Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Woodward–Hoffmann rules wikipedia , lookup
Discodermolide wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Asymmetric induction wikipedia , lookup
Asymmetric hydrogenation wikipedia , lookup
Kinetic resolution wikipedia , lookup
Enantioselective synthesis wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
George S. Hammond wikipedia , lookup
1,3-Dipolar cycloaddition wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
Polythiophene wikipedia , lookup
Ene reaction wikipedia , lookup
Petasis reaction wikipedia , lookup
Cracking (chemistry) wikipedia , lookup
Stille reaction wikipedia , lookup
Strychnine total synthesis wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
Hydrogenation wikipedia , lookup
Fischer–Tropsch process wikipedia , lookup
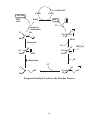
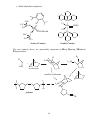
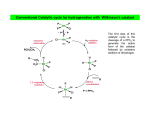
PART 3 Principles and Applications of Organometallics in Catalysis 1. Oxidative Addition and Reductive Elimination These processes are of great importance in synthesis and catalysis using organometallics. A oxidative addition L nM + A B reductive elimination ∆ OS=+2 ∆ CN=+2 L nM B ∆ VE=+2 18 VE 16 VE In the OA we break the (2-electron) A—B bond and form two (2-electron) bonds to the metal, i.e. M—A and M—B. Hence, there are some requirements for OA to occur. (i) A vacant coordination site must be available on the metal complex, and (ii) the metal must have an accessible oxidation state 2 units higher. Conversely, for RE to occur, there must be a stable oxidation state 2 units lower than that in the starting complex. The resulting complex will necessarily have a free coordination site which may make it reactive or perhaps unstable. The position of the OA/RE equilibrium is determined by the overall thermodynamics of the system, i.e. the relative stabilities of the two oxidation states, the relative bond strengths of A—B, M—A and M—B, and entropy terms. 2. Insertion and Elimination X Once we have arranged our ligands by addition M A B (either oxidative or simple) we can combine 18 V.E. and transform them within the coordination sphere by the process of insertion and its reverse X elimination. The two A M main types of insertion B are shown here. In principle, the reactions are 18 V.E. reversible, but usually only one direction is observed. 1,1-migratory insertion X M A B 16 V.E. X 1,2-migratory insertion A M B 16 V.E. 1 3. Catalysis; basic principles what catalysts do and don't do The effect of a catalyst is to change the rate of conversion of a substrate into products, but they do not change the position of an equilibrium. The thermodynamics of the reaction concerned have to be favourable at the outset; catalysts can't perform the miracle of pushing a reaction up the thermodynamic hill! The catalyst is not consumed by the reaction (although it may be changed subtly) but goes on to mediate the same reaction successively a number of times. Sometimes, the thermal reaction and the catalysed reaction give different products. This is because the catalyst has accelerated a reaction that is normally kinetically unfavourable. Generally, we will be interested in catalysts that increase the rate of reaction, but it should be noted that inhibitors which slow down certain reactions are also of great commercial and academic importance (e.g. flame retardents, corrosion inhibitors, antiknock additives in fuel, antioxidants in food). "catalyst" or "reagent"? In catalysis by organometallic systems, the catalyst brings together substrates within the coordination sphere. This means that the catalyst must have a free coordination site, or at least be able to free-up a coordination site by ligand dissociation or isomerisation. The substrates are activated in some way by the process of coordination and a reaction takes place. Often the most difficult step is that of decomplexation of the transformed substrate. Indeed, although many "organic" processes can be mediated by organometallics, relatively few are truly catalytic. This is largely because the transformed fragment refuses to de-complex from the metal centre in order to free-up a coordination site for the next substrate molecule. Hence the distinction between the organometallic as a catalyst or reagent. heterogeneous catalysis The "vacant coordination site" is located at a phase boundary. Only some (often very few) of the surface atoms are active catalysts. Advantages • The catalyst can be separated from the reaction mixture readily (it is not in the same phase) • Amenable to continuous processes (bed of catalyst in a tube). 