromosome. If this were the case, the mini-chromosome should be imprinted when transmitted by XXY females. This hypothesis was tested by comparing variegation of genotypically identical XIDp progeny from XXY and standard XX females. The phenotype of the progeny of these crosses was identical (Figure 36). Thus the minichromosome responds to the sex of the parent rather than the presence or absence of a Y chromosome. 2. Allele specific interactions. Imprinting of the mini-chromosome could be a consequence of an interaction between g5 allele on the d the wild type allele of garnet on the mini-chromosome and the 3 regular X chromosome. If this were the case, a parental imprinting effect would not be seen with other garnet alleles. To determine if the imprinting effect was allele specific, the variegation of the mini-chromosome was examined in strains heterozygous for the wild type garnet gene on the mini-chromosome and eight different garnet alleles on the regular X chromosome. In every case variegation was evident when the minichromosome was introduced paternally but not when introduced maternally (Table 27). This suggests that the imprinting effect is not due to some combination of the garnet alleles. This result and the imprinting of the nearby narrow abdomen and tiny genes also argue against specific allelic interaction as the cause of the imprint. 3. Maternal effect of the garnet gene. It is possible that the differential expression of the garnet gene (and the other two genes) results from maternal rescue of the mutant phenotype rather than paternal 238 Figure 36. The Y chromosome does not cause the imprint. The possibility that the imprint was caused directly by the Y chromosome, as opposed to the sex of the parent, was tested by examining the variegation of the standard test 53 when the mini-chromosome was derived from g genotype, yza d/Dp(1;f)LJ9, females with or without a Y chromosome. The first experimental column shows the results of transmission of the mini-chromosome from males (which necessarily possess a Y chromosome). The next two experimental columns show the level of 53 progeny in which g garnet expression in genotypically identical yza d/Dp(1;f)LJ9 the mini-chromosome was derived from females with and without a Y chromosome, respectively. The values shown in the middle of the figure give an indication of the level of expression of the garnet gene as an indicator of the imprint. The expression of the garnet gene was monitored both visually and quantitatively by microflourimetric assay. The eye phenotype is indicated schematically at the bottom of the figure. Values are taken from Table 34. Crosses: Paternal: X’Y/Dp(1;f)LJ9 ‘ d/y 5 3 za 3 g5 —>y za d/Dp(1;f)LJ9cJ d 53 g ® y za g Maternal : WITH Y CHROMOSOME X’X/Dp(1;f)LJ9 53 g 0 any male y za d/yf ‘I, X’X/Y/Dp(1;f)LJ9 53 g ®y za d/y ‘I 53 g y za d/Dp(1;f)LJ9f Maternal: WITHOUT Y CHROMOSOME X’X/Dp(1;f)LJ9 d/Y 5 g & 3 y za d/Dp(1;f)LJ9 53 g 0 y za 0 239 The imprint is not caused by the Y chromosome F’ )CkY xY Dp(1 ;f)U9 Dp(1 ;f)LJ9 0 I 1 y zag 53d,Dpu9 visual estimate pigment assay zag53d,Dpg 0. 10 ± 0.02 Dp(1 ;f)LJ9 zag53d,p9 1.0±0 0.92 ± 0.01 94±4 100± 3 VARIEGATED WILD TYPE WILD TYPE (IMPRINTED) (NOT IMPRINTED) 41 ±2 S 240 (NOT IMPRINTED) Table 27. The effect of different garnet alleles on the imprint. The first column lists the garnet allele. The second and third columns indicates variegation when the Dp(1;f)LJ9 mini-chromosome is transmitted maternally or paternally, respectively. Cross: Maternal cross: X’X/Dp(1;f)LJ9 Paternal cross: X’Y/Dp(1;f)LJ9cJ ® ® g*/yl > g*/Dp(1;f)LJgf g*/g* > g*/Dp(’1;f)LJ9c/ where g* is the given allele of garnet. 241 The effect of different garnet alleles on the imprint garnet allele on the regular X chromosome phenotype of g/Dp Maternal phenotype of g/Dp Paternal 1 g wild type variegated 2 g wild type variegated 3 g ND variegated 4 g ND variegated Oe 5 g wild type variegated d 3 g5 wild type variegated 61 g wild type variegated gEMS wild type ND gP wild type ND gS3 wild type variegated 242 regardless of parental origin, but if the mini-chromosome were the only source of wild type garnet+ product, and if the mother were able to deposit wild type garnet product in the egg, a conventional maternal effect could be misinterpreted as paternal imprinting. This explanation is unlikely. Firstly, more than fifty different garnet alleles have been examined and none show a maternal effect (Lindsley and Zimm 1992). Secondly, the tissue profile of the garnet gene failed to reveal high levels of garnet mRNA in ovaries and eggs (Figure 27) as would be expected for a maternally deposited product. Finally this argument would have to be extended to the other imprinted genes, narrow abdomen and tiny. Nevertheless, to completely exclude this argument I genetically tested the garnet gene for a maternal effect on eye pigmentation. No maternal effect was detected (Table 28). I also directly tested the mini-chromosome for maternal effect rescue by a wild-type copy of the garnet gene. If the imprinting effect was due to maternal rescue of the garnet mutant phenotype, providing a wild type garnet allele (elsewhere than on the mini-chromosome) should eliminate the imprinting effect. Specifically, female parents with a wild type garnet allele should deposit enough wild-type product in the egg cytoplasm to rescue the variegation of the garnet allele on the Dp(1;f)L9 mini-chromosome derived from the paternal parent. This was not the case (Table 28). Thus a maternal effect for the garnet gene can be excluded as a cause of the imprint. 4. Physiological compensation. The physiological compensation model is a variant of the maternal effect hypothesis. This model posits that the female parent produces, and transmits to the egg, factors capable of suppressing variegation, but only in response to the variegating Dp(1;f)LJ9 mini-chromosome in her cells. This scenario is formally equivalent to that suggested to explain the imprinting effect in the imprinted diseases Huntington’s Chorea (Bird, Caro and Pillins 1974) and fragile X syndrome (Van Dyke and Weiss 1986). Adapted to this 243 Table 28. Maternal effect of the garnet gene. The potential maternal effect of the garnet gene was examined in two ways. The first 53 males (where the Dp g test involved generating genotypically identical za d/Dp chromosome was paternally derived) produced from females heterozygous (column 2) or homozygous (column 3) for the garnet gene. The amount of variegation for garnet was assayed visually (first row of data) as described in the legend to Figure 34 and by microflourometric measurement of pteridine pigments (second data row, the values are expressed as percent wild type pigmentation). Cross: X’Y/Dp(1;f)LJ9cf 0 ÷÷+/y za g53d d/y 5 3 za g5 d 3 or y za g 153 g y za d/Dp(1;f)LJ9cf The lower portion of the table shows a direct test for a maternal effect of the garnet gene. The amount of pigment was quantitated in homozygous daughters (first row of data) and hemizygous sons (second row of data) derived from females heterozygous (column 2) or homozygous for garnet (column 3). All values are again given as percent wild type pigment levels. d/y 5 3 za g53d or +/y za g53d g53d/y ® y za g f Cross: y za 1 1 53 g y za d/Dp(1;f)LJ9 244 Maternal effect of the garnet gene 1. Effect on imprint 53 g y za d/Dp(1;f)LJ9 visual estimate pigment assay 2. Effect on garnet pigment 53 za g53d g y za d/y d/Y 53 yzag heterozygous mother 53 g + /y za d 0.14 ± 0.03 73±8 homozygous mother d 53 y zag53d/yzag 0.10 ± 0.02 41 ±2 heterozygous mother d 53 +/yzag 4±1 6±1 homozygous mother d 53 yzag53d/yzag 10±1 12±2 245 example of imprinting, this model posits that flies bearing the variegating minichromosome experience a physiological stress. They compensate for this stress by producing substances that reduce the level or frequency of variegation. (These hypothetical substances would have to be germ line specific as the female soma shows variegation when bearing a paternally transmitted mini-chromosome, Figure 37b.) It these substances were additionally transmitted cytoplasmically then females that possessed a mini-chromosome would produce and transmit substances capable of rescuing the variegation, and thus their progeny would appear wild type. Males bearing a mini-chromosome would obviously be unable to transmit such substances to their progeny. Initially, this is seemed a plausible explanation for the imprinting effect. Variegation can be deleterious (unpublished observations) and many modifiers of position effect variegation are early acting and are maternally deposited (Sinclair et al 1992, Dorn et al. 1993). To examine this model, crosses were performed in such a way that genotypically identical progeny were generated, all of which receive the minichromosome from their paternal parent. The crosses differ in whether or not the maternal parent bore a mini-chromosome and thus might possess and transmit the 53 progeny from normal mothers g hypothetical compensatory molecules. y za d/Dp with a paternally derived mini-chromosome had an arbitrary visual score of variegation of 0.05 ± 0.01 corresponding to pigment levels of 48±2% (see figure legend). Genotypically identical progeny from mothers possessing a variegator had a score of 0.07 ± 0.04, corresponding to pigment levels of 46 ± 9% (Figure 37). These values are not satirically different. Thus there is no evidence that the imprint is due to maternal transmission of substances capable of suppressing variegation. 246 Figure 37. Test of the physiological compensation model. A. The top portion of this figure diagrams the physiological compensation model which has been invoked to the explain reduced severity of Huntington’s chorea when the disease gene is maternally inherited. The model essentially postulates that maternal transmission compensatory substances (shown as smiling faces in the leftmost figure) produced in response to the abnormal condition, mitigates the impact of the disease in the offspring of these “conditioned” females. In genetic terms this is essentially modifier gene products with a maternal effect. This models has been proposed implicitly by Bird, Caro and Pillings (1975) and Van Dyke and Weiss (1980) and explicitly by David Baillie (personal communication). B. The lower figure shows a test of the Drosophila equivalent of this model. Essentially the model postulates that mini-chromosome-bearing mothers will transmit cytoplasmic factors to the embryo which are capable of mitigating the extent of variegation. The first two rows show the maternal and paternal genotypes. The third row indicates the parent contributing the mini-chromosome to the embryo. The fourth row shows the potential for contribution of maternal modifiers which might modify the imprint. The last rows show the phenotype and amount of variegation of the diagnostic y za 3 progeny as a measure of the imprint, assessed both by d/Dp(1;f,)LJ9 5 g microflourimeter and visual assay. Pigment values for the microflourimeter assay are expressed as percent wild type levels. 247 Crosses. Experimental 1. X’X/Dp(1;f)LJ9 0 d 3 g5 ....> /y y za d/Dp(1,.f)LJ9f 53 g ® y za & 53 za d/Dp(1;f)LJ9 g 53 g Experimental 2*. y za d/y ® XY/Dp(1;f)LJ9cf 53 g —>y za d/Dp(1;f,)LJ9cf Experimental 3. X’Y/Dp(1;f)LJ9 53 za g5 g d 3 cf ® y za d/y 53 g >y za d/Dp(1;f)LJ9 * Two mini-chromosomes are lethal due to presence of the male diplo-lethal region. As 53 must have a paternally derived mini-chromosome as g a result the y za d/Dp(1;f,)LJ9 the y za g53d homolog must come from the mother. 248 The physiological compensation model A. MOTHER +1+ LESS (later onset) B. MATERNAL PATERNAL GENOTYPE y zag5sq,Y MATERNAL GENOTYPE SOURCE OF MINI-CHROMOSOME MATERNAL CONTRIBUTION percent wild type pigment visual estimate PHENOTYPE MORE PATERNAL MINI-CHROMOSOME MATERNAL CYTOPLASM 53 z y d,yIDp XXIDpLJ9 XX/Dp mother (earlier onset) PATERNAL XYIDp a53d 53d yzg iyzg father father potential modifiers potential modifiers none 98±1 46±9 48±2 0.82 ± 0.02 0.07 ± 0.04 0.05 ± 0.01 WILD TYPE VARIEGATED 249 VARIEGATED 5. Unusual structure of the euchromatic region encompassing the garnet gene. It is possible that the garnet gene possesses some unusual feature which would permit the ingress of heterochromatin when the mini-chromosome was received from the male. The DNA sequence of the garnet gene itself is generally unremarkable (Figure 26). Nevertheless, it is possible that there is some gross structural abnormality in the region surrounding the garnet gene which could allow the aggressive invasion of heterochromatin, although in a parental specific manner. Localized changes in somatic copy number of genes showing position effect variegation have been observed in polytene tissue (Karpen and Spradling 1990). If altered copy number were the cause of the differential expression of the garnet gene, one would predict that individuals with maternally, versus paternally, derived minichromosomes would differ in DNA content in the vicinity of the garnet gene. To test this, DNA was extracted from flies which had either a paternally or a maternally derived mini-chromosome and an X chromosome marked with the g 2 allele, which has a restriction polymorphism 2 kb from the 3’ end of the garnet gene. These individuals were tested for alteration in copy number of the garnet gene by Southern blot analysis. Figure 38 shows that while there is considerable under-representation of DNA at the garnet locus, the degree of under-representation does not correlate with the amount of variegation at the phenotypic level, nor with the parental origin of the minichromosome. Thus the under-representation seems unrelated to the imprinting and may simply reflect the limited amount of euchromatin present in the mini-chromosome. The control of under representation at the garnet locus of this mini-chromosome is complex. The results shown in Figure 38 are derived from males bearing an X chromosome marked with y e(g) cv g 2 and the mini-chromosome. Very few males of this genotype are obtained because the mini-chromosome appears to variegate for a diplo-male-lethal locus identified in this region (Stewart and Merriam 1973, Belote and 250 Figure 38. Under-representation of the garnet gene in the Dp(1;f)LJ9 minichromosome. This figure shows Southern analysis of whole, newly eclosed, y e(g) cv g / Dp(1;f)LJ9 2 males where the mini-chromosome was inherited maternally (first lane paternally (second lane - - MAT.) or PAT.). DNA was extracted from (0-1 day) males of the appropriate genotype, restricted with Eco RI, separated on a 1% agarose gel, transferred to nylon membrane and probed with the garnet c-DNA derived from the imaginal disc library. Whole flies were used as a source of DNA as the garnet gene seems to be expressed in a variety of tissues (Figure 27) and males of this genotype were sufficiently rare to exclude analysis of isolated tissues. The g 2 allele which marks the normal X chromosome is associated with a Eco RI restriction polymorphism 2 kb 3’ to the garnet gene and thus serves as an internal control. This generates the lower mobility, 8.5 kb band. The wild type garnet gene on the mini-chromosome produces the higher mobility, 6.6 kb, band. The lowest band is a 0.2 kb Eco RI band generated by both the normal X chromosome and the mini-chromosome. 