Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Polycomb Group Proteins and Cancer wikipedia , lookup
Maximum parsimony (phylogenetics) wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Koinophilia wikipedia , lookup
Metagenomics wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Non-coding DNA wikipedia , lookup
Protein moonlighting wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Human genome wikipedia , lookup
Microevolution wikipedia , lookup
Gene expression programming wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
Genome evolution wikipedia , lookup
Helitron (biology) wikipedia , lookup
Expanded genetic code wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Genome editing wikipedia , lookup
Frameshift mutation wikipedia , lookup
Genetic code wikipedia , lookup
Sequence alignment wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Point mutation wikipedia , lookup
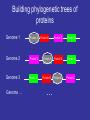
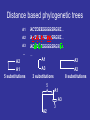
http://creativecommons.org/licens es/by-sa/2.0/ Multiple Alignments & Molecular Evolution Prof:Rui Alves [email protected] 973702406 Dept Ciencies Mediques Basiques, 1st Floor, Room 1.08 Website of the Course:http://web.udl.es/usuaris/pg193845/Courses/Bioinformatics_2007/ Course: http://10.100.14.36/Student_Server/ Part I: Multiple Alignments Pairwise Alignment • We have seen how pairwise alignments are made. • Dynamic programming is an efficient algorithm for finding the optimal alignment. – Break problem into smaller subproblems – Solve subproblems optimally, recursively – Use optimal solutions to construct an optimal solution for the original problem • Alignments require a substitution (scoring) matrix that accounts for gap penalties. Sub. Matrix: Basic idea • Probability of substitution (mutation) PAM Matrices • A family of matrices (PAM-N) • Based upon an evolutionary model • The score for a substitution of nucleotides/amino acids is based on how much we expect that substitution to be observed after a certain length of evolutionary time • The scores are derived by a Markov model – i.e., the probability that one amino acid will change to another is not affected by changes that occurred at an earlier stage of evolutionary history Nucleic acid PAM matrices • PAM = point accepted mutation • 1 PAM = 1% probability of mutation at each sequence position. • A uniform PAM1 matrix for a familiy of closely related proteins: A G T C A 0.99 0.00333 0.00333 0.00333 G 0.00333 0.99 0.00333 0.00333 T 0.00333 0.00333 0.99 0.00333 C 0.00333 0.00333 0.00333 0.99 How did they get the values for PAM-1? • Look at 71 groups of protein sequences where the proteins in each group are at least 85% similar (Why these groups?) • Compute relative mutability of each amino acid – probability of change • From relative mutability, compute mutability probability for each amino acid pair X,Y– probability that X will change to Y over a certain evolutionary time • Normalize the mutability probability for each pair to a value between 0 and 1 Transitions and transversions • Transitions (A G or C T) are more likely than transversions (A T or G C) • Assume that transitions are three times as likely: A G T C A 0.99 0.006 0.002 0.002 G 0.006 0.99 0.002 0.002 T 0.002 0.002 0.99 0.006 C 0.002 0.002 0.006 0.99 PAM-N Matrices • N is a measure of evolutionary distance • PAM-1 is modeled on an estimate of how long in evolutionary time it would take one amino acid out of 100 to change. That length of time is called 1 PAM unit, roughly 10 million years (abbreviated my). • Values in a PAM-1 matrix show the probability that an amino acid will change over 10 my. • To get the PAM-N matrix for any N, multiply PAM-(N-1) by PAM-1. Distant relatives • If a family of proteins is say, 80% homologous use a PAM 2. A G T C A 0.98014 0.011888 0.003984 0.003984 G 0.011888 0.98014 0.003984 0.003984 T 0.003984 0.003984 0.98014 0.011888 C 0.003984 0.003984 0.011888 0.98014 Computing Relative Mutability – A Measure of the Likelihood that an Amino Acid Will Mutate For each amino acid • changes = number of times the amino acid changed into something else • exposure to mutation = (percentage occurrence of the amino acid in the group of sequences being analyzed) * (frequency of amino acids changes in the group) • relative mutability = (changes/exposure to mutation) / 100 Computing Relative Mutability of A: changes = # times A changes into something