Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Cell nucleus wikipedia , lookup
Tissue engineering wikipedia , lookup
Endomembrane system wikipedia , lookup
Cell encapsulation wikipedia , lookup
Signal transduction wikipedia , lookup
Extracellular matrix wikipedia , lookup
Cell culture wikipedia , lookup
Programmed cell death wikipedia , lookup
Cytokinesis wikipedia , lookup
Organ-on-a-chip wikipedia , lookup
Cell growth wikipedia , lookup
Cellular differentiation wikipedia , lookup
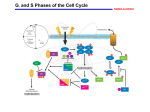
Cardiovascular Research 64 (2004) 395 – 401 www.elsevier.com/locate/cardiores Review Therapeutic opportunities for cell cycle re-entry and cardiac regeneration Kelly M. Regula, Marek J. Rzeszutek, Delphine Baetz, Charit Seneviratne, Lorrie A. Kirshenbaum* The Institute of Cardiovascular Sciences, St. Boniface General Hospital Research Centre, and the Department of Physiology, Faculty of Medicine, University of Manitoba, Rm. 3016, 351 Taché Avenue, Winnipeg, Manitoba, R2H 2A6, Canada Received 17 June 2004; received in revised form 20 August 2004; accepted 3 September 2004 Time for primary review 20 days Abstract Over the last two decades, considerable effort has been made to better understand putative regulators and molecular switches that govern the cell cycle in attempts to reactivate cell cycle progression of cardiac muscle. Rapid advancements on the field of stem cycle biology including evidence of cardiac progenitors within the adult myocardium itself and reports of cardiomyocyte DNA synthesis, which each suggest that the adult myocardium may in fact have the capacity for de novo myocyte regeneration. Augmenting cardiomyocyte number by targeting specific cell cycle regulatory genes or by stimulating cardiac progenitor cells to differentiate into cardiac muscle may be of therapeutic value in repopulating the adult myocardium with functionally active cells in patients with end-stage heart failure. Advancements in the area of cardiomyocyte cell cycle control and regeneration and their therapeutic potential are discussed. D 2004 European Society of Cardiology. Published by Elsevier B.V. All rights reserved. Keywords: Cell cycle; DNA synthesis; Ventricular myocytes; Regeneration 1. Introduction Heart disease represents a major cause of morbidity and mortality and is reaching pandemic proportions worldwide. In particular, the increased incidence of patients with heart failure has largely been attributed to the widespread adoption of western diet in developing and third world countries. Risk factors including uncontrolled hypertension, diabetes, smoking, sedentary life style, and viral infection have been identified as key underlying factors that contribute to the demise of ventricular performance and heart failure. Given the meager and limited ability of cardiac muscle for repair and/or self-renewal after injury, an inordinate loss of working ventricular muscle cells has been suggested to be a predominant underlying cause of contractile failure in patients with ischemic heart disease or impaired coronary reserve. * Corresponding author. Tel.: +1 204 235 3661; fax: +1 204 233 6723. E-mail address: [email protected] (L.A. Kirshenbaum). Following myocardial injury, a series of biochemical and local neurohormonal events are initiated resulting in release and activation of bioactive molecules such as angiotensin II, endothelin, aldosterone, and inflammatory cytokines. These act in a paracrine or autocrine manner to reprogram the myocardium at the cellular and molecular levels [1]. Distinct phenotypic changes at the cellular and subcellular levels result in both structural and functional changes to the myocardium resulting in heart hypertrophy. However, in a number of individuals for reasons unknown, these adaptive physiological processes eventually fail, resulting in a diminished cardiac pump function and overt heart failure. While the molecular events and signaling pathways that underlie heart failure remain cryptic, there is general consensus that heart failure results from one or more mutually related events. In this context, defects in calcium handling proteins of the sarcoplasmic reticulum [2,3], increased oxidative stress, from a reduction in antioxidant reserve [4,5], mitochondrial defects [6,7], genetic alterations to key myofibrillar proteins [8] and extracellular matrix proteins [9] have been identified to be crucial events in the 0008-6363/$ - see front matter D 2004 European Society of Cardiology. Published by Elsevier B.V. All rights reserved. doi:10.1016/j.cardiores.2004.09.003 396 K.M. Regula et al. / Cardiovascular Research 64 (2004) 395–401 remodeling process that underlie ventricular dysfunction and heart failure. Recently, there has been considerable interest in the role of apoptosis in heart failure. Undoubtedly, the acute loss of working ventricular myocytes in the absence of de novo myocyte regeneration could be seen as a major clinical impediment and underlying event leading to heart failure. There is now considerable evidence from studies in animals as well as in humans that documents an increased apoptotic index in the failing heart [10,11]. Whether the loss of cells through an apoptotic process is adaptive, maladaptive, or contributes to ventricular remodeling and failure is less clear and remains an active area of inquiry. However, the development of new therapeutic strategies to improve cardiac cell number has been identified as a means to improve cardiac function after injury. Although recent studies have detected stem cells within the myocardium, the impact of myocardial infarction outweighs the repair capacity of these cells [12–14]. Pharmacological and surgical revascularization is a cytoprotective treatment, but so far, they cannot compensate for cell loss. Inasmuch as myocyte number can directly influence cardiac pump performance, the definitive therapeutic goal in treating patients with heart failure would be to either preserve the number of pre-existing myocytes or to increase the number of functionally active force generating cells through de novo regenerative processes. Many exciting approaches have been postulated in efforts to achieve this therapeutic objective. These include the inhibition of apoptosis [15,16], the transdifferentiation of nonmuscle cells to muscle cells [17], the exogenous grafting of skeletal and cardiac myoblasts into infarcted myocardium [18,19], the incorporation of pluripotent bone marrow progenitors [20] into the myocardium, and the genetic manipulation of key cell cycle regulator factors to effects of cell cycle progression and DNA synthesis in the postmitotic adult myocardium [21,22]. 2. Regulators of cell cycle—the basics Organogenesis and the renewal of differentiated cell types depend upon a delicate balance between the proliferation of cells and cell death. While this is true of most cell types throughout their life, cardiomyocytes, among few other cell types, withdraw from the cell cycle during the perinatal period of development. Thereafter, cardiomyocyte hypertrophy prevails as the primary mechanism of myocardial growth to support increases in myocardial demand and/ or to compensate for cardiomyocyte cell loss during cardiac disease states [23–25]. Cell proliferation is a tightly controlled process mediated by internal and external signals. Growth factors stimulate the replication process, but it is an intricate intrinsic control mechanism that determines whether the cell is ready to proceed through the ordered set of events, the cell cycle, which culminate in the production of two daughter cells. The cell cycle consists of four phases (Fig. 1): M (Mitosis), G1 (Gap 1), S (Synthetic), G2 (Gap 2), and M phase or mitosis, the period of the cell cycle during which nuclear division occurs. In a carefully orchestrated manner, the nuclear envelope breaks down, chromosomes condense and align at the metaphase plate, and duplicated chromosomes are segregated to opposite poles of the cell. Cytokinesis marks the end of mitosis. The period between one mitosis and the next is known as interphase and consists of the G1, S, and G2 phases of the cell cycle. Immediately following mitosis and in response to appropriate mitogenic signals, the Fig. 1. The mammalian cell cycle. Cyclins/Cdk complexes control entry and exit through the 4 phases of the cell cycle. Growth factors through the action of Ras, Raf, and ERK trigger the synthesis of D-type cyclins which complex with Cdk4 or Cdk6 to regulate cell cycle progression through Go. The phosphorylation of pRb by G1 cyclins (D1, D2, D3, and E)/Cdk complexes promotes the release of E2F-1 from Rb. Through its