251 I 0, Lucchesi 1980). The lethality seems to occur principally at the pupal stage, structurally normal pharate adults form but do not eclose. This diplo-lethality appears sensitive to the zeste gene as it is alleviated by za and z 1 alleles. When this experiment was repeated using a za g /Dp progeny for molecular 2 2 strain, to increase the number of g analysis, the under representation was not observed (data not shown). Thus the under-representation appears to be zeste sensitive. Whether there is a causal relationship between zeste mediated pairing, transcriptional repression, the male diplo-lethality and sequence under-representation is unclear. The question of underrepresentation is further complicated by recent results which suggest that the observed “under-representation” is an artifact of DNA modification which inhibits transfer to solid support during Southern blotting (Glasser and Spradling 1994). Whether this is also true for the under-representation around the garnet gene in the Dp(7;f)LJ9 minichromosome is currently being investigated by the Glasser lab. 6. The role of heterochromatin. There are a number of features shared by imprinting and position effect variegation which might suggest a mechanistic link between the two phenomena. As heterochromatin formation has been posited to cause position effect variegation and as heterochromatin seems widely involved in imprinting, I decided to test factors known to modify position-effect variegation for both their effect on the variegation, which is the mode of the expression of the imprint, and also on the transmission of the variegation, that is, the imprint itself. There are three general groups of factors that affect position effect variegation. They are: environmental factors such as temperature, chemical factors, such as sodium butyrate and genetic factors such as the presence of extra heterochromatin in the cell, usually in the form of an additional Y chromosome. I tested all these factors for their 253 Table 29. Variegation of the Dp(1,i)LJ9 mini-chromosome: The effect of developmental temperature. Cultures generating g/Dp(1;f)LJ9 males, where the mini-chromosome was transmitted either maternally or paternally, were raised at 18, 22, 25 and 290. The amount of pigment in the eyes of these progeny was determined by microflourimeter assay. Non specific effects of temperature on pigment (such as effects on fly size or number) were controlled by assaying siblings. No such effects were noted (data not shown). The top row indicates the temperature of the culture. MAT and PAT indicate the parental origin of the mini-chromosome. Values derived from the paternal cross are shown in italics 50 or g g for contrast. The garnet allele of the non-Dp bearing parent, either g53d, 0 , is 2 indicated in the first column. ND = not done. All values are given as percent wild type pteridine pigments. Crosses: Maternal cross: X”X/Dp(1;f)LJ9 d/y 5 3 or y za 5 g O e/yf or ® yz g /Yo’ 2 y e(g) cv g yza g”/Dp(1,’f)LJ9o’ d/y 5 3 za g53d Paternal cross: X”Y/Dp(1;f)LJ9o’ ® y za g Oe 5 Oe/y za g 5 y za g or or /y e(g) cv g 2 2 y e(g) cv g ‘I yza 254 The effect of culture temperature on variegation of Dp(1;f)LJ9. g5 d 3 MAT PAT Oe 5 g MAT PAT 2 g MAT PAT culture temperature 22° 18° 25° 29° 72±8 44± 14 73+8 37± 13 64±6 36±7 39±5 28±4 70±8 47±17 80±9 50±6 71±8 52±9 44±7 35±5 ND 83±9 93±10 89±9 105±8 84±15 90±11 84±7 255 Table 30. Variegation of the Dp(1,i)LJ9 mini-chromosome: The effect of sodium butyrate. 3 males, where the mini-chromosome was d/Dp(1;f)LJ9 5 Cultures which generated g transmitted either maternally or paternally, were supplemented with no, 100, 150, 200, 250 or 300 mM sodium butyrate (top row). The amount of pigment in the eyes of these progeny was determined by microflourimeter assay (row five and six, all values expressed as percent wild type pteridine levels). Non specific effects of temperature on pigment (such as effects on fly size or number) were controlled by assaying phenotypically wild type XX//Y siblings. These results are shown in the second row. Flies variegating for In(1)wm4 were grown alongside the Dp(1;f)LJ9 crosses on identical sets of supplemented media as a control for the effectiveness of butyrate treatment (row three and four). ND not done. Crosses: Maternal cross: X’X/Dp(1;f)LJ9 f 1 ® y za g53d/y 13 d/Dp(1;f)LJ9cf 5 y za g 53 za g53d g Paternal cross: X’Y/Dp(1;f)LJ9o’ ® y za d/y ‘I 3 d/Dp(1;f)LJ9 5 y za g m4 cross: 4 /wm wm /Ycf> m4 4 ® wm 256 and ci’. The effect of butyrate on variegation of Dp(1;f)LJ9. X’XhY wm4 ’ 0 wm4 d/DpLJ9 53 yzag (maternal) d/DpLJ9 53 yzag (paternal) 0 mM 98±4 14±3 1±1 butyrate concentration 100 mM 150 mM 200 mM 250 mM 300 mM ND ND 98±4 91±3 87±3 18±4 23±3 40±11 65±10 34±4 19±6 18±6 9±1 18±13 13±10 87±6 81 ±4 71 ±3 64±6 ND ND 40±2 44±5 38±3 31±2 34±2 34±3 257 Table 31. Variegation of the Dp(1;f)LJ9 mini-chromosome: The effect of Y chromosome dosage: Effect of Y chromosome dosage on garnet variegation of Dp(1;f)LJ9 is shown for individuals in which the mini-chromosome was derived either maternally or paternally. The first and second columns show the amount of variegation assayed both visually (row 1) and by microflourimeter assay (row 2, all values expressed as percent wild type pteridine levels) for genotypes which are X/Dp and X/Dp + Ywhere the minichromosome is paternally derived. The third and fourth columns show the equivalent genotypes with a maternally derived mini-chromosome. 53 That of the X/Dp g Genotype of the X/Dp flies is yza d/Dp(1;f)LJ9. + Yflies is 53 g y za d/Dp(1;f)LJ9/y. Crosses: 53 g ® y za d/y Maternal cross: X’XJY/Dp ‘I 53 and y za d/Dp(1;f)LJ9/ycJ g 53 g y za d/Dp(1,.f)LJ9 These progeny were separated by progeny testing. 53 g Paternal cross: y za d/y/Dp(1;f)LJ9 ‘® d/y 5 3 za g53d y za g ‘I, yza d/Dp(1;f)LJ9cJ 53 and yza d/Dp(1;f)LJ9/ycf g 53 g These progeny were separated by progeny testing. 258 The effect of Y chromosome dosage on variegation of Dp(1;f)LJ9. Paternal visual estimate pigment assay Maternal X/Dp X/Dp 0.07 ± 0.03 37 ± 9 0.97 ± 0.03 99 ±4 + Y 259 X!Dp X/Dp 1.0 ± 0 79±5 1.0±0 94±4 + Y ability to affect both the extent and the transmission of variegation; that is the maintenance and establishment of the imprint. All these factors which modify classical position effect variegation, also modified the variegation associated with this instance of imprinting. Tables 29, 30, and 31 show the effect of temperature, sodium butyrate, and the presence of an extra Y chromosome on variegation, respectively. The effect of developmental temperature and butyrate was the reverse of the canonical response. While a Hreverseu response to temperature is hardly unprecedented (Spofford 1976) the biological significance is unclear. Growth on butyrate supplemented media also enhanced the variegation of the garnet gene on the minichromosome, again, an unconventional response. The effect of these genetic modifiers of position effect variegation was, with the exception of the Y chromosome, not particularly dramatic. However, in general, the modifiers of position-effect variegation did modify the variegation of the garnet gene on the mini-chromosome. I next tested these same modifiers to determine if they could affect the imprint, that is the meiotic transmission of the variegation. Table 32, 33, and 34 show the results of these tests. In no case did any of these conditions, which alter the variegation, alter the parental imprint. This conclusion is naturally subject to the criticism that heterochromatin is a complex, and largely uncharacterized structure. Those factors involved in the establishment of the imprint may in fact be components of heterochromatin but simply different from those tested here. The effect of the Y chromosome is particularly striking. Although the strongest modifiers of variegation, capable of completely obliterating variegation of a paternally derived mini-chromosome, it had no effect on the transmission of the variegation the - imprint. The effect of the Y chromosome on imprinting was examined in four ways: A 260 Table 32. Imprinting of the Dp(1;f)LJ9 mini-chromosome: The effect of developmental temperature. Cultures generating g*/Dp(1;f)LJ9 males, where the mini-chromosome was transmitted both maternally and paternally, were raised at 18, 22, 25 and 29°. In addition the parents were raised throughout their lives (from egg onwards) at 18, 22, 25 and 29° The amount of pigment in the eyes of these progeny was determined by microflourimeter assay, all values are expressed as percent wild type pteridine levels. Non specific effects of temperature on pigment (such as effects on fly size or number) were controlled by assaying siblings. No such effects were noted (data not shown). Data from experiments with three different garnet allele in the non-Dp bearing parent, d, g 3 Oe or g 5 either g5 2 is shown separately in each table. The temperature at which the parents were raised is shown on the left. The culture temperature of the cross which generated the diagnostic g/Dp(1;f)LJ9 progeny is shown at the top. Values derived from the paternal cross are shown in italics for contrast. ND = not done. There is limited information for the 290 series of paternal crosses as the XY/Dp males were generally sterile when raised at this temperature. Crosses: Maternal cross: X’X/Dp(1;f)LJ9 g5 d /yf or ® y za 3 1 or y e(g) cv g 0 /YQ’ 2 y za g53d/y d/y 5 3 za g5 d 3 Paternal cross: X’Y/Dp(1;f)LJ9o’ ® y za g Oe 5 Oe y za g 5 y za g 261 or or y e(g) cv g /y e(g) cv g 2 2 Imprinting of the Dp(1;f)LJ9 mini-chromosome: The effect of developmental temperature. d 5 g 3 temperature 22° 89±3 37±2 87±3 36±3 96±4 34±4 92±30 16±4 25° 63±5 50±3 61±2 54±3 58±3 27±2 85±4 ND 29° 33± 10 39±2 49±2 30±3 54±1 41±2 59±4 ND MAT PAT MAT PAT MAT PAT MAT PAT g O 5 e culture temperature 18 22 96±1 87±2 37±2 69±4 89±4 89±3 81±5 56±3 88±4 95±3 58±7 73±3 108±8 89±3 ND ND 25° 69±4 58±2 75±4 53±3 60±3 52±2 83±3 ND 29° 61±2 41±3 60±2 33±4 58±2 52±1 59±5 45±6 18° MAT parental PAT 22 MAT temperature PAT 25 MAT PAT 29° MAT PAT culture temperature 18° 22° 112±6 98±3 67± 10 96±3 102±3 103±6 80±5 79±4 112± 10 107±5 ND ND ND 105±5 ND ND 25° 99±9 ND 108±3 87±4 100±3 114± 30 113±6 ND 29° 96±4 99±5 99±3 92±3 104±2 100± 3 133±30 ND 180 MAT PAT parental 22° MAT PAT temperature 25° MAT PAT 29° MAT PAT parental temperature 18° 22 25° 29° culture 18 94±3 42±4 85±4 27±3 80±4 30±2 91±5 ND 262 Table 33. Imprinting of the Dp(1,’f)LJ9 mini-chromosome: The effect of sodium butryrate. Cultures which generated g/Dp(1;f)LJ9 males, where the mini-chromosome was transmitted either maternally or paternally, were raised on media supplemented with 0 or 200 mM sodium butyrate. In addition the parents were also raised on either 0 or 200 mM sodium butyrate. The amount of pigment in the eyes of these progeny was determined by microflourimeter assay. All values are expressed as percent wild type pteridine levels. The first set of numbers reflects the amount of pigment in yza 53 progeny with a maternally inherited mini-chromosome. The second g d/Dp(1;f)LJ9 set shows pigment levels of this genotype when the mini-chromosome is paternally inherited. ND = not done. Crosses: Maternal cross: X’X/Dp(1;f)LJ9 53 g f 1 ® y za d/y —* 53 g y za d/Dp(1;f)LJ9cJ d/y 5 3 za g53d Paternal cross: X’Y/Dp(1;f)LJ9o’ ® y za g — 263 yza d/Dp(1;f)LJ9cJ 53 g Imprinting of the Dp(1;f)LJ9 mini-chromosome: The effect of sodium butyrate. parent concentration progeny concentration 0 mM 0 mM 0 mM 200 mM 200 mM 200 mM 200 mM 0 mM Maternal minichromosome Paternal minichromosome 87±6 64±6 67±3 77±3 40±2 31 ±2 ND 71 ±5 264 Table 34. Imprinting of the Dp(1;f)LJ9 mini-chromosome: The effect of Y chromosome dosage. Four tests of the effect of the Y chromosome on imprinting of Dp(1;f) LJ9 were performed. 