else = 4 % occurrence of A in group = 10 / 63 = 0.159 frequency of all amino acid changes in group = 6 * 2 = 12 (Note: Count changes backwards and forwards.) exposure to mutation = (% occurrence of A in group) * (frequency of all amino acid changes in group) = 12 * 0.159 relative mutability = (changes / exposure to mutation) / 100 = (4 / (12 * 0.159)) = 2.09 / 100 = 0.0209 Example from Fundamental Concepts of Bioinformatics by Krane and Raymer. How can we understand relative mutability intuitively? relative mutability = changes / exposure to mutation = the number of times A changed in proportion to the the probability that it COULD have changed exposure to mutation – that were 6 times when something changed in the tree. Each time, that change could have been A changing to something else, or something else changing to A – 12 chances for a change involving A. But A appears in a sequence only .159 of the time. Computing Mutability Probability Between Amino Acid Pairs For each pair of amino acids X and Y: r = relative mutability of X c = num times X becomes Y or vice versa p = num changes involving X mutability probability of X to Y = (r * c) / p Computing Mutability Probability that A will change to G: r = relative mutability of A = .0209 c = num times A becomes G or vice versa = 3 p = num changes involving A = 4 mutability probability of A to G = (r * c) / p = (0.0209 * 3) / 4 = 0.0156 Normalizing Mutability Probability, X to Y • For each Y among all amino acids, compute mutability probability of X to Y as described above • Get a total of these 20 probabilities. Divide them by a normalizing factor such that the probability that X will NOT change is 99% and the sum of probabilities that it will change to any other amino acid is 1% • These are the numbers that go in the PAM-1 matrix! Converting Mutability Probabilities to Log Odds Score for X to Y • Compute the relative frequency of change for X to Y as follows: – Get the X to Y mutability probability – Divide by the % frequency of X in the sequence data – Convert to log base 10, multiply by 10 • In our example, we get log10(0.0156/0.1587) = log10(.098) • To compute log10(.098) solve for x: – 10x = 0.098 x = -1.01 10-1.01 = 1/101.01 = 0.098 • Compute log odds score for Y to X • Take the average of these two values Usefulness of Log Odds Scores • A score of 0 indicates that the change from one amino acid to another is what is expected by chance • A negative score means that the change is probably due to chance • A positive score means that the change is more than expected by chance • Because the scores are in log form, they can be added (i.e., the chance that X will change to Y and then Y to Z) Disadvantages of PAM Matrices • An alignment tree must be constructed first, implying some circularity in the analysis • The original PAM-1 matrix was based on a limited number of families, not necessarily representative of all protein families • The Markov model does not take into account that multi-step mutations should be treated differently from single-step ones Most Commonly-Used Amino Acid Subtitution Matrices • PAM (Percent Accepted Mutation, also called Dayhoff Amino Acid Substitution Matrix) • BLOSUM (BLOcks amino acid SUbstitution Matrix) BLOSUM Scoring Matrices • Based on a larger set of protein families than PAM (about 500 families). The proteins in the families are known to be biochemically related. • Focuses on blocks of conserved amino acid patterns in these families • Designed to find conserved domains in protein families • BLOSUM matrices with lower numbers are more useful for scoring matches in pairs that are expected to be less closely related through evolution – e.g., BLOSUM50 is used for more distantly-related proteins than BLOSUM62. (This is the opposite of the PAM matrices.) BLOSUM Matrices • Target frequencies are identified directly and not by extrapolation • Sequences more than x% identical are collapsed into a single sequence –BLOSUM 50: >=50% Identity –BLOSUM 62: >=62% Identity Building a BLOSUM Matrix • BLOSUM 62: – Collapse Sequences that have more than 62% identity into one – Calculate probability of a given pair of AAs being in same column (qij) – Calculate the frequency of a given AA (fi) – Calculate log odds ratio sij=log2(qij/fi). This is the value that goes into the BLOSUM matrix BLOSUM50 What matrix to choose? • BLOSUM Matrices perform better in local similarity searches • BLOSUM 62 is the default matrix used for database searching Gap Penalty (Gap Scoring) Gap Penalties • Gaps in the alignment are necessary to increase score. • They must be penalized; however if penalty is to high no gaps will appear • On the other hand if they are too low, gaps everywhere!!! • The default settings of programs are usually ok for their default scoring matrices Once a gap, can we widen it? >gi|729942|sp|P40601|LIP1_PHOLU Length = 645 Lipase 1 precursor (Triacylglycerol lipase) Score = 33.5 bits (75), Expect = 5.9 Identities = 32/180 (17%), Positives = 70/180 (38%), Gaps = 9/180 (5%) Query: 2038 IYSLYGLYNVPYENLFVEAIASYSDNKIRSKSRRVIATTLETVGYQTANGKYKSESYTGQ 2097 +++ YGL+ Y+ ++ Y D K +R ++ + N + G+ Sbjct: 441 VFTAYGLWRY-YDKGWISGDLHYLDMKYEDITRGIVLNDW----LRKENASTSGHQWGGR 495 Query: 2098 LMAGYTYMMPENINLTPLAGLRYSTIKDKGYKETGTTYQNLTVKGKNYNTFDGLLGAKVS 2157 + AG+ + + +P+ + KGY+E+G + + Y++ G LG ++ Sbjct: 496 ITAGWDIPLTSAVTTSPIIQYAWDKSYVKGYRESGNNSTAMHFGEQRYDSQVGTLGWRLD 555 Query: 2158 SNINVNEIVLTPELYAMVDYAFKNKVSAIDARLQGMTAPLPTNSFKQSKTSFDVGVGVTA 2217 +N P ++ F +K I + + + S KQ + +G+ A Sbjct: 556 TNFG----YFNPYAEVRFNHQFGDKRYQIRSAINSTQTSFVSESQKQDTHWREYTIGMNA 611 Real gaps are often more than one letter long. Affine gap penalty LETVGY W----L -5 -1 -1 -1 • Separate penalties for gap opening and gap extension. • This requires modifying the DP algorithm to store three values in each box. Scoring Gap Penalties • Linear Gap Penalty Score gap gap (opening penalty score) • Affine Penalty Score gap (opening penalty score) (extension penalty ) ( gap 1) • Opening a gap is costly; extending it not so much (open=12; extension=1) Multiple Sequence Alignment MSA Introduction • Goal of protein sequence alignment: – To discover “biological” (structural / functional) similarities • If sequence similarity is weak, pairwise alignment can fail to identify important features (eg interaction residues) • Simultaneous comparison of many sequences often find similarities that are invisible in PA. Why do we care about sequence alignment? • Identify regions of a gene (or protein) susceptible to mutation and regions where residue replacement does not change function. • Information about the evolution of organisms. • Orthologs are genes that are evolutionarily related, have a similar function, but now appear in different species. • Homologous genes (genes with share evolutionary origin) have similar sequences. • Paralogs are evolutionarily related (share an origin) but no longer have the same function. • You can uncover either orthologs or paralogs through sequence alignment. Multiple Sequence Alignment • Often applied to proteins (not very good with DNA) • Proteins that are similar in sequence are often similar in structure and function • Sequence changes more rapidly in evolution than does structure and function. Work with proteins! If at all possible — • Twenty match symbols versus four, plus similarity! Way better signal to noise. • Also guarantees no indels are placed within codons. So translate, then align. • Nucleotide sequences will only reliably align if they are very similar to each other. And they will require extensive hand editing and careful consideration. Overview of Methods • Dynamic programming – too computationally expensive to do a complete search; uses heuristics • Progressive – starts with pair-wise alignment of most similar sequences; adds to that (LOCAL OPTIMIZATION) • Iterative – make an initial alignment of groups of sequences, adds to these (e.g. genetic algorithms) (GLOBAL OPTIMIZATION) • Locally conserved patterns • Statistical and probabilistic methods Dynamic Programming • Computational complexity – even worse than for pair-wise alignment because we’re finding all the paths through an ndimensional hyperspace (Remember matrix, now add many dimensions) • Can align less than 20 relatively short (200-300) protein sequences in a reasonable amount of time; not much beyond that A Heuristic for Reducing the Search Space in Dynamic Programming • Consider the pair-wise alignments of each pair of sequences. • Create alignments from these scores. • Consider a multiple sequence alignment built from the individual pairwise alignments. • These alignments circumscribe a space in which to search for a good (but not necessarily optimal) alignment of all n sequences. The details • Create an “alignment of alignments” (AOA) based on pair-wise alignments (Pairs of sequences that