2 Disadvantages • Low specificity • High reaction temperatures • Mechanisms hard to determine homogeneous catalysis The catalyst and substrate are in the same phase (usually liquid but sometimes gas). Disadvantages • Separation of catalyst from the reaction mixture often problematic. • Less amenable to continuous processes. Advantages • High specificity (tailoring) • Low reaction temperatures • Mechanisms can be studied more easily heterogenised homogeneous catalysts Many attempts have been made to marry the advantages of both homo- and heterogeneous processes, e.g. by attaching a homogeneous catalyst on a polymer support or by performing the reaction in a two-phase liquid/liquid system using a water soluble catalyst. H P Ar3P Rh PAr3 Ar3P CO P Ph Ph Ph3P Rh Cl Ph P Ar= Ph Cl Rh Ph3P PPh3 PPh3 SO3-Na+ A water soluble hydroformylation catalyst (see Section 7) Polymer supported Wilkinson's Catalyst (see Section 4) 3 4. Catalytic Hydrogenation of Alkenes Hydrogenation catalysts add H2 to the double bond of an alkene to give alkanes. We will look at two types, defined according to how they activate the H2 molecule. (i) Oxidative addition Wilkinson's Catalyst [RhCl(PPh3)3] is the best known of this or any type of hydrogenation catalyst. The activation of H2 is accomplished by its oxidative addition to the coordinatively unsaturated, 16 VE Rh(I) centre An outline mechanism for the hydrogenation of alkenes by Wilkinson's catalyst (L = PPh3):H L L H2 Cl Rh L Cl Rh H Identify the reaction L L type in each step, L count electrons and calculate oxidation states in all the -L irrev. intermediates. H H H Cl L H H Rh Cl L Rh H L L L L What does the reaction below tell us about the regio- and stereoselectivity of Wilkinson's Catalyst? O O D D D2, [Rh(PPh3)3Cl] O O (ii) Heterolytic H2 activation 4 [RuCl2(PPh3)3] is believed to activate H2 by a heterolytic process. Cl H2 RuCl(PPh3)3 RuCl2(PPh3)3 -HCl RuHCl(PPh3)3 H H How might we accelerate this process? Note that we have not drawn any Ru(IV) species. Why is this? The catalytic cycle is different from that in Wilkinson's catalyst in that there is not an OA/RE cycle. Instead, the Ru(II) centre prefers to mediate σ-bond metathesis and insertion steps: Identify the reaction type in each step, count electrons and calculate oxidation states in all the R intermediates. H H R RuCl(PPh3)3 HRuCl(PPh3)3 R R H H R RuCl(PPh3)3 RuCl(PPh3)3 H H2 H 5 H 5. Fischer-Tropsch Chemistry The "reformation" of natural gas (see below) provides a C1 feedstock called variously "water-gas" or "synthesis gas" or plain "syngas" which is used in the production of a variety of chemicals. Synthesis gas can be converted to methanol (a useful feedstocksee later) or long-chain alkanes and alcohols via the Fischer Tropsch reaction. CO + 3H2 heat CH4 + H2O het. cat. CH3OH Synthesis Gas homogeneous F.-T. Fischer-Tropsch CH3(CH2)nCH3 + CH3(CH2)nCH2OH + H2 O HOCH2CH2OH A wide variety of heterogeneous catalysts are used, but perhaps the most common is Fe or Fe oxides. Some "models" of what is occurring at the surface of the catalyst in this reaction have been developed in homogeneous systems, but it is hard to relate these systems to the real heterogeneous catalyst. See section 28-3 of Cotton & Wilkinson 5th Edition. 