1. The direct paternal test was intended to determine if an additional Y chromosome in 53 g the Dp bearing father affected imprinting. Genotypically identical y za d/Dp(1,’f)LJ9 progeny, both with a paternally transmitted mini-chromosome, where generated from an XY + Dp and an XYY + Dp fathers (first and second data columns). The results were assessed by visual inspection and by microflourimeter pigment assay (first and second data row, values are expressed as percent wild type pteridine levels). y za 53 progeny were distinguished from their y za g g d/Dp(1,.f)LJ9 d/Dp(7;f)LJ9/y 5 3 siblings by progeny testing. Cross: XX/Dp ® )Q’Y (any male) X’)QY/Dp 0 X’Y/Dpo’ ‘I, 53 za g5 g d 3 XY/Dp or X”Y/Y/Dpo’ ®y za d/y 53 g y za d/Dp(1;f)LJ9f 2. A direct maternal effect was tested by examining progeny derived from XX vs. XXY d/Dp(1;f)LJ9 5 3 progeny, duplication bearing mothers. Genotypically identical y za g both with a maternally transmitted mini-chromosome, where generated from an XX Dp and an XXY + + Dp mothers (first and second data columns). The results were assessed by visual inspection and by microflourimeter pigment assay, the latter values d/Dp(1;f)LJ9 5 3 progeny are expressed as percent wild type pteridine levels. y za g d/Dp(7,.f)LJ9/y 5 3 siblings by progeny testing. were distinguished from their yza g 265 Cross: XX/Dp ® any male X”X/Dp or X”XJY/Dp d/yçj& 5 3 y za d/Dp(1;f)LJ9cf 53 g ®y za g 3. The third test was of a paternal effect on the mini-chromosome variegation 53 g unrelated to the mini-chromosome. Genotypically identical y za d/Dp(1;f)LJ9 progeny, both with a maternally transmitted mini-chromosome, where generated from an XY and an XYY (no mini-chromosome) fathers (first and second data columns). The results were assessed by visual inspection and by microflourimeter pigment assay, the 53 g latter values are expressed as percent wild type pteridine levels. y za d/Dp(1;f)LJ9 53 siblings by progeny g progeny were distinguished from their yza d/Dp(1,.f)LJ9/y testing. Cross: X’Y/O XY/Y ‘I d/y 5 3 za d/Dp 53 g X’Y/O vs. X’Y/Yd ® y za g _> 53 g y za d/Dp(1;f)LJ9c 4. The final test was of a maternal effect on the mini-chromosome variegation 53 g unrelated to the mini-chromosome. Genotypically identical y za d/Dp(7;f)LJ9 progeny, both with a paternally transmitted mini-chromosome, where generated from an XX and an XXY (no mini-chromosome) mothers (first and second data columns). The results were assessed by visual inspection and by microflourimeter pigment assay, the latter values are expressed as percent wild type pteridine levels. y za 53 siblings g d/Dp(1;f)LJg 5 g 3 progeny were distinguished from their y za d/Dp(7;f)LJ9/y by progeny testing. d/y 5 3 za g53d cross: X’Y/Dp ® y za g 1 y za g53d/y za g53d or y za g53d/xy 53 g ® X’Y/Dpo’— y za d/Dp(7;f)LJ9 266 Imprinting of the Dp(1;f)LJ9 mini-chromosome: The effect of Y chromosome dosage. 1. Direct paternal effect XJY/Dp vs. X/Y/Y/Dp visual estimate pigment assay XY+Dp father XY+Y+Dp father 0.10±0.02 0.12±0.02 41 ±2 48±3 3. Paternal Y effect XYvs. XYYx Dp females visual estimate pigment assay 2. Direct maternal effect X’X’/Dp vs. X’X//X’X/ XX+Dp mother XX+Y+Dp mother visual 0.92±0.01 estimate pigment 100 ±3 assay 1.0±0 94±4 4. Maternal V effect XXvs. XXY x Dp males XY ® Dp mother XYY 0 Dp mother 0.73 ± 0.03 1.0±0 75 ± 10 81 ± 10 XX 0 Dp XXY 0 Dp father father visual 0.05 ± 0.02 estimate pigment 25 ±5 assay 267 0.03 ± 0.03 34 ± 11 direct effect in the male, and female (both the Y chromosome and mini-chromosome present in the same individual); and as an indirect maternal, and paternal effect (Y chromosome and mini-chromosome not present in the same parent). The presence of an extra Y chromosome did not alter the imprint regardless of whether it was present with or without the mini-chromosome, or in males or females. There was no evidence of a maternal or paternal Y chromosome effect on variegation as reported by Khesin and Bashirov (1978). If we accept for the moment, that these modifiers of position effect variegation act via heterochromatin, then it seems that heterochromatin is involved in the somatic propagation of the imprint. Clearly an additional Y chromosome can eliminate the garnet variegation, which is the signature of the imprint of the Dp(1;f)LJ9 mini-chromosome. 268 DISCUSSION-imprinting In this chapter I have described a mini-chromosome in Drosophila melanogaster which exhibits genomic, parent-specific or gamete-specific imprinting. The imprint is independent of the sex of the progeny and is completely reversible. This imprint is manifest as parent-dependent expression of the garnet eye colour gene, and of at least two other genes on the mini-chromosome, tiny and narrow abdomen. Imprinting of the mini-chromosome was first noted as the parent-specific expression of the garnet gene. While other genes on the mini-chromosome also appear to be imprinted, the imprint at the garnet gene is the most dramatic expression of the imprint because of its striking mosaic phenotype in the eye. This mosaic phenotype arises because the garnet gene is cell autonomous and the inactivation of the paternally derived garnet allele is not complete. Imprinting of a non-cell autonomous gene would be manifest as a quantitative difference in the level of gene expression which would be less readily apparent. The Dp(1;f)LJ9 mini-chromosome shows extensive variegation (inactivation) for the garnet+ gene when it is transmitted by a male. When this mini-chromosome is transmitted by a female the garnel gene is fully expressed. Thus according to the terminology of Reik (1992) the mini-chromosome is paternally imprinted. It should be noted that this terminology is arbitrary. There is no data to indicate whether imprinting acts to inactivate otherwise active genes or visa versa or whether the ground state of the garnet+ gene on the mini-chromosome is variegated or fully expressed. The immediate cause of disruption of garnet expression is position effect variegation. That the mosaic imprinted phenotype is due to conventional position effect variegation, is shown by the response of this variegator to the standard factors which modify 269 position effect variegation. The variegation is unusual in that is dependent of the sex of the transmitting parent thus the variegation itself is imprinted. As this example of genomic imprinting involves position effect variegation, the mechanism whereby position effect variegation causes gene inactivation is of considerable importance. While number of models of the mechanism of position effect variegation have been proposed (Fran kham 1988, Karpen 1994), there is experimental evidence to support three general classes of models; the somatic elimination model, the nuclear compartmentalization model and chromatin or heterochromatin formation models. I have shown that neither the expression or the imprint of the garnet gene correlates with under-representation of this gene (Figure 38), arguing against a role for somatic elimination. Of the two other models, experimental evidence slightly favours the chromatin formation model although a role for functional nuclear localization is intriguing and can not be ruled out. These two models are discussed in more detail below, in context of the mechanism of imprinting. If we accept, for the moment, that position effect variegation is due to the illicit spread of heterochromatin than this striking phenotype could arise from either, a parent dependent ability to induce variegation, or from a parent-dependent difference in the distance of spread or aggressiveness of the invading heterochromatin. The latter is more likely for the following reasons. There is unlikely to be a parent specific difference in the ability of the mini-chromosome to form centric heterochromatin. If this were the case, increased non-disjunction of paternally derived mini-chromosome would be expected. Frequent non-disjunction has been observed with another mini chromosome which was broken near the centromere and may have variegated for centromere function (Wines and Henikoff 1992). However, such mitotic non-disjunction was not observed with the Dp(1;f)LJ9 mini-chromosome used in this study. Nor is the imprint likely to result from parent specific recognition of heterochromatic boundary 270 sequences (if in fact such sequences exist) as the same sequence would be present or absent on the mini-chromosomes whether it was derived from males or females. Finally, the effect of temperature on the imprint suggests that the maternally derived mini-chromosome can variegate under some circumstances, If individuals with a maternally derived mini-chromosome are raised at high temperature a few individuals show limited inactivation of the garnet gene (Table 29). Thus I propose that variegation occurs on the mini-chromosome regardless of its parental origin, but the distance over which heterochromatin spreads, and thus the likelihood of inactivating the garnet reporter gene is dependent on the parental origin. This situation is diagrammed in Figure 39. Implicit in this model is that the imprint is evident only within a limited range along the mini-chromosome. Also implicit in this model is the proposal that the imprint is nucleated with in the centric heterochromatin and spreads distally. The resulting parent-dependent differences in heterochromatin formation become less pronounced with distance from the centromere. This is in accordance with the extent of the imprinting of the three neighboring genes, garnet, narrow abdomen and tiny (Figure 35). I tested a number of factors to determine the cause of the imprinting. The imprinting is not allele specific. Nor is it a trivial artifact of maternal action of the marker gene, garnet. More interestingly, the imprint does not seem to be due to maternallycontributed compensatory substances, induced by physiological stress, as has been proposed for the imprinting effects associated with Huntington’s Chorea and fragile X mental retardation (Bird, Caro and Pilling 1974, VanDyke and Weiss 1986, respectively). Investigation of the role of heterochromatin in imprinting initially seemed promising given the widely observed involvement of heterochromatin in imprinting and position effect variegation. Tests of the stability and generation of the imprint in the 271 Figure 39. Model of the parent-dependent spread of heterochromatin responsible for imprinting of the Dp(1;f)LJ9 mini-chromosome. This figure diagrams the percent of cells showing phenotypic inactivation due to the spread of heterochromatin as a function of distance from the heterochromatic boundary. The effect of culture temperature is shown as altering the distance of this spread rather than the occurrence of variegation. 272 Model of the mechanism of parent-dependent variegation of Dp(1;f)LJ9 100% variegation of maternally derived mini-chromosome variegation of paternally derived mini-chromosome percent inactive cells 0% Distance from heterchromatic boundary garnet tiny narrow abdomen 273 presence of environmental, chemical and genetic factors which modify position effect variegation, and are proposed to modify heterochromatin formation and integrity, suggest that at least in this instance of imprinting, the role of heterochromatin is restricted to the somatic expression of the imprint but is not involved in the establishment of the imprint. While these data might challenge the role of chromatin structure in initiation the imprint, heterochromatin formation is clearly necessary for the somatic manifestation or maintenance of the imprint. This was shown by the response of the variegation of the mini-chromosome to conventional modifiers of position effect variegation. Three factors which generally modify position effect variegation (temperature, butyrate, and Y chromosome dosage) altered the variegation of this mini-chromosome. The effects of these modifiers deserve additional comment. The effect of temperature is the reverse of the conventional effect. Usually high temperature suppresses position effect variegation. A “reverse” response to high temperature is not unprecedented by any means (Spofford 1967), however, the other examples of reverse temperature sensitivity have been attributed to temperature sensitive hypomorphic alleles on the non-variegating homolog. This cannot be the case for the garnet variegation on the mini-chromosome as the temperature effect is seen with several alleles, none of which are temperature sensitive. While the mechanism of position effect variegation is unknown, the canonical response to temperature suggests that the process of heterochromatin formation is limiting. The simplest interpretation of the reverse response of the Dp(1;f)LJ9 variegation to temperature might suggest that it is euchromatin formation which is limited in this chromosome. Why or how this might occur is not clear. The unconventional response to butyrate supplemented media is also interesting. Sodium butyrate has been proposed to suppress position effect variegation by altering the de-acetylation of histones and so disrupting chromatin 274 condensation. If this is in fact occurring, it is not clear why this would enhance heterochromatin formation on the mini-chromosome. Finally, the moderation of variegation by temperature and sodium butyrate was quite modest. This difference in effectiveness between modifiers of position effect variegation is in contrast to their effect on conventional (non-parental) variegators (Clegg et al. 1992). In contrast, the effect of an extra Y chromosome (X/Y/Dp versus X/Dp) was much more dramatic, essentially eliminating variegation even for paternally derived minichromosomes. It