have the best scores are paired first in the tree.) • Do a “first-cut” msa by incrementally doing pair-wise alignments in the order of “alikeness” of sequences as indicated by the AOA. Most alike sequences aligned first. • Use the pair-wise alignments and the “first-cut” msa to circumscribe a space within which to do a full msa that searches through this solution space. • The score for a given alignment of all the sequences is the sum of the scores for each pair, where each of the pair-wise scores is multiplied by a weight є indicating how far the pair-wise score differs from the first-cut msa alignment score. Heuristic Dynamic Programming Method for MSA • Does not guarantee an optimal alignment of all the sequences in the group. • Does get an optimal alignment within the space chosen. Progressive Methods • Similar to dynamic programming method in that it uses the first step (i.e., it creates an AOA, aligns the most-alike pair, and incrementally adds sequences to the alignment.) • Differs from dynamic programming method for MSA in that it doesn’t refine the “first-cut” MSA by doing a full search through the reduced search space. (This is the computationally expensive part of DP MSA.) Progressive Method: the details • Generally proceeds as follows: – Choose a starting pair of sequences and align them – Align each next sequence to those already aligned, one at a time • Heuristic method – doesn’t guarantee an optimal alignment • Details vary in implementation: – How to choose the first sequence to align? – Align all subsequence sequences cumulatively or in subfamilies? – How to score? ClustalW • Based on phylogenetic analysis • A AOA is created using a pairwise distance matrix and nearest-neighbor algorithm • The most closely-related pairs of sequences are aligned using dynamic programming • Each of the alignments is analyzed and a profile of it is created • Alignment profiles are aligned progressively for a total alignment • W in ClustalW refers to a weighting of scores depending on how far a sequence is from the root on the AOA ClustalW Procedure AOA “Once a gap, always a gap” Basic Steps in Progressive Alignment “Once a gap, always a gap” Problems with Progressive Method • Highly sensitive to the choice of initial pair to align. If they aren’t very similar, it throws everything off. • It’s not trivial to come up with a suitable scoring matrix or gap penalties. Part II: Molecular Evolution Theory of Evolution • Evolution is the theory that allows us to understand how organisms came to be how they are •In probabilistic terms, it is likely that all living beings today have originated from a single type of cells •These cells divided and occupied ecological niches, where they adapted to the new environments through natural selection How did the first cell create different cells? Neutral Mutation (e.g. by error in genome replication) How did the first cell create different cells? Neutral Mutation Mutation (e.g. by error in genome replication) How did the first cell create different cells? Neutral Mutation Mutation (e.g. by error in genome replication) How did the first cell create different cells? Deleterious Mutation (e.g. by error in genome replication) How did the first cell create different cells? Deleterious Mutation Mutation (e.g. by error in genome replication) How did the first cell create different cells? Deleterious Mutation Mutation (e.g. by error in genome replication) How did the first cell create different cells? Advantageous Mutation (e.g. by error in genome replication) How did the first cell create different cells? Neutral Mutation Mutation (e.g. by error in genome replication) And then there was sex… Why Sex??? • Asexual reproduction is quicker, easier, more offspring/individual. • Sex may limit harmful mutations – Asexual: all offspring get all mutations – Sexual: Random distribution of mutations. Those with the most harmful ones tend not to reproduce. • Generate beneficial gene combinations – – – – Adaptation to changing environment Adaptation to all aspects of constant environment Can separate beneficial mutations from harmful ones Sample a larger space of gene combinations What drives cells to adapt? New Niche/ New conditions in old niche What drives cells to adapt? New (better addapted) mutation How do New Genes and Proteins appear? • Genes (Proteins) are build by combining domains • New proteins may appear either by intradomain