6. Carbonylation of Methanol Another important industrial process involving CO is the carbonylation of methanol to give acetic acid. The feedstocks may all be produced from coal or methane by virtue of a combination of Syngas and Fischer-Tropsch technology. Spectroscopic evidence for all the key intermediates has been collected. The iodomethane promoter is key to the success of this reaction. [Rh] CH3 OH + CO MeI 6 CH3COOH The cycle (below) uses familiar processes in each step: feedstock product CH3COOH HI CH3OH H 2O CH3I promoter CH3COI CO CO I Rh I I Rh I CO I Me CO catalyst CO I I O CO I Rh I - CO Me I O Rh I Me CO Monsanto Acetic Acid Process Classify the organometallic intermediates and the reactions that interconvert them. Why is it necessary to convert the MeOH to MeI? 7 Although rate-determining step is the oxidative addition, the reaction is usually still performed under a high pressure (15 atm.) of CO. This presumably prevents catalyst decomposition. There are severe problems with corrosion of reactor vessels by HI and MeI and so expensive alloys or glass reactors have to be used. Also, MeI is very volatile and seriously carcinogenic, and must be continuously recycled from effluent gases. The process can be tuned to produce acetic anhydride (BP-Monsanto Process). Many of the uses of acetic anhydride are based on acetylation of alcohols and phenols. The side products from such processes such as acetic acid and methyl acetate can be recycled using a further adaptation of the Monsanto process. 7. Hydroformylation of Alkenes This was one of the first commercial processes to use a homogeneous catalyst. Millions of tons of aldehydes p.a. are produced in this way. R catalyst R + H2/CO R CHO CHO branched linear The original process (1930s) used [Co2(CO)8] and this is still the most popular. There are some problems with this system - HCo(CO)4, which is the active species, is volatile and unstable - it is also a good hydrogenation catalyst - large amount of branched aldehydes produced (linear aldehydes are the most valuable) - no spectroscopic "handle" except IR \ mechanistic studies difficult. The proposed catalytic cycle is shown overleaf. 8 H CO OC Co CO CO CO O H C R CO R Co H feedstock CO OC product catalyst O C R H H R CO CO Co Co H CO CO CO CO H2 feedstock O R Co Co OC CO R CO C OC CO CO CO CO Co R CO CO Hydroformylation of an Alkene Catalysed by [Co(CO4)H] 9 CO feedstock Most of the problems associated with the cobalt catalyst can be circumvented by use of the Rh catalyst [Rh(PPh3)2(CO)H], which only produces linear aldehydes: H PPh3 Ph3P Rh PPh3 CO PPh3 O C R R H Ph3 P Rh OC H PPh3 feedstock product catalyst O R H H C Rh H R Ph3P OC PPh 3 PPh3 Rh PPh3 CO H2 R feedstock O R C Ph3P Rh OC Ph3P Rh OC PPh3 PPh3 R CO Ph3P Rh OC PPh3 CO feedstock Classify the reactions in this cycle Which step decides whether a linear or branched aldehyde is formed? How might we increase the proportion of linear product? 10 8. Wacker Process Complexes of alkenes often undergo nucleophilic attack to give metal alkyls which may then rearrange to give other products. M M .. Nu Nu This is used as the basis for production of millions of tons of aldehydes p.a. in the Wacker Process. It was established in the 19th century that aqueous PdCl2 oxidises ethylene to acetaldehyde. The reaction consumes the PdCl2 (and is thus hideously expensive) and deposits black Pd metal. The key discovery that made this reaction catalytically feasible is that acidified Cu(II) will oxidise the Pd(0) before it has a chance to precipitate. As the Pd(II) is reduced by the ethylene (mechanism later) it is quickly re-oxidised by two moles of Cu(II). The Cu(I) product is airsensitive and is reoxidised itself to Cu(II). Pd(0) is not oxidised by air at a significant rate under these conditions. C2 H4 PdCl2/O2 /H2 O/Cu(II) CH3CHO Mechanistic work on the process suggested the following rate equation: Rate = k[PdCl4 2-][C2H4] [Cl-]2[H+] What can you say about the mechanism from this data? Hints: Which species are involved, and how many moles of them? Are they adding to or leaving the active site? Because the reaction takes place in water, it is not feasible to determine the order of reaction with respect to [H2O]. 