may be that the Y chromosome effects variegation by mechanisms distinct from that of the other modifiers. In this regard, Talbert and Henikoff (1994) have argued that the suppressing effect of the Y chromosome on position-effect variegation is due to occlusion of the heterochromatin-forming compartment of the nucleus rather than directly on heterochromatin formation. The Y chromosome might also alter the somatic pairing of the mini-chromosome. In contrast to these models, Zuckerkandel (1974) proposed that the Y chromosome suppresses position effect variegation by acting as a binding site for heterochromatic-specific proteins. It is possible that the Y chromosome is a particularly potent suppresser of Dp(1;f)LJ9 variegation because it is a powerful competitor for the specific heterochromatic proteins which bind to the minichromosome. One possible candidate might be the proteins which bind to the Stellate gene cluster or proteins which bind to repetitive sequences near to the garnet gene (see below). In the absence of data on the mechanism of position effect variegation, any number of possibilities can be envisioned. Why do only some variegators show parental effects? All published parental effects in Drosophila involve position effect variegation (Table 26). This may indicate an obligatory requirement for the large scale chromosome 275 rearrangements associated with position effect variegation. This scenario is hard to justify conceptually. Absence of published reports of parental imprinting of transgenes in Drosophila, as is seen in mice, may reflect nothing more than investigators’ aversion to non-Mendelian expression which defeats the purpose for which transgenes are usually generated. There have been sporadic unpublished reports of parent dependent transgene expression as seen in mice (C. Bazinet, personal communication, C. Berg, personal communication). Although all published parental effects in Drosophila (excluding conventional maternal effects) involve variegating rearrangements, by no means is every example of position effect variegation associated with parental effects (Spofford 1976). Another oddity evident from Table 26 is that all the examples of parental effects in Drosophila involve the X chromosome. They do not however share any other common chromosomal element nor is the heterochromatin of the X chromosome always involved. Thus this may be simply a spurious coincidence. As imprinting amongst variegating rearrangements is decidedly a rarity, this raises the question of what features distinguish those variegating rearrangements which do show parent-specific variegation. The existence of cloned and characterized DNA sequences from the vicinity of the garnet gene made this an obvious candidate to investigate any sequence or larger scale peculiarities which might explain the imprinting. Specific sequence motifs within the garnet gene are unlikely to be the cause of the parental imprinting. Although the 3’ region does contain a polyglutamine repeat sequence which has been associated with some imprinted genes in humans (Green 1993) the repeat seems too short and inconsequential to explain the imprinted spread 276 of heterochromatin through at least three genes. The trinucleotide repeats found in the fragile X syndrome, spinal and bulbomuscular atrophy, Huntington’s disease, spinocereballar ataxia type I and myotonic dystrophy are present as between 5-50 copies in the normal alleles and 40-4000 copies in the imprinted disease causing alleles. Large scale structural features of the mini-chromosome might also be associated with the imprint. The Steilate genes are highly repetitive gene clusters present on the X and Y chromosomes and degenerate members are present in the proximal heterochromatin of the X chromosome (Shevelyov 1992, E. V. Benevolenskaya, personal communication). The Stellate genes are highly transcribed in spermatogenesis but are of unknown function (Livak 1990, Palumbo et al. 1994). The proximity of Stellate sequences to the garnet gene, as well as possible degenerate Steilate sequences in the X heterochromatin, may meant that the garnet gene is flanked by multiple Stellate repeats. Given the propensity of heterochromatin to engage in non-homologous pairing (Eberl, Duyf and Hilliker 1993, Talbert, LeCiel and Henikoff 1994), the ability of repeated sequence to nucleate heterochromatin formation (Dorer and Henikoff, 1994), the fact that many imprinted transgenes in mammals are present as multiple repeats (Surani, Reik and Allen 1988) and the presence of repeat sequence within a region of an imprinted transgene defined as the cis-acting imprinting signal (Chaillet at al. 1995), the Stellate sequences might induce aberrant heterochromatin structure or nuclear localization of the mini-chromosome which could lead, somehow, to imprinting. Intriguingly, the euchromatic and heterochromatic Stellate sequences can be seen to form ectopic fibres, which indicate illicit pairing, in polyene chromosome preparations (Palumbo et al. 1994). The many Stellate repeats on the Y chromosome might explain the remarkable effectiveness of the Y chromosome in suppressing variegation of the mini-chromosome it these sequences compete for binding proteins, Of course the presence, number and function of the degenerate heterochromatic Stellate sequences in the mini-chromosome 277 remains to be confirmed. A similar argument could be made for any other middle or highly repetitive sequences present near garnet and on the Y chromosome. In this regard it is interesting that the 12DE region adjacent to garnet is peppered with repetitive sequences (Leung et al. 1987). Although the consequences of imprinting are dramatically manifest in development, neither the evolutionary forces which lead to imprinting or the mechanisms are known. The former issue has occupied the attention of many biologists, but in the absence of data on the mechanism, all evolutionary scenarios remain completely speculative. The mechanism of imprinting is of considerable interest in itself, and is also of medical importance given the number of human diseases in which imprinting has been implicated. Mechanism of imprinting: While it is evident that differences in packaging genetic material in eggs and sperm may facilitate differential gene expression in early embryogenesis, a simple response to the different physiology and morphology of the germ cell formation and structure may not suffice as a mechanism for imprinting. In any case, this explanation fails to address the question of why and how some genes show parent specific expression whereas others do not. Based on observations that pairing, recombination and gene conversion events coincide with condensation at pachytene stage of meiosis, Monk and Grant (1990) and Hall (1990) have proposed that imprinting is a side effect of these events. Details of how or why this might occur are unclear and as such this model is difficult to test empirically. Functional differences in nuclear localization, in various guises, has been proposed as a potential basis for imprinting. 278 Modulation of gene expression by nuclear compartmentalization has been inferred from both cytological and genetic evidence. The existence of nuclear compartments is indisputable but ascribing specific functions to them remains problematic. For example, the nucleolus is an obvious example of a specialized nuclear compartment. It has been suggested that other structures found in the nucleus, such as the coiled bodies, perichromatin fibrils and interchromatin granules, correspond to other functional compartments associated with splicing. Recent evidence, however, suggests that this is not the case (Mattaj, 1994). The first intimation that nuclear compartments might have specific functions in gene regulation stemmed from observation of the specific orientation of chromosomes in the nucleus, with telomeres and centromeres apposed to the nuclear membrane (RabI 1885). A similar chromosome organization in yeast is correlated with variable gene inactivation resembling position effect variegation (Laurenson and Rhine 1992), however, a causal relationship between either telomere-nuclear membrane apposition in S. cerevisiae or centromere-nuclear membrane apposition in S. pombe remains to be shown. Heslop Harrison (1990) has proposed that imprinting results from differential location of the two parental or species haploid sets of chromosomes in the zygotic nuclei. There are many well documented and striking examples of differential location of the two chromosome sets in plant inter-specific hybrids (summarized by Heslop-Harrison 1990) however, these studies did not correlate the location of the two genome sets with gene expression. Several investigators have argued for a similar effect of nuclear localization on gene expression in position effect variegation (Hessler 1957, Wakimoto and Hearn 1990, Talbert, LeCiel and Henikoff 1994) however cytological correlations are generally lacking. If a causal relationship between nuclear localization and gene expression could be shown in position effect variegation, it might serve to support hypotheses suggesting a connection between nuclear location and gene expression in imprinting. 279 In mammals and plants, DNA methylation has long been known to be linked to gene inactivation. There are correlations between methylation and the gene inactivation resulting from imprinting in mammals. Unfortunately the convenience of determining the methylation status of a gene has led to methylation being treated synonomously with imprinting without other evidence of genetic activity. But, the correlation between methylation and gene activity is by no means absolute. Such a correlation is seen in only one of six examples of imprinted transgenes (Surani, Reik and Allen 1988) and it has been shown in a number of cases that the decision to inactivate a gene occurs before methylation is evident. The Hprt gene on the imprinted X chromosome is inactivated several days before differential methylation is established (Lock, Takagi and Martin 1987). Likewise, differential methylation of the imprinted genes H19, Igf2 and Igf2r does not generally persist through meiosis, thus can not constitute the initial imprinting signal. For example, the parent-specific methylation associated with imprinted expression of H19 occurs late in embryogenesis and is not propagated through the male meiosis (Ferguson-Smith et al 1993). Most of the parent-specific methylation pattern in the promoter and coding region of Igf2r/Mpr is established late in embryogeneis and is erased in meiosis. There is, however, in this case, one site in an intron which maintains its methylation status though the female meiosis (Stoger et al 1993). In contrast, the imprinted gene Igf2 seems devoid of parent specific methylation in the immediate vicinity of the gene, and the gene is identically methylated in eggs and sperm (Sasaki et al 1992, Brandeis et al 1993). Thus methylation seems not to constitute the initial decision or even an early event in imprinting. The role of methylation in imprinting may be relegated to maintenance of the decision. Finally, methylation is not causally involved paramutation in maize (Patterson, Thorpe and Chandler 1993), a phenomenon that resembles imprinting, nor does it correlate with either imprinting or heterochromatin formation in coccids 280 (Scarbrough, Hattman and Nur 1984) nor is it found in Dipteran insects where there are striking examples of imprinting. Thus methylation is unlikely to play a role in this case, or others, of imprinting in Drosophila. The role of chromatin structure in imprinting is less experimentally tractable than that of methylation, but, its near universal involvement in imprinting and the involvement of position effect variegation in imprinting in Drosophila, makes it a logical candidate for investigation. The role of heterochromatin formation in position effect variegation has been extensively investigated. Evidence for a chromatin-based model of position effect variegation stems from cytological, genetic and molecular work. A correlation between gene inactivation and the cytological appearance of heterochromatin in the corresponding region of the chromosome (Hartmann-Goldstein 1967, Spofford 1976) has been observed for many genes. A role for chromatin formation in position effect variegation is further suggested by the sensitively of position effect variegation in Drosophila (and the seemingly related telomere mediated position effect in S. cerevisiae) to alterations in histone levels. A number of other genetic modifiers of position effect variegation have also proven to encode chromatin associated proteins (reviewed by Orlando and Paro, 1995). Finally, alterations in nucleosome positioning, phasing and chromatin accessibility have been found associated with position effect variegation (Wallrath and Elgin, 1995). Thus there is reasonable evidence to support the assertion that gene inactivation associated with position effect variegation is related to changes in chromatin structure. Tartof and Bremer (1990) have extended these observations to imprinting and have made the rather stringent prediction that imprinted regions will correspond to regions of intercalary heterochromatin. No ectopic pairing sites, a sign of intercalary heterochromatin have been reported around the garnet gene. Nevertheless, the role of chromatin structure in imprinting can not be disregarded. The results presented in chapter 3 of this thesis imply that 281 heterochromatin in Drosophila may perform a maintenance