mutation of by combining existing domains of other proteins Cell Cell Division Division … … The Coalescent •This model of cellular evolution has implications for molecular evolution •Coalescent Theory: •a retrospective model of population genetics that traces all alleles of a gene in a sample from a population to a single ancestral copy shared by all members of the population, known as the most recent common ancestor Why is the coalescent the de facto standard today? Alternatives? Current sequences have evolved from the same original sequence (Coalescent) Current sequences have converged to a similar sequence from multiple origins of life Back of the envelop support for divergence ? ACDEFGHIKLMNPQRSTVWY A EDYAHIKLMNPQRGTVWY AAi AAk AAi AAk AAk AAk Log[ p1] 0 Log[ p 2] 0 Log[ p1] 0 Log[ p 2] 0 AAi ptot p1 p 2 p1 p 2 p1 p 2 20 20 Convergence ptot14 p16 p214 p120 Divergence p 214 p16 Which is more likely? Convergence p114 ()1 Divergence About the mutational process Point mutations: • Transitions (A↔G, C↔T) are more frequent than transversions (all other substitutions) • In mammals, the CpG dinucleotide is frequently mutated to TG or CA (possibly related to the fact that most CpG dinucleotides are methylated at the C-residues) • Microsatellites frequently increase or decrease in size (possibly due to polymerase slippage during replication) Gene and genome duplications (complete or partial), may lead to: • pseudogenes: function-less copies of genes which rapidly accumulate (mostly deleterious) mutations, useful for estimating mutation rates! • new genes after functional diversification Chromosomal rearrangements (inversions and translocation), may lead to • meiotic incompatibilities, speciation Estimated mutation rates: • Human nuclear DNA: 3-5×10-9 per year • Human mitochondrial DNA: 3-5×10-8 per year • RNA and retroviruses: ~10-2 per year Consequences of the coalescent model? So what if we accept the coalescent model? A1-6 TSRISEIRR A7 PSRISEIRR A8-9 PKRISEVRR A10-11 PQRISAIQR A12-13 PQRISTIQR A14 ASHLHNLQR A15-17 TKHLQELQR A18 SKHLHELQR A19 PKNLHELQK A20 SKRLHEVQS A1 TSRISEIRR A2 TSRISEIRR A3 TSRISEIRR A4 TSRISEIRR A5 TSRISEIRR A6 TSRISEIRR A7 PSRISEIRR A8 PKRISEVRR A9 PKRISEVRR A10 PQRISAIQR A11 PQRISAIQR A12 PQRISTIQR A13 PQRISTIQR A14 ASHLHNLQR A15 TKHLQELQRE A16 TKHLQELQRE A17 TKHLQELQRE A18 SKHLHELQRD A19 PKNLHELQKD A20 SKRLHEVQSE So what if we accept the coalescent model? A1-6 TSRI SEI RR A7 PSRI SEI RR A8-9 PKRI SEVRR A10-11 PQRI SAI QR A12-13 PQRI STI QR A14 ASHLHNLQR A15-17 TKHLQELQR A18 SKHLHELQR A19 PKNLHELQK A20 SKRLHEVQS A’1-7 A’10-13 A1-6 A7 A10-11 A12-A13 So what if we accept the coalescent model? A’1-7 (p-t) SRI S E I RR A8-9 P KRI S E VRR A’10-13 P QRI S(a-t)I QR A14 A SHLH N LQR A15-17 T KHLQ E LQR A18 S KHLH E LQR A19 P KNLH E LQK A20 S KRLH E VQS 4 3324 5 323 Phylogenetic trees Rooted tree satisfying “molecular clock” hypothesis: all leaves at same distance from the root. Rooted tree root 6 8 root 7 time 7 6 3 1 8 5 2 4 1 2 3 4 5 Unrooted tree: 3 4 8 2 6 1 7 5 Note: 1-5 are called leaves, or leave nodes. 6-8 are inferred nodes corresponding to ancestral species or molecules. Branches are also called edges. The edge lengths reflect evolutionary distances. Phylogenetic trees A tree is a graph reflecting the approximate distances between a set of objects. A tree is also called a dendrogram. There are different types of trees: Unrooted versus rooted trees: A rooted tree has an additional node representing the origin, in molecular phylogeny the last common ancestor of the sequences analyzed. In general, the root cannot be directly inferred from the data. It may be inferred from the paleontological record, from a trusted outlier, or on the basis of the molecular clock hypothesis. Scaled and unscaled trees: In an unscaled tree, the length of the branches are not important. Only the topology counts. In phylogeny, trees are usually scaled. Binary trees: each node branches into two daughter nodes. Other trees are usually not considered in phylogeny as they can easily be approximated by binary trees with very short edges between nodes. Note: A rooted (or unrooted) tree connecting n objects (leaves) has 2n–1 (or 2n–2) nodes altogether and 2n–2 (or 2n–3) edges Phylogenetic tree reconstruction, overview Computational challenge: There is an enormous number of different topologies even for a relatively small number of sequences: 3 sequences: 1 4 