11 O2/H2 O/H+ Cu(II) Cu(I) CH3CHO HCl H2O PdCl2 Pd(0) Cl- reductive elimination Cl Cl Me Cl Pd Cl Pd O H H2 O H2O 1,2 insertion ClCl Pd H2O HO Cl H Cl Pd H2O OH H+ β-elimination Cl- - Cl Cl Pd H Cl Pd H2 O OH H2O Proposed Catalytic Cycle for the Wacker Process 12 OH 9. Alkene Metathesis. Discovered in the mid-1950s by Dupont, Standard Oil and Phillips Petroleum: H2 C CHR + + CHR CH2 CHR H2C CHR CH2 Catalysed by heterogeneous and homogeneous systems. Mechanism proposed by Chauvin in 1970 involves a metallocyclobutane complex: R'2 C M CR2 + R'2C CR'2 M M CR'2 Schrock alkylidene CR'2 + R'2C CR2 C R2 " " from e.g. WCl6 + ZnMe2 " " Me W W CH2 Me Homogeneous Alkene Metathesis Catalysts • Transient alkylidene complexes: Cl Ti AlMe2 -AlClMe2 CH2 Tebbe's Reagent 13 Ti CH2 • Stable alkylidene complexes: P CH3 F3C O F3C Cl N Ru Mo H3 C F3C O CHPh Cl P CHCMe2Ph CF3 Grubbs Catalyst Schrock Catalyst The two catalysts above are particularly important in Ring Opening Metathesis Polymerisation:R R R L nM L nM + L nM norbornene metallocyclobutane + L nM n polymer R 14 10. Alkene Polymerisation; Ziegler-Natta Catalysis (see MPC Organometallic Option) Ziegler and Natta shared the Nobel Prize in 1963 for their earlier work on the heterogeneous polymerisation of ethene and propene. The catalysts used are similar to those active as metathesis catalysts (early TM in high oxidation state, see Section 9), the earliest and most popular is a mixture of TiCl3 and the alkylating agent AlEt2Cl. Homogeneous analogues of the Ziegler-Natta system have been very widely studied. The parent complex of several generations of catalyst, zirconocene dichloride, was popularised by Sinn and Kaminsky in 1976. Zr Cl excess MAO very high activity for olefin polymerisation Cl MAO= [MeAlO]n AlMe 3 + H2O! Me Me probable structure of MAO Al O Al Me O Al Me n Me Development of the mechanism of this process has a long history and is still going on today (see later). Cossee-Arlman Mechanism (1964) The polymer chain grows by successive insertion of alkene into the M–C bond at the catalyst centre. M CH2 P + H2C CHR M CH2 CHR P CH 2 This rather simple approach has some problems: (i) Why does the polymer not chain-terminate by a β-elimination step? α β P M H (ii) Insertion of alkenes into M—C bonds was (and still is) rare. 15 P M H Green-Rooney Mechanism (1978) Green suggested that both of the above problems could be solved by the polymer chain performing an α-elimination to give a alkylidene hydride. The reason why the chain does not β-eliminate is because it α-eliminates! This alkylidene could react with the incoming alkene in a metathesis-like step and then reductively eliminate the intermediate alkyl hydride to give the expected polymer. H M H α β P M α P β C 2H4 β H H M M P α P Holes in this mechanism were obvious from the outset: (i) The metallacycle could just as easily eliminate the other M—C bond to give the unobserved ethyl-branched polymer (draw this out!!). (ii) The mechanism could not apply to a d0 or d1 catalyst because the α-elimination requires ∆OS = +2. 16 Hence, the modified Green-Rooney mechanism was developed; the alkylidene hydride was replaced by an agostic interaction (see earlier!) H M H α C2H4 P M α P H M olefin insertion P The idea behind this was two-fold. First, the formation of the agostic bond would swing the growing polymer chain around so as to not only present the M—C bond to the incoming alkene, but also open up the coordination site. Secondly, it will also weaken the M—C bond (remember, the agostic is a three-centre two-electron bond). Most researchers still broadly agree with the modified Green-Rooney mechanism. Recent Developments ACTIVE SPECIES CH2 Zr+ Zr+ CH3 H 14e alkyl cation Ethene coordination and insertion ‡ Zr+ Zr+ Zr+ CH2 CH2 H H 17 CH2 H Generation of Alkyl Cations Involves the use of strong Lewis Acids to abstract an alkyl group (as R- ) from the metal centre: Catalyst System Cl Zr Me MAO Zr Cl MAO Zr Me alkylation + CH3 [Me-MAO]- alkyl abstraction Model Systems Me Zr [CPh3]+X- Zr Me + CH3 + Ph3CMe XX= [B(C6F5)4]- how do you make this? Me Zr B(C6F5)3 Zr+ CH3 Me [MeB(C6F5)3]- 18 CATALYSIS PROBLEM CLASS Answer ALL the questions in the catalysis handout!! 19