function, similar to that played by methylation in mammals. There is evidence that the maintenance stage in mammals can be resolved into early and late stages. Intriguingly, preliminary evidence suggests that the early maintenance stage may be mediated by chromatin binding proteins (Monk 1988, Barlow 1994, Ohlsson, Barlow and Surani, 1994). This may suggest a parallel between the maintenance mechanisms of the imprint between mammals and Drosophila. Different mechanisms of late somatic memory might simply reflect differences in the life cycles of mammals versus Drosophila. Most cells of a mammal continue to undergo cell division throughout the adult portion of life. The continuation of cell division requires a high fidelity memory mechanism. Covalent modification of DNA based by methylation, clearly, would fulfill this function. In contrast, adult flies are mitotically quiescent, thus there might be less rigorous requirement for the memory mechanism and a “chaotic” system such a chromatin partitioning might suffice. A major impediment to resolution of the precise role of heterochromatin in imprinting is that the structure of chromatin and molecular architecture of heterochromatin remains undefined, leaving any models about the role of heterochromatin necessarily vague. One approach to resolving the mechanism of imprinting is to isolate genetic modifiers of the imprinting process by screening for mutations which enhance, eliminate or switch the parental specificity of imprinted genes. Genes which modify imprinting would be expected to encode products which are components of the machinery which recognizes the sexual context of the imprinted region and thus might act in one sex only (which depends on whether gene activity or inactivity is the default state). Some of these gene products would also have to recognize and isolate the region to be imprinted, make the initial decision to imprint and then, at least initially, propagate and enforce the imprinted state. Some progress has been made in 282 Table 35. Effect of attached versus free sex chromosomes on imprinting of Dp(1;f)LJ9. 53 progeny, with either a paternally g Genotypically identical y za d/Dp(1;f)LJ9 transmitted mini-chromosome (columns 2 and 3) or a maternally transmitted minichromosome (columns 4 and 5), were generated from stocks with either attached sex chromosomes (column 2 and 4) or freely segregating sex chromosomes (column 3 and 5). The results were assessed by visual inspection (first data row) and by microflourimeter pigment assay (second data row, values are expressed as percent 53 progeny were distinguished from their g wild type pteridine levels). yza d/Dp(1;f)LJ9 d/Dp(1,.f)LJ9/y 5 3 siblings by progeny testing. y za g Crosses: d/y 5 3 za g53d 53 ®y za g g paternal: X’Y/Dp or y za d/y/Dpo’ 53 d/Dp g 53 g maternal: X’X/Dp or yza d/yza yza d/yor 53 X’Y/OO’ g 283 Effect of attached versus free sex chromosomes on imprinting of Dp(1;f)LJ9. Paternal cross attached chromosomes visual 0.10 ± 0.02 estimate pigment 41±2 assay free chromosomes Maternal cross attached chromosomes free chromosomes 0.071 ± 0.03 0.92 ± 0.01 0.73 ± 0.03 37±9 100±3 85±3 284 identifying the location of imprinting genes (imprinting genes are those genes which control the imprinting process as opposed to imprinted genes) in mice, (Allen, Norris and Surani 1990, Babinet et al 1990, Cattanach and Beechey 1990, DeLoia and Softer 1990, Reik, Howlett and Surani 1990, Surani et al 1990, Engler et al 1991, Foreijt and Gregorova 1992 and Sapeinza et al 1992, Chaillet et al. 1995) and humans (Sapienza -personal communication). However these genes have not been otherwise characterized. As imprinting in mammals is early acting and an important developmental process, the genes which effect it are likely to be both pleiotropic and lethal when mutant. Such genes cannot easily be detected by crossing different mice strains each homozygous for potential modifier genes. Human genetics poses similar but more extreme problems. For the purpose of identifying, cloning and characterizing such genes, the sophisticated genetic and molecular tools available in Drosophila would make this organism an excellent system for examining and dissecting imprinting phenomenon. The existence of genetic modifiers of imprinting in Drosophila is suggested by the data shown in Table 35. This table shows that the extent of the imprint differs slightly, but reproducibly, when the mini-chromosome is transmitted by females with free versus attached X chromosomes. This difference in the extent of variegation may represent segregation of sex linked imprinting genes with minor effects in the genetic background of these two strains. In summary, the garnet gene has been used as a tool to examine various biological processes. This chapter describes a mini-chromosome which is subject to parent specific imprinting. This imprint is manifest as a parent-dependent variegation of the garnet gene. The result is imprinted expression of the garnet gene. The genetic and molecular information on the garnet gene allowed me to test several factors which have been postulated to cause imprinting. The primary finding is that heterochromatin, which is implicitly involved in imprinting in a wide variety of organisms, may act as a 285 memory mechanism, not a primary determinant of the imprint. If heterochromatin is relegated to a “memory” mechanism, as is methylation in mammals, then there must be an independent system which is involved in establishing the imprint. This process is undoubtedly complex but isolation of modifiers of imprinting should help to illuminate the mechanics and possibly the evolutionary and developmental rational of the phenomenon of genomic imprinting. 286 Bibliography Allen, N. D., D. G. Cran, S. C. Barton, S Hettle, W. Reik and M. A. Surani. 1988. Transgenes as probes for active chromosomal domains in mouse development. Nature 333: 852-855. Allen, N. D., M. L. Norris and M. A. Surani. 1990. Epigenetic control of transgene expression and imprinting by genotype-specific modifiers. Cell 61: 853-861. Altenburg, L. S. and E. Alternberg. 1959. The mutagenicity of 2,5-bis-ethylene hydroquinone in Drosophila. Genetics 44: 498. Babinet, C., V. Richoux, J.-L. Guénet and J.-P. Renard. 1990. The DDk inbred strain as a model for the study of interactions between parental genomes and egg cytoplasm in mouse pre implantation development. p. 81-87 In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Baker, W. K.. 1963. Genetic control of pigment differentiation in somatic cells. Am. Zool. 3: 57-69. Baker, W. K. and J. B. Spofford. 1959. Heterochromatic control of position-effect variegation in Drosophila. Biological Contributions. Univ. Texas PuIb. 5914:135-154. Barlow, D. P. 1994. Imprinting: a gamete’s point of view. Trends Genet. 10: 194199. Bartolomei, M. S., A. C. Webber, M. E. Brunkow and S. M. Tilghman. 1993. Epigenetic mechanisms underlying the imprinting of the mouse H19 gene. Genes and Devel. 7: 1663-1673. Beadle, G. W. 1937a. Development of eye colors in Drosophila: Fat bodies and malpighian tubes in relation to substances. Genetics 22:587-611. Beadle, G. W. 1 937b. The inheritance of the color of malpighian tubes in Drosophila melanogaster. Amer. Nat. 71: 277-279. Beadle, G. W. and B. Ephrussi. 1935a. Différenciation de Ia couleur de l’oeil cinnabar chez a Drosophile (Drosophila melanogaste,). Comptes Rendus Acad. Sci. Paris 201: 642-646. Beadle, G. W. and B. Ephrussi. 1935b. Transplantation in Drosophila. Proc. Nat. Acad. Sci. (USA) 21:642-646. Beadle, G. W. and B. Ephrussi. 1936. The differentiation of eye pigments in Drosophila as studied by transplantation. Genetics 21: 225-247. 287 Beadle, G. W. and B. Ephrussi. 1937. Development of eye colors in Drosophila: Diffusible substances and their interrelations. Genetics 22: 76-86. Bel, Y. and J. Ferré. 1986. Biosynthesis of pteridines and metabolism of aromatic amino acids in Drosophila melanogaster. p. 335-338 In: Chemistry and biology of pteridines. Walter de Greuyer and Co. Berlin, N.Y. Belote, J. M. and J. C. Lucchesi. 1980. Male-specific lethal mutations of Drosophila melanogaster. Genetics 96: 165-186. Benzer, S. 1955. Fine structure of a genetic region in bacteriophage. Proc. NatI. Acad. Sci. USA. 41 :344-354. Bingham, P. M., and B. H. Judd. 1981. A copy of the copia transposable element is very tightly linked to the wa allele of the white locus of D. melanogaster. Cell 25: 705-711. Birchier, J. A., U. Bhadra, L. Rabinow, R. Linsk and A. T. Nguyen-Huynh. 1994. Weakener of white (Wow), a gene that modifies the expression of the white eye color locus and that suppresses position effect variegation in Drosophila melanogaster. Genetics 137: 1057-1070. Bird, E. D., A. J. Caro and J. B. Pilling. 1974. A sex related factor in the inheritance of Huntington’s chorea. Ann. Human Genet. 37: 255-260. Blakeslee, R. W. 1938. Report of the Director. Carnagie Institute of Washington Yearbook. 42: p156-i 61. Bonse, A.. 1967. Untersuchungen uber die chemische Natatur und die Bildung der Harnkong lomerate in die Malpighischen Gefassen der Mutante rosy von Drosophila melanogaster. Z. Naturforsh. 22: 1027-1029. Brandeis, M., T. Kafri, M. Ariel, J. R. Chaillet, J. McCarrey, A. Razin and H. Cedar. 1993. The ontogeny of allele-specific methylation associated with imprinted genes in the mouse. EMBO J. 12: 3669-3677. Brehme, K. S. and M. Demerec. 1942. A survey of malpighian tube color in the eye color mutants of Drosophila melanogaster. Growth 6: 351-355. Bridges, C. B. 1916. Non-disjunction as proof of the chromosome theory of heredity. Genetics 1:1-52 and 107-163. Bridges, C. B. and K. A. Breme. 1944. The mutants of Drosophila melanogaster. Carnagie Inst. Wash. PubI. 552. Brown, G. M. 1989. Biosynthesis of H4Biopterin and related compounds. In Biology and Chemistry of Pteridines. Ed: H.-Ch. Curtius, S. Ghisla, N. Blau. Walter de Gruyter and Co. Berlin, N. York. p. 199-21 2. 288 Brown, G. M. and C. L. Fan. 1975. The synthesis of pterins catalyzed by enzymes from Drosophila melanogaster. In: Chemistry and Biology of Pteridines. Ed: W. Pfleiderer. Walter de Gruyter, Berlin, N. York. Brown, G. M., G. G. Krivi, C. L. Fan, and T. R. Unnasch. 1978. The biosynthesis of pteridines in Drosophila melanogaster. In Biology and Chemistry of Pteridines. Ed: Kisliuk and Brown. Elsevier North Holland, Inc. p. 81-86. Brun, L. 0., P. Borsa, V. Gaudichon, J. J. Stuart, K. Aronstein, C. Coustau and R. H. ffrench Constant. 1995. Functional haplodiploidy. Nature 374:506. Calatayud, M. T., D. A. Jacobson and J. Ferré. 1989. New eye colour mutants affecting the biosynthesis of pteridine and ommochromes in Drosophila melanogaster. In Chemistry and Biology of Pteridines. Ed: H.-Ch. Curtius, S. Ghisla and N. Blau. Walter de Gruyter and Co. N. York, Berlin. p. 591 -594. Campos-Ortega, J. A. 1988. Cellular interactions during early neurogenesis of Drosophila melanogaster. Trends Neurosci. 11: 400-405. Carlson, E. A. 1959. Comparative genetics of complex loci. Quart. Rev. Biol. 34:33-67. Casteel, D. B. 1929. Histology of the eyes of X-rayed Drosophila. J. Exp. Zool. 53: 373-385. Castro-Sierra, E. and S. Ohno. 1968. Allelic inhibition at the autosomally inherited gene locus for liver alcohol dehydrogenase in chicken-quail hybrids. Biochem. Genet. 1: 323-335. Cattanach, B. M. and C. V. Beechey. 1990. Autosomal and X-chromosome imprinting. p. 53-72. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Cattanach, B. M. and J. N. Perez. 1970. Parental influence on X-autosome translocation-induced variegation in the mouse. Genet. Res. Camb. 15:43-53. Chaillet, J. R., D. S. Badar and P. Leder. 1995. Regulation of genomic imprinting by gametic and embryonic processes. Genes Dev. 9: 1177-1187. Chandra, H. S. and S. W. Brown. 1975. Chromosome imprinting and the mammalian X chromosome. Nature 253: 165-168. Chandra, H. S. and V. Nanjundiah. 1990. The evolution of genomic imprinting. p. 47-53. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Chovnick, A. 1957. Pseudoallelism at the garnet locus in Drosophila melanogaster. Genetics 42:365. 289 Chovnick, A. 1958. Aberrant segregation and pseudoalleleism at the garnet locus in Drosophila melanogaster. Proc. Nati. Acad. Sd. USA. 44:333-337. Chovnick, A. 1958. Structural and functional aspects of pseudoallelism in Drosophila melanogaster. Proc. 10th mt. Cong. Genetics. 2:49-50. Chovnick, A. 1961. The garnet locus in Drosophila melanogaster. 1. Pseudoallelism. Genetics 46:493-507. Chovnick, A. 1989. Intragenic recombination in Drosophila: The rosy locus. Genetics 123:621-624. Chovnick, A., A. Schalet, R. P. Kemaghan and M. Krauss. 1964. The rosy cistron in Drosophila meIanogaster genetic fine structure analysis. Genetics 50:12451259. Chovnick, A., R. J. Lefkowitz and D. R. McQuinn. 1956. Complexity at the garnet locus in Drosophila melanogaster. Genetics 41:637. Christensen, H. N. 1973. On the development of amino acid transport systems. Fed. Proc. 32: 19-37. Clarke, A. 1990. Genetic imprinting in clinical genetics. p. 131-139. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Cohen, J.. 1962. Position-effect variegation at several closely linked loci in Drosophila melanogaster. Genetics 47:647-659. Cremer-Bartels, G. 1975. Pteridines in the mammalian retina and light effects. p. 861-870. In: The chemistry and Biology of Pteridines. Ed: W. Pfleiderer. Walter de Gruyter, Berlin, N. York. Crouse, H. V.. 1960. The controlling element in sex chromosome behavior in sciara. Genetics 45: 1429-1443. DeLoia, J. A. and D. Solter. 