sequences: 3 5 sequences: 15 10 sequences: 2,027,025 20 sequences: 221,643,095,476,699,771,875 Consequence: Most tree construction algorithm are heuristic methods not guaranteed to find the optimal topology. Input data for two major classes of algorithms: 1. Input data distance matrix, examples UPGMA, neighbor-joining 2. Input data multiple alignment: parsimony, maximum likelihood Distance matrix methods use distances computed from pairwise or multiple alignments as input. Building phylogenetic trees of proteins Genome 1 Protein A Genome 2 Protein C Genome 3 Genome … Protein D Protein B Protein A Protein B … Protein C Protein D Protein B Protein D Protein A Protein C Distance based phylogenetic trees A1 A2 A3 … A2 A1 5 substitutions ACTDEEGGGGSRGHI… A-TEEDGGAASRGHI… ACFDDEGGGGSRGHL… … A1 A3 A3 3 substitutions A2 8 substitutions 5 A1 A3 3 A2 Maximum likelihood phylogenetic trees Alignment Probability of aa substitution A - E D … ACTDEEGGGGSRGHI… 0.01 0.2 0.09 … A-TEEDGGAASRGHI… A 1 ACFDDEGGGGSRGHL… - 0.01 1 0.0001 0.0001 … … E 0.2 0.0001 1 0.5 D … 0.09 0.0001 0.5 1 Maximum likelihood phylogenetic trees A2 Alignment p(1,2) ACTDEEGGGGSRGHI… A-TEEDGGAASRGHI… ACFDDEGGGGSRGHL… … p(1,3) A1 5 substitutions A1 A3 3 substitutions p(2,3)>p(1,2)>p(1,3) A3 A2 A1 p(2,3) A3 A3 A1 A2 A2 8 substitutions Maximum Likelyhood:Parsimony • • • • Goal: To explain the MSA with a minimal number of mutational events – to find the tree with the minimal cost Input: a multiple sequence alignment (MSA): Major components: • A cost function for a tree given an MSA which simultaneously defines the branch lengths • An algorithm which finds a tree with the minimal cost Output: • an un-rooted tree (topology plus branch-lengths) • A total cost • an ancestral sequences for each non-terminal node Statistical evaluation of trees: bootstrapping 5 1 2 4 6 7 8 3 Motivation: Some branching patterns in a tree may be uncertain for statistical reasons (short sequences, small number of mutational events) Goal of bootstrapping: To assess the statistical robustness for each edge of the tree. Note that each edge divides the leave nodes into two subsets. For instance, edge 7–8 divides the leaves into subsets {1,2,3} and {4,5}.However, is this short edge statistically robust ? Method: Try to generate tree from subsets of input data as follows: • Randomly modify input MSA by eliminating some columns and replacing them by existing ones, This results in duplication of columns. • Compute tree for each modified input MSA. • For each edge of the tree derived from the real MSA, determine the fraction of trees derived from modified MSAs which contain an edge that divides the leaves into the same subsets. This fraction is called the bootstrap value. Edges with low bootstrap values (e.g. <0.9) are considered unreliable. Statistical evaluation of trees: bootstrapping Other Trees • Use genomes • Use Enzymomes • Use whatever group of molecules are important for a given function To Do • Take the HKs and RRs you have annotated. • Go to • http://bioinf.cs.ucl.ac.uk/dompred/DomPre dform.html • Identify the different domains in each individual protein. To Do • Take the HKs and RRs you have annotated. • Create phylogenies for the complete HKs and RR sequences • Create phylogenies for the individual homologous domains of the different proteins. • Use either clustal or STRAP for protein alignment and tree building. Distance measures for phylogenetic tree construction Distance measures respect the following constraints: d = 0 if the sequences are identical, d > 0 if the sequences are different Distances between molecular sequences are computed from pair-wise alignment scores. For closely related DNA sequences, one could simply use f , the fraction of nonidentical residues (readily computed from the % identity value returned by an alignment program). For more distantly related sequences, the Jukes-Cantor distance, d = –¾log(1–4f/3) is preferred. This measure is supposed to be proportional to evolutionary time. It takes into account that the percent identity value saturates at 25% over time. For protein sequences aligned with the aid of a substitution matrix, an approximate distance is often computed as follows: D log S obs S rand S max S rand Sobs Smax Srand observed pairwise alignment score maximum score (average of alignment scores of each sequence against itself) expected score for random sequences of same length and composition