1990. A transgene insertional mutation at an imprinted locus in the mouse genome. p. 73-79. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Demakova, 0. V. and E. S. Belyaeva. 1988. Effect of mating direction on the 7 in Drosophila melanogaster. Dros. position effect variegation of T(1;2)dorvar Info. Service. 67:19-20. Dobzhansky, T. 1946. Genetics of natural populations. XIII. Recombination and variability in populations of Drosophila pseudoobscura. Genetics 31: 269-290. 290 Dorer, D. R. and S. Henikoff. 1994. Expansions of transgene repeats cause heterochromatin formation and gene silencing in Drosophila. Cell 77:993-1002. Dorn, R., J. Szidonya, G. Korgo, M. Schnert, H. Taubert, E. Archoukieh, B. Tschiersch, H. Morwietz, G. Wustmann, G. Hoffman and G. Reuter. 1993. P transposon-induced dominant enhancer mutations of position-effect variegation in Drosophila melanogaster. Genetics 133: 279-290. Dorn, R., V. Krauss, G. Reuter and H. Saumweber. 1993. The enhancer of position-effect variegation of Drosophila, E(var)3-93D, codes for a chromatin protein containing a conserved domain common to several transcriptional regulators. Proc. Nat. Acad. Sci. USA 90:11376-11380. Dorsett, D. L., J. J. Yim and K. B. Jacobson. 1978. Biosynthesis of drosopterins in the head of Drosophila melanogaster. In: The chemistry and Biology of Pteridines. Ed: R. L. Kisliuk and G. M. Grown. Elsevier, North Holland. p. 99-104. Dressen, T. D., D. H. Johnson and S. Henikoff. 1988. The brown protein of Drosophila melanogaster is similar to the white protein and to components of active transport complexes. Mol. Cell Biol. 8:5206-5215. Dryja, T. P., S. Mukai, R. Petersen, J. M. Rapaport, D. Walton and D. W. Yandell. 1989. Parental origin of mutations of the retinoblasoma gene. Nature 339: 556558. Eberl, D. F., B. J. Duyf and A. J. Hilliker. 1993. The role of heterochromatin in the expression of a heterochromatic gene, the rolled locus of Drosophila melanogaster. Genetics 134: 277-292. Engler, P., D. Haasch, C. A. Pinkert, L. Doglio, M. Glymour, R. Brinster and U. Storb. 1991. A strain-specific modifier on mouse chromosome 4 controls the methylation of independent transgene loci. Cell 65: 939-947. Ephrussi, B. and G. W. Beadle. 1935a. La transplantation des disques imaginaus chez Ia Drosophile. Comptes Rendus Acad. Sci. Paris. 201: 98-99. Ephrussi, B. and G. W. Beadle. 1935b. La transplantation des ovaires chez Ia Drosophile. Bull. Biol. 69: 492-502. Ephrussi, B. and G. W. Beadle. 1935c. Sur les conditions de l’autodifferenciation des carateres mendeliens. Comptes Rendus Acad. Sci. Paris. 201: 1148-1150. Ephrussi, B. and G. W. Beadle. 1937a. Development of eye colors in Drosophila: Production and release of cn+ substance by the eyes of different eye color mutants. Genetics 22: 479-483. 291 Ephrussi, B. and G. W. Beadle. 1 937b. Development of eye colors in Drosophila: Transplantation experiments on the interaction of vermilion with other eye colors. Genetics 22: 65-75. Ephrussi, B. and J. L. Herold. 1944. Studies of eye pigments of Drosophila. I. Methods of extraction and quantitative estimation of the pigment components. Genetics 29: 148-175. Ferguson-Smith, A. C., H. Sasaki, B. M. Cattanach and M. A. Surani. 1993. Parental-origin-specific epigenetic modification of the mouse H19 gene. Nature 362: 751-755. Ferré, J., F. Silva, M. D. Real and J. L. Mensua. 1983. Comparative study of the eye colour mutants of Drosophila meIanogaster Quantitation of the eyepigments and related metabolites. In Chemistry and Biology of Pteridines. Ed: J. A. Blair. Walter de Gruyter and Co. N. York, Berlin. p. 669-673. Ferré, J., F. Silva, M. D. Real and J. L. Mensua. 1986. Pigment patterns in mutants affecting the biosynthesis of pteridines and xanthomatin in Drosophila melanogaster. Biochem. Genet. 24: 545-569. Flavell, R. B. and M. O’Dell. 1990. Variation in inheritance cytosine methylation patterns in wheat at the high molecular weight glutinin and ribosomal RNA gene loci. p. 15-20. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Forejt, J. and S. Gregurova. 1992. Genetic analysis of genomic imprinting: An imprintor- 1 gene controls inactivation of the paternal copy of the mouse Tme locus. Cell 70: 443-450. Frankham, R. 1988. Molecular hypotheses for position-effect variegation: antisense transcription and promotor occlusion. J. Theor. Biol. 135: 85-1 07. Fuyama, Y. 1984. Gynogenesis in Drosophila melanogaster. Jpn. J. Genetics 59: 91-96. Gilchrist E. J. and D. G. Moerman. 1992. Mutations in the sup-38 gene of Caenorhabditis elegans suppress muscle-attachment defects in unc-52 mutants. Genetics 132: 431-442. Glaser, R. L, and A. C. Spradling. 1994. Unusual properties of genomic DNA molecules spanning the euchromatic heterochromatic junction of a Drosophila minichromosome. Nuc. Acids. Res. 22: 5068-5075. - Glass, H. B. 1934. A study of dominant mosaic eye-color mutants on Drosophila. I. Phenotypes and loci involved. Amer. Nat. 68: 107-114. Glassman, E. 1956. Kynurenine formamidase in mutants of Drosophila. Genetics 41: 5666-574. 292 Goldberg, A., A. Schalet and A. Chovnick. 1962. On the lethality of double 3 and various ry mutant alleles. Dros. Info. Serv. 36: 67-68. mutants of Hnr Green, H. 1993. Human genetic diseases due to codon reiteration: Relationship to an evolutionary mechanism. Cell 74: 955-956. Green, M. M. 1959. Spatial and functional properties of pseudoalleles at the white locus in Drosophila melanogaster. Heredity 13:303-315. Gumaratne, P. H., A. Mansukhani, S. E. Lipari, H. C. Loiu, P. W. Matindale and M. L. Goldberg. 1986. Molecular cloning, germ-line transformation and transcriptional analysis of the zeste locus of Drosophila melanogaster. Proc. Nat. Acad. Sd. USA 83: 701-705. Gunaratne, P. H., M. Nakao, D. H. Ledbetter, J. S. Sutcliffe and A. C Chinault. 1995. Tissue-specific and allele-specific replication timing controls in the imprinted human Prader-Willi syndrome region. Genes Dev. 9: 808-820. Haas, 0. A., A. Argyriou-Tirita and T. Lion. 1991. Parental origin of the chromosomes involved in the translocation t(9;22). Nature 359: 414-41 6. Hadchouel, M., H. Farza, D. Simon, P. Tiollais and C. Pourcel. 1987. Maternal inhibition of hepatitis B surface antigen gene expression in transgenic mice correlates with de novo methylation. Nature 329: 454-456 Hadorn E. 1962. Fractionating the fruitfly. Sci. Am (April): 101-110. Hadorn, E. and H. K. Mitchell. 1951. Properties of mutants of Drosophila melanogaster and changes during development as revealed by paper chromatography. Proc. Nat. Acad. Sci. 37: 650-665. Hall, J. G. 1990. How imprinting is relevant to human disease. p. 141-148. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Hardy, R. W., D. L. Lindsley, K. J. Livak, B. Lewis, A. L. Siversten, G. L. Joslyn, J. Edwards and S. Bonaccorsi. 1984. Cytogenetic analysis of a segment of the Y chromosome of Drosophila melanogaster. Genetics 107: 591-610. Harnly, M. H. and B. Ephrussi. 1937. Development of eye colors in Drosophila: Time of action of body fluid on cinnabar. Genetics 22:393-401. Hartmann-Goldstein, I. J.. 1967. On the relationship between heterochromatinization and variegation in Drosophila, with special reference to temperature sensitive periods. Genet. Res. 10: 143-1 59. Hayman, D. L. and R. H. Maddern. 1969. Report-D. melanogaster-new mutants. Dros. Info. Service 44:69. 293 Hearl, W. G., D. Dorsett and K. B. Jacobson. 1983. The common precursor of sepiapterin and drosopterin in Drosophila: enzymatic and chemical synthesis. In Chemistry and Biology of Pteridines. Ed: J. A. Blair. Walter de Gruyter and Co. N. York, Berlin. p. 397-401. Hearl, W. G., K. B. Jacobson. 1984. Eye pigment granules of Drosophila melanogaster. Isolation and characterization for synthesis of sepiapterin and precursors of drosopterin. Insect Biochem. 14:329-335. Henikoff, S., D. Nash, R. Hards, J. Bleskan, J. F. Wodford, F. Naguib and D. Patterson. 1986. Two Drosophila melanogaster mutations block successive steps of de novo purine synthesis. Proc. Nat. Acad. Sci. USA 83: 3919-3923. Heslop-Harrison. 1990. Gene Expression and parental dominance in hybrid plants. p. 21-28. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Hess 0. 1970. Independence between modification of genetic position effects and formation of Iampbrush loops by the Y chromosome of Drosophila hydel. Mol. Gen. Genet. 107: 224-242. Hessler, A. 1958. V-type position effects at the light locus in Drosophila melanogaster. Genetics 43: 395-403. Hessler, A. Y.. 1961. A study of parental modification of variegated position effects. Genetics 46: 463-484. Hexter, W. H. 1956. Pseudoallelism at the g locus. Dros. Info. Serv. 30:121. Hexter, W. M. 1958a Probable gene conversion in Drosophila. Proc. 10th Int. Cong. Genetics. 2:120. Hexter, W. M. 1 958b. On the nature of the garnet locus in Drosophila melanogaster. Proc. NatI. Acad. Sci. USA. 44:768-771. Hilliker, A. J., B. Duyf, D. Evans and J. P. Phillips. 1992. Urate-null rosy mutants of Drosophila melanogaster are hypersensitive to oxygen stress. Proc. Nat. Acad. Sci. 89: 4343-4347 Holliday R. 1990. Genomic imprinting and allelic exclusion. p. 125-129. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Holliday, R. 1987. The inheritance of epigenetic defects. Science 238: 163-170. Hopkins, F. G. 1889. Note on yellow pigments in butterflies. Nature 40: 335. 294 Howland, R., E. A. Glancy and B. P. Sonnenblick. 1937. Transplantation of wild type and eye disks among four species of Drosophila. Genetics 22:434-442. Janning, W. 1970. Bestimmung des heterochromatisierungsstodiums beim white-positionseffekt mittles röntgen induzierter mitotischer rekombination in der augenanlage von Drosophila melanogaster. Mol. Gen. Genet. 107: 128-149. Johannsen, A. 0. 1924. Eye structure in normal and eye-mutant Drosophila. J. Morph. 39: 337-350. Jones, W.K. and J. M. Rawls. 1988. Genetic and molecular mapping of chromosome region 85A in Drosophila melanogaster. Genetics 120: 733-742. Jowett, T. 1986 Preparation of nucleic acids p.275-286. In: Drosophila a practical approach. Ed. D. E. Roberts. IRL Press, Oxford, Washington. - Kajii, T. and K. Ohama. 1977. Androgenic origin of hydatidiform mole. Nature 268: 633-634. Kamdor, K. P., M. E. Shelton and V. Finnerty. 1994. The Drosophila molybdenum cofactor gene cinnamon is homologous to three EscherichIa coli cofactor proteins and to the rat protein gephyrin. Genetics 137: 791 -801. Karpen, G. H.. 1994. Position-effect variegation and the new biology of heterochromatin. Curr. Op. Genet. and Dev. 4: 281-291. Karpen, G. H. and A. C. Spradling. 1990. Reduced DNA polytenization of a minichromosome region undergoing position-effect variegation in Drosophila. Cell 63: 97-107. Keith, T.P., M. A, Riley, M. Kreitman, R. C. Lewantin, D. Curtis and G. Chambers. 1987. Sequence of the structural gene for xanthine dehydrogenase (rosy locus) in Drosophila melanogaster. Genetics 116: 67-73. Kermicle, J. L. and M. Alleman. 1990. Gametic imprinting in maize in relation to the angiosperm life cycle. p. 9-14. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Khesin, R. B., and V. N. Bashkirov. 1978. Maternal influence upon the V-type gene position effect in Drosophila melanogaster. Molec. Gen. Genet. 163: 327334. KIar, A. J. 1987. Differentiatied parental DNA strands confer developmental asymmetry on daughter cells in fission yeast. Nature 326: 466-470. KIar, A. J. 1990. Regulation of fission yeast mating-type interconversion by chromosome imprinting. p3-8. In: Genomic imprinting (development 1990 295 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Kuhn, D. T. 1972. Report-D. melanogaster-new mutants. Dros. Info. Service 49:38. Laird, C. D. 1987. Proposed mechanism of inheritance and expression of the human fragile-X syndrome of mental retardation. Genetics. 117: 587-599. Laird, C. D. 1990. Huntington’s disease: proposed mechanism of mutation, inheritance and expression. Trends Genet. 6: 242-247. Laurenson, P. and J. Rine. 1992. Silencers, silencing and heritable transcriptional states. Microbiol. Rev. 56: 543-560. Lederer, E. 1940. Les pigments des invertebres. Biol. Rev. Camb. Phil. Soc. 15: 273-306. Lewis, E. B. 1951. Pseudoallelism and gene evolution Cold Spring Harbor Symp. Quant. Biol. 16:159-174. Linder, D., B. McCaw, X. Kaiser and F. Hecht. 1975. Parthenogenetic origin of benign ovarian teratoma. New EngI. J. Med. 292: 63-66. Lindsley, D. L. and G. G. Zimm. 1992. The genome of Drosophila melanogaster. Academic Press. N.Y. Livak, K. J.. 1990. Detailed structure of the Drosophila melanogaster Stellate genes and their transcripts. Genetics 124: 303-31 6. Lock, L. F., N. Takugi and G. R. Martin. 1987. Methylation of the Hprt gene in the inactive X occurs after chromosome inactivation. Cell 48: 39-46. Lucchesi, J. C. 1968. Synthetic lethality and semi-lethality among functionally related mutants of Drosophila melanogaster. Genetics 59: 37-44. Lyon, M. F. 1993. Epigenetic inheritance in mammals. Trends Genet. 9: 123128. Mackay, W. J. and J. M. O’Donnell. 1983. A genetic analysis of the pteridine biosynthitic enzyme, guanosine triphosphate cyclohydrolase, in Drosophila melanogaster. Genetics 105: 35-53. Mainx, F. 1938. Analyse der genwirkung durch faktoren kombination. Versuche mit den augenfarbenfaktoren von Drosophila melanogaster. Z. lnductiv. Abstammungs-Vererbungslehre (Mol. Gen. Genet.) 75: 256-276. Mattaj, I, W.. 1994. Splicing in space. Nature 372:727-728. 296 Matzke, M. A. and A. J. M. Matzke. 1995. Homology-dependent gene silencing in transgenic plants: what does it really tell us? Trends Genet. 11: 1-3. McCarthy, A. and H. Nickla. 1980. Morphology of the carnation-light synthetic lethal focus in Drosophila melanogaster. Experimentia 36:1361-1362. McClintock, B. 1944. The relation of homozygous deficiencies to mutations and allelic series in maize. Genetics 29: 478-502. McLean, J. R., R. Boswell and J. O’Donnell. 1990. Cloning and molecular characterization of a metabolic gene with developmental functions in Drosophila. Genetics 126: 1007-1019. Metz, C. W.. 1938. Chromosome behavior, inheritance and sex determination in Sciara. Am. Naturalist 72:485-520. Meyerowitz, E. M. and D. R. Kankel. 1978. A genetic analysis of visual system development in Drosophila melanogaster. Dev. Biol. 62: 112-142 Modelell, J., W. Bender and M. S. Messelson. 1983. Drosophila melanogaster mutations suppressible by the suppressor-of-Hairy-wing are insertions of a 7.3 kb mobile element. Proc. Nat. Acad. Sci. USA 80: 1678-1 682. Monk, M. 1988. Genomic imprinting. Genes and Dev. 2: 921-925. Monk, M. and M. Grant. 1990. Preferential X-chromosome inactivation, DNA methylation and imprinting. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Moore, T., and D. Haig. 1991. Genomic imprinting in mammalian development: A parental tug-of-war. Trends Genet. 7: 45-49. Mori, K. 1937. A study on the development of pigments in various eye color mutants of Drosophila. Japan. J. Genet. 13: 81-99. Moses, K., M. C. Ellis and G. M. Rubin. 1989. The glass gene encodes a zincfinger protein required by Drosophila photoreceptor cells. Nature 340: 531-536. Mottus, R., R. Reeves and T. A. Grigliatti. 1980. Butyrate suppression of position effect variegation in Drosophila melanogaster. Mol. Gen. Genet. 178: 465-469. Muller, H. J. 1958. An androgenic homozygous male. Dros. Info. Serv. 32: 140. Nájera, C. 1985. Report-D. melanogaster-new mutants. Dros. Info. Service 61 :21 5. Narayanan, Y. and J. A. Weir. 1964. Paper chromatography of pteridines of prune and clot stocks of Drosophila melanogaster. Genetics 50: 387-392. 297 Nash, W. G. 1971. deep orange and carnation: Another lethal gene combination in D. m. Dros. Info. Serv. 47:73. Nicholls, R. B., J. H. M. Knoll, M. G. Butler, S. Karam and M. Lalande. 1989. Genetic imprinting suggested by maternal heterodisomy in non-deletion Prader Willi syndrome. Nature 342: 281 -285. Nickla, H. 1977. Maternal effects determine effect lethal phase of carnation-light synthetic lethal in Drosophila melanogaster. Nature 268:638-639. Nickla, H., T. Lilly and A. McCarthy. 1980. Gene activity in the carnation-light synthetic lethal in Drosophila melanogaster. Experimentia 36:402-403. Nolte, D. J. 1943. Appearance of unexpected eye colors. Dros. Info. Serv. 17:63. Nolte, D. J. 1944. White of y wan interaction-product. Dros. Info. Serv. 18: 54. Nolte, D. J. 1950. The eye pigmentary system of Drosophila: The pigment cells. J. Genetics 50:79-99. Nolte, D. J. 1 952a. The eye pigmentary system of Drosophila: II. Phenotypic effects of gene combinations. J. Genetics 51:130-141. Nolte, D. J. 1952b. The eye pigmentary system of Drosophila: III. The action of eye-colour genes. J. Genetics 51:142-186. Nolte, D. J. 1 954a. The eye pigmentary system of Drosophila: IV. The pigments of the vermilion group of mutants. J. Genetics 52:111-126. Nolte, D. J. 1954b. The eye pigmentary system of Drosophila: V. The pigments of the light and dark groups of mutants. J. Genetics 52:127-135. Nolte, D. J. 1955. The eye pigmentary system of Drosophila: VI. The pigments of the ruby and red groups of mutants. J. Genetics 53:1-10. Nolte, D. J. 1959a. The eye pigmentary system of Drosophila: VII. The white locus. Heredity 13:219-231. Nolte, D. J. 1959b. The eye pigmentary system of Drosophila: VIII. Series of multiple alleles. Heredity 13:233-241. Nolte, D. J. 1 959c. The eye pigmentary system of Drosophila: IX. Heterozygous effects of eye-colour genes. Heredity 13:219-231. Nur, U. 1970. Translocations between eu- and heterochromatic chromosomes, and spermatocytes lacking a heterochromatic set in male mealy bugs. Chromosoma 29: 42-61. 298 Nur, U. 1990. Heterochromatinization and euchromatinization of whole genomes in scale insects (Coccoidea: Homoptera). P. 29-34. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Nur, U., J. H. Werren, D. G. Eickbush, W. D. Burke and T. H. Eichbush. 1988. A “selfish” B chromosome that enhances its transmission by eliminating the paternal genome. Science 240: 512-514. Ohlsson, R., D. Barlow and A. Surani. 1994. Impressions of imprints. Trends Genet. 10: 41 5-41 7. Orlando, V. and R. Paro. 1995. Chromatin multiprotein complexes involved in the maintenance of transcription patterns. Curr. Op. Genet. and Dev. 5: 174-179. Pak, W. L., Grabowski, S. R. 1980. Physiology of the visual and flight systems. In: Genetics and Biology of Drosophila, Ed. M. Ashburner and E. Noviski. Academic Press, London, New York, Sidney, Toronto and San Francisco. Palumbo, G., S. Bonaccorsi, L. G. Robbins and S. Pimpinelli. 1994. Genetic analysis of Stellate elements of Drosophila melanogaster. Genetics 138: 11811197. Parkhurst, S. M. and V. G. Corces. 1985. forked, gypsys and suppressors in Drosophila. Cell 41: 429-437. Paro, R. 1990. Imprinting a determined state into the chromatin of Drosophila. Trends in Genetics 6: 116-121. Patterson, G. I., C. J. Thorpe and V. L. Chandler. 1993. Paramutation, an allelic interaction, is associated with a stable and heritable reduction of transcription of the maize b regulatory gene. Genetics 135: 881 -894. Payne, F. and M. Denny. 1921. The heredity of orange eye color in Drosophila melanogaster. Am. Nat. 55: 377-381. Pfleiderer, W. 1993. Natural pteridines A chemical hobby. In: Chemistry and biology of pteridines and folates. p. 1-16. Eds: J. E. Ayling, M. G. Nair and C. M. Baugh. Plenum Press, N. York. - Phillips, J. P and H. S. Forrest. 1976. Ommochromes and Pteridines. p. 541617. In: Genetics and Biology of Drosophila. Ed. Ashburner and Noviski, Academic Press, N.Y. Phillips, J. P, H. S. Forrest and Kulkarni. 1973. Terminal synthesis of xanthommatin in Drosophila melanogaster. Ill. Mutational pleiotropy and pigment granule association of phenoxazinone synthesis. Genetics 73: 45-56. 299 Pirrota, V., H. Stellar and M. P. Bozzetti. 1985. Multiple upstream regulatory elements control the expression of the Drosophila white gene. EMBO J. 4: 3501-3508. Prokofyeva-Belgovskaya, A. A.. 1947. Heterochromatinization as a change of chromosome cycle. J. of Genetics 48:80-98. Rabinow, L., A. T. Nguyen-Huyah and J. A. Birchler. 1991. A transacting regulatory gene that inversely affects the expression of the white, brown and scarlet loci in Drosophila Genetics 129: 463-480. Rabi, C. 1885. Uber Zelltheilung. Morphologisches Jahrbuch 10: 214-330. Rangunathang, R., W. A. Harris and C. S. Zuker. 1991. The molecular genetics of invertebrate phototransduction. Trends Neurosci. 14: 486-493. Ready, D. P. 1989. A multifaceted approach to neural development. Trends Neurosci. 12: 102-110. Real, M. D., J. Ferré and J. L. Mensua. 1985. Methods for the quantitative estimation of the red and brown pigments of Drosophila melanogaster. Dros. Info. Serv. 61:198-1 99. Reame, A. G., D. A. Knecht and A. Chovnick. 1991. The rosy locus in Drosophila melanogaster Xanthine dehydrogenase and eye pigments. Genetics 129: 1099-1109. Reedy, J. J. and F. P. Cavalier. 1971. Epistasis in eye colors of Drosophila melanogaster. J. Heredity 62:131-134. Reik, W. 1992. Genomic imprinting in mammals. p. 203-229. In: Results and problems in cell differentiation 18. Ed. W. Hennig. Springer-Verlag. Berlin, Heidelberg. Reik, W., S. K. Howlett and M. A. Surani. 1990. Imprinting by DNA methylation: From transgenes to endogenous gene sequences. p. 99-106. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Renfranz, D. J. and S. Benzer. 1989. Monoclonal antibody probes discriminate early and late mutant defects in development of the Drosophila retina. Dev. Biol.. 136: 411-429. Reuter, G., I. Wolff and B. Friede. 1985. Functional properties of the heterochromatic sequences inducing m4 position-effect variegation in Drosophila melanogaster. Chromosoma 93: 132-139. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: A laboratory manual. Ed: C. Nolan, Cold Spring Harbor Laboratory Press, N.Y. 300 Sapienza, C. 1990. Sex-linked dosage-sensitive modifiers as imprinting genes. p. 107-113. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Sapienza, C., J. Paquette, P. Pannunzio, S. Albrechtson and K. Morgan. 1992. The polar-lethal Ovum Mutant gene maps to the distal portion of the mouse chromosome 11. Genetics 132: 241-246. Sasaki, H., B. A. Jones, R. J. Chaillet, A. C. Ferguron-Smith, S. C. Barton, W. Reik and M. A. Surani. 1992. Paternal imprinting: potentially active chromatin of the repressed maternal allele of the mouse insulin-like growth factor II (IGF2) gene. Genes Dev. 6: 1843-1 856. Scarbrough, K., S. Hattman and U. Nur. 1984. Relationship of DNA methylation level to the presence of heterochromatin in mealy bugs. Mol. Cell Biol. 4: 599603. Schalet, A. 1957. Spontaneous mutations at specific X chromosome loci in Drosophila melanogaster. Genetics 42:393. Schalet, A. P. 1986. The distribution of, and complementation relationships between spontaneous X-linked recessive lethal mutations recovered from crossing long-term laboratory stocks of Drosophila melanogaster. Mutation Res. 163: 115-144. Schmidtke, J. P. KuhI and W. Engel. 1976. Transitory hemizygosity of paternally derived alleles in hybrid trout embryos. Nature 260: 31 9-320. Schott, D. R., M. C. Baldwin and V. Finnerty. 1986. Molybdenum hydroxylases in Drosophila. Ill. Further characterization of the low xanthine dehydrogenase gene. Biochem. Genet.. 24:509-527. Schultz, J. 1935. Aspects of the relation between genes and development in Drosophila. Am. Nat. 69: 30-54. Schwinck, I and L. Schwinck. 1972. Report-D. melanogaster-new mutants. Dros. Info. Service 49:38. Schwinck, I. 1975. Aurodrospterins in eye colour mutants of Drosophila melanogaster. In: The chemistry and Biology of Pteridines. Ed: W. Pfleiderer. Walter de Gruyter, Berlin, N. York. p. 919-929. Schwink, I. 1978. Drosopterins and the in vivo modulation of their synthesis by implantation of metabolites in eye colour mutants of Drosophila melanogaster. In: The chemistry and Biology of Pteridines. Ed: R. L. Kisliuk and G. M. Grown. Elsevier, North Holland. p. 141-146. 301 Searles, L. L. and R. A. Voelker. 1986. Molecular characterization of the Drosophila vermillion locus and its suppressible alleles. Proc. Nat. Acad. Sci. USA 83: 404-408. Shepherd, J. C. W., U. Walldorf, P. Hug and W. J. Gehring. 1989. Fruit flies with additional expression of elongation factor EF-1 oc live longer. Proc. Nat. Acad. Sci. USA 86: 7520-7521. Shevelyov, Y. Y.. 1992. Copies of a Stellate gene variant are located in the X heterochromatin of Drosophila melanogaster and are probably expressed. Genetics 132: 1033-1037. Shoup, J. R. 1966. The development of pigment granules in the eye of wildtype and mutant Drosophila melanogaster. J. Cell Biol. 29: 223-249. Sinclair, D. A., A. A. Ruddell, J. K. Brock, N. J. Clegg, V. K. Lloyd and T. A. Grigliatti. 1992. A cytogenetic and genetic characterization of a group of closely linked second chromosome mutations that suppress position-effect variegation in Drosophila melanogaster. Genetics 130: 333-344. Spofford, J. B. 1967. Single-locus modification of position-effect variegation in Drosophila melanogaster. I. white variegation. Genetics 57: 751-766. Spofford, J. B.. 1959. Parental control of position-effect variegation: I. Parental heterochromatin and expression of the white locus in compound-X Drosophila melanogaster. Proc. Nat. Acad. Sci. 45: 1003-1007. Spofford, J. B.. 1961. Parental control of position-effect variegation. II. Effect of sex of parent contributing white-mottled rearrangement in Drosophila melanogaster. Genetics 46:1151-1167. Spofford, J. B.. 1976. Position-effect variegation in Drosophila. p. 955-1018. In Genetics and Biology of Drosophila. Ed. Ashburner and Novitski. Academic Press. N.Y. Stewart, B. and J. R. Merriam. 1973. Segmental aneuploidy of the X chromosome. Dros. Info. Service 50: 167-170. Stoger, R., P. Kubicka, C.-G. Lui, T. Katri, A. Razin, H. Cedar and D. P. Barlow. 1993. Maternal-specific methylation of the imprinted mouse lgf2r locus identifies the expressed locus as carrying the imprinting signal. Cell 73:61 -71. Sturtevant, A. H. 1932. The use of mosaics in the study of the developmental effects of genes. Proc. 6th Int. Cong. Genet., Vol. 1: 304-307. Sturtevant, A. H. 1956. A highly specific complementary lethal system in Drosophila melanogaster. Genetics 41: 118-123. 302 Sturtevant, A. H. and E. Novitski. 1941. The homologies of the chromosome elements in the genus Drosophila. Genetics 26: 517-541. Sturtevant, A. H., C. B. Bridges, T. H. Morgan, L. V. Morgan and J. U. Chili. 1929. Garnet. p. 24 In: Contributions to the genetics of Drosophila simulans and Drosophila melanogaster. Carnagie Inst. Wash. PubI. 399. Carnagie Inst. Washington. Sullivan, D. T. and M. C. Sullivan. 1975. Transport defects as the physiological basis for eye color mutants of Drosophila melanogaster. Biochem. Genet. 13: 603-613. Sullivan, D. T., R. J. Kitos and M. C. Sullivan. 1973. Developmental and genetics studies on kynurenine hydroxylase from Drosophila melanogaster. Genetics 75: 651-661. Sullivan, D. T., S. L. Grillo, and R. J. Kitos. 1974. Subcellular localization of the first three enzymes of the ommochrome synthetic pathway in Drosophila melanogaster. J. Exp. Zool. 188: 225-234. Surani, M. A., R. Kothary, N. D. Allen, P. B. Singh, R. Fundel, A. C. Reguson Smith and S. C. Barton. 1990. Genomic imprinting and development in the mouse. p. 89-98. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge Surani, M. A., W. Reik and N. D. Allen. 1988. Transgenes as molecular probes for genomic imprinting. Trends. Genet. 4: 59-62. Taira, T. 1960. Is the double recessive Hflr3 ry homozygote a synthetic lethal? Dros. Info. Service 34: 107. Talbert, P. B., C. D. S. LeCiel and S. Henikoff. 1994. Modification of the Drosophila heterochromatic mutation brownDomlflaflt by linkage alterations. Genetics 136: 559-571. Tartof, K. D. and M. Bremer. 1990. Mechanisms for the construction and developmental control of heterochromatin formation and imprinted chromosome domains. p35-45. In: Genomic imprinting (development 1990 supplement). Ed: M. Monk and A. Surani. Company of Biologists Ltd., 1990, Cambridge. Tautz, D. and C. Pfeifle. 1989. A non-radioactive in situ hybridization method for the localization of specific RNAs in Drosophila embryos reveals transitional control of the segmentation gene hunchback. Chromosoma 98:81-85. Tearle, R. G., J. M. Belote, M. Mckewan, B. S. Baker and A. J. Howells. 1989. Cloning and characterization of the scarlet gene of Drosophila melanogaster. Genetics 122: 595-606. 303 Tearle, R.. 1991. Tissue specific effects of ommochrome pathway mutations in Drosophila melanogaster. Genet. Res. Camb. 57:257-266. Teng, D. H. F., C. M. Engele and T. ft Venkatesh. 1991. A product of the prune locus of the Drosophila is similar to mammalian GTPase-activating proteins. Nature 353: 437-440. Thaker, H. M., Kankel, D. R. 1992. Mosaic analysis gives an estimate of the extent of genomic involvement in the development of the visual system in Drosophila melanogaster. Genetics 131:883-894. Toguchida, J., K. Ishizaki, M. S. Sasaki, Y. Nakumara, M. Ikenaga, M. Kato, M. Sugimot, Y. Kotoura and T. Yamamuro. 1989. Preferential mutation of the paternally derived RB gene as the initial event in sporadic osteosarcoma. Nature 338: 156-158. Tomlinson, A. 1985. Cellular interactions in the developing Drosophila eye. Development 104: 183-193. Tower, J., G. H. Karpen, N. Craig and A. C. Spradling. 1993. Preferential transposition of Drosophila P-elements to nearby chromsomal sites. Genetics 133: 347-359. Valencia, R. M. 1966. Report-D. melanogaster-new mutants. Dros. Info. Service. 41:58. Van Dyke, D. L., and L. Weiss. 1986. Maternal effect on intelligence in fragile X males and females. Am. J. Med. Genet. 23: 723-737. Varmuza, S. and M. Mann. 1994. Genomic imprinting time bomb. Trends. Genet. 10: 118-123. - defusing the ovarian Venkatesh, T. R., Zipursky, S. L., Benzer, S., 1985. Molecular analysis of the development of the compound eye in Drosophila. Trends in nuerosci. 8:251257. Wakimoto, B. and M. Hearn. 1990. The effects of chromosome rearrangements on the expression of heterochromatic genes in chromosome 2L of Drosophila melanogaster. Genetics 125: 141-151. Walirath, L. L. and S. C. R. Elgin. 1995. Position effect variegation in Drosophila is associated with an altered chromatin structure. Genes and Dev. 9:1263-1277. Warner, C. K., D. T. Watts and V. Finnerty. 1980. Molybdenum hydroxylases in Drosophila. I. Preliminary studies of pyridoxal oxidase. Mol. Gen. Genet. 180: 449-453. Wennberg, R. A. 1988. A novel requirement for the X-chromosome in P-M hybrid dysgenesis and the interaction of the garnet and enhancer of garnet loci 304 in Drosophila melanogaster. MSc. Thesis. University of British Columbia. Canada. Wiederrecht, G. J. and G. M. Brown. 1984. Purification and properties of the enzymes from Drosophila melanogaster that catalyze the conversion of dihydroneopterin triphosphate to the pyrimidodiazepine precursor of the drosopterins. J. Biol. Chem. 259: 14121-14127. Wiederrecht, G. J., D. R. Paton and G. M. Brown. 1984. Enzymatic conversion of dihydroneopterin triphosphate to the pyrimido diazepine intermediate involved in the biosynthesis of the drosopterins of Drosophila melanogaster. J. Biol. Chem. 259:2 1 95-2200. Wiley, K. and H. S. Forrest. 1981. Terminal Synthesis of xanthommatin in Drosophila melanogaster. IV. Enzymatic and non enzymatic catalysis. Biochem. Genet. 19:1211-1221. Wines, D. R. and S. Henikoff. 1992. Somatic instablity of a Drosophila chromosome. Genetics 131: 683-691. Wright, 5. 1932. Complementary factors for eye color in Drosophila. Am. Nat. 66: 282-283. Yamamoto, A. H., D. J. Komma, C. D. Shaffer, V. Pirrotta and S. A. Endow. 1989. the claret locus in Drosophila encodes products required for eye color and for meiotic chromosome segregation. EMBO J. 8: 3543-3552. Zipursky, S. L. 1989. Molecular and genetic analysis of Drosophila eye development: sevenless, bride of sevenless and rough. Trends in Neurosci. 12:183-1 89. Zipursky, S. L., T. R. Vankatesh, D. B. Teplow and S. Benzer. 1984. Neuronal development in the Drosophila retina: monoclonal antibodies as molecular probes. Cell 36: 15-26. Zuckerkandl, E. 1974. Recherches sur les proprietes et l’activité biologique de Ia chromatine. Biochimie 56: 937-954. 305 Appendix 1. Determination of eye pigment levels. As the principal phenotype of the garnet and other eye colour genes described in this thesis is alterations in eye pigments, it was necessary to accurately and precisely quantify the amount of eye pigments. A. Efficiency and Selectivity of pigment extraction. The first step in quantitation of eye pigments is to extract these pigments efficiently and specifically from the eye. The different biochemical properties of the pteridine (red) and ommochrome (brown) pigments have been exploited to differentially extract those pigments from fly eyes. A number of such procedures have been published, that of Real, Ferré and Mensua (1985) was adapted for this work. It was necessary to show that these procedures efficiently extracted the intended pigments, either the pteridines or xanthommatin, without contamination by the other class of pigment. Figure A shows the pigments extracted from wild type (Oregon R), brown and vermilion mutants as a test of the efficiency and selectivity of the extraction method. The brown mutant should have no pteridine pigments whereas the vermilion should not have the ommochrome pigment. The top series of data show that the procedure for pteridine pigment extraction is both efficient and highly selective. Essentially no ommochrome pigment is extracted. The second data series show that the procedure for ommochrome pigment extraction is less selective, some pteridine pigments are extracted. It also appears less efficient as the levels of the 306 ommochrome pigment, xanthommatin, extracted from the brown mutant are consistently lower than from wild type. The extraction method for pteridine pigments was not only more specific and reproducible but considerably less time consuming than the method for the ommochrome pigment. Consequently, only the pteridine pigments were assayed in most experiments. A. Efficiency of red pigment extraction Q %O.R 100 <1% 98±5 Efficiency of brown pigment extraction O.R. average reading 106±8 73±3 5±2 %O.R. 100±8 69±8 5±2 B. Contribution of the different pteridine pigments to total pteridine pigment level. Unlike the single ommochrome pigment, there are numerous (28+) pteridine pigments, each of which may contribute to the reading obtained for the total pteridine pigments. To determine the degree to which each of these pigments contributed to the total fluorescence recorded for the pteridines, 307 chromatography plates on which the pteridine pigments had been partially separated was scanned under UV illumination using the same conditions used to quatitate total pteridine pigment levels. The percent of total fluorescence attributed to each pigment is shown in Figure B. The major contribution is from the drosopterin pigments (70%) and the second largest is from the unseparated residue (15%). Based on the distinctive orange colour, florescence of the residue is probably due largely to drosopterins. Thus approximately 85% of total fluorescence is due to drosopterin pigments. While the system could probably be optimized to preferentially detect other pigments by changing the extraction technique and the UV illumination wavelength, the drosopterins are stable molecules and a convenient indicator of pteridine pigment levels. B. Fluorescence of separated pteridine pigments. percent total pigment pigment residue drosopterin mystery spot 1 mystery spot 2 isoxanthopterin mystery spot 3 xanthopterin sepiapterin 2-amino-4-hydroxypteridine biopterin isosepiapterin 15 70 2 1 1 2 2 1 1 1 5 C. Comparison between different methods of pteridine pigment quantification. Most published methods of pigment quantification rely on a change in UV absorbance as measured by spectrophotometer. I found this method to be too cumbersome and slow to be suited for large numbers of pigment level 308 determinations. A method involving quantification of fluorescence by a microflourimeter allowed rapid scanning of multiple spots of extracted pigments. The top portion of Figure C shows a comparison between pigment levels determined by spectrophotometric assay (Y axis) and by microflourometric assay (X axis). Each reading represents the results of pigment quantification from one group of frozen fly heads. The graph incorporates data from two separate experiments. One set of data was produced by Kevin Swanson, and undergraduate research assistant. The relationship between the two methods seems to be slightly sinusoidal. This indicates that at high pigment levels the spectrophotometric method provides more discrimination. However, the lower portion of the graph shows that for low pigment levels (<20% by the microflourometric method) that the microflourometric method is the more accurate. These results are not unexpected as the two methods exploit different properties of the pigments. Nevertheless, the relationship between the two methods is roughly linear. The greater speed afforded by the microflourometric assay and the lower variability made the microflourometric assay the method of choice for pigment quantitation. Although the microflourometric assay was more sensitive than the spectrophotometric assay, it still involved combining five heads to extract sufficient pigment. Combining heads posses no problem for genotypes where all individuals should have the same amount of pigment. For mosaic phenotypes, such as the flies with variegating genotypes studied in chapter 3, each individual has different amounts of pigmentation. As combining heads would obscure these differences, I adapted the microflourometer assay to 309 measure pigment from individual heads. The lower figure shows a comparison of microflourometer readings using one head (Y axis) and 5 heads (X axis). Each assay was again performed on the same set of frozen heads and incorporate two independent sets of experiments. One set of data was provided by David Dyment, an undergraduate student working on a directed studies project. The results of the two assays are roughly comparable. There is however, discrepancy between the two assays systems at higher pigment levels. The deviation appears as a random scatter about the mean suggesting that it represents genuine differences between the pigment of individual heads and the average reading of five heads. Data derived from uniformly pigmented heads shows far less variation (data not shown). 310 Comparison between microflou rimeter and spectrophotometer assays for pteridine pigment levels 10090 U, c V (1$ a, I L. a, a, E a, 0 €0 0. (I) — — — n 0’)— 4 — 70 — n_ - — I— — — . ‘— — — — . — — — — 50— — — — — — ).4 40— 30--—-.‘.1 —— —I- _I 2” — — I 10— - 0— — 0 — — 10 20 30 40 50 60 70 80 90 100 microflourimeter readings (5 head assay) + Comparison between five and one head + + microflourimeter assay 100 + I ::41 90 ÷ , - —— *4 * — 80 70 60 50 0 E a) -c 1 40 30 20 10 0 : - - - --k-- + + + + —-÷- L± — I’ll — — — — — II 0102030405060708090100 5 head microflourimeter assay 311. + Appendix 2. Cloning of the garnet gene. The cloning of the garnet gene forms the basis of the molecular analysis recorded in chapter 2 of this thesis. This appendix is included to record the cloning of the gene. The garnet gene was cloned by Donald A. Sinclair using a P-element induced garnet gene isolated by Richard Wennberg (Wennberg 1988). A P-element induced allele of garnet, and four revertants thereof, was isolated from a naturally occurring P-element strain, S6-1 (see Figure 15). After replacement of the autosomes and most of the X-chromosome by recombination, two P-elements remained, one at cytological position 1 2B, the location of the garnet gene, and one at 1 2E. Genomic DNA isolated from the gP mutation, restricted with Eco RI and probed with the P-element containing Hind Ill fragment of the p1125.1 plasmid revealed an approximately 8.5 kb fragment. A library made from size fractionated DNA from the gP strain yielded eight P element containing clones when probed with the same P-element containing probe. Hybridization of these clones to polytene chromosmes showed that seven of these clones represented the 1 2E P-element and the last one, at cytological position 12B, potentially identified the garnet gene. Using the non P element containing DNA from this clone, a series of lambda phage clones where isolated from a wild type genomic (EMBL 3) library (Figure 17 and 30). A 6.6 Eco RI fragment common to all the phage clones was subcloned into puclg. This fragment was used to probe two c-DNA libraries (Figure 27) and a number of spontaneous garnet mutations (Figure 19 and 22). 312