association with p300, E2F-1 activates the transcription of genes important for the G1 transition to S phase. K.M. Regula et al. / Cardiovascular Research 64 (2004) 395–401 cell enters G1. G1 is one of two important periods of cell growth that occur prior to the next mitosis. During G1, the cell produces enzymes necessary for nucleotide metabolism, which will take place during the next phase of the cell cycle, S phase. If the cell is not committed to entering S phase, it can enter a state of quiescence known as Go. Depending on the cell type and environmental factors, Go can vary in length from days to years. Once exited from Go, cells progress to S phase during which DNA replication occurs. G2 is the second important period of cell growth and precedes mitosis. G2 ensures that DNA replication is complete before mitosis can occur and is the period during which the chromosomes start to condense. Cell cycle progression is controlled by a complex system of proteins that coordinate the biochemical events within the cell necessary for cell division. These proteins include cyclins, cyclin-dependent protein kinases (Cdk), Cdkactivating kinase (CAK), Cdk inhibitors, and members of the Retinoblastoma (Rb) family which function at different stages of the cell cycle. Cyclins are the regulatory subunit for the activity of Cdks. Each cyclin contains a region called the cyclin box which is involved in the binding of specific Cdks [26]. In complex with cyclins, they phosphorylate proteins at critical serine and threonine residues to drive the cell cycle. The G1 cyclins include cyclin D1, D2 and D3, and cyclin E. They are expressed consecutively throughout G1 and are required for entry into S phase. Complexes of the D-type cyclins with Cdk4 and Cdk6 play a critical role in a cell’s transition from Go to G1. Growth factors induce synthesis of D-type cyclins via the Ras/Raf/ERK signaling pathway. In cycling cells, cyclin D is constitutively produced but continuously turned over as a result of phosphorylation of a conserved threonine by glycogen synthase kinase-3g, followed by proteasome degradation [27]. Cyclin E is expressed later in G1 and together with Cdk2 promotes entry into S phase. Moreover, G1 cyclins facilitate phosphorylation of members of the Retinoblastoma family of pocket proteins (pRb, p107, and p130), promoting the activation of cellular transcription factors that are required for cell proliferation. Other cyclins, cyclin A and B, are expressed during S phase and G2/M. DNA synthesis is contingent upon complexes of cyclin A and Cdk2. Cyclin B synthesis begins in S phase, and the protein accumulates through to G2 phase as it forms a complex with Cdc2. The assembly, activation, and disassembly of cyclin B/Cdc2 complexes modulate entry and exit from mitosis. 397 loss or inactivation of pRb correlated with the development of a variety of human malignancies. These include retinoblastoma, a rare inherited childhood eye tumor [29] as well as breast, bladder, and prostate carcinomas as well as glioblastoma and leukemia [30,31]. The biochemical property of pRb, which enables it to suppress growth, has largely been attributed to its ability to negatively regulate the cell cycle by sequestering E2F transcription factors, particularly E2F-1, via its large A/B pocket domain (amino acid residues 379–869). Hypophosphorylated pRb sequesters E2F1 in G0 and G1. The phosphorylation of pRb by G1 cyclins (D1, D2, D3, and E)/Cdk complexes promotes the release of E2F-1 from the inhibitory pocket. E2F-1 is then free to modulate the transcription of genes important for the G1 transition to S phase, including DNA polymerase a, c-myc, dihydrofolate reductase, and thymidine kinase. In S phase, E2F-1 interacts with and becomes phosphorylated by cyclin A/Cdk2 resulting in its inactivation [32], as shown in Fig. 1. Subsequently during M phase, Rb becomes dephosphorylated, enabling it to reassociate with E2F during Go. To date, seven homologues of E2F (E2F-1 to E2F-7) have been identified. pRb interacts with E2F-1 to -3, while E2F-4 and E2F-5 primarily associate with p107 and p130 [33,34]. E2F-6 does not interact with pocket proteins [35]. An association between E2F-7 and a pocket protein is undetermined. In cells, E2F-1 to E2F-6 bind to DNA in heterodimeric complex with a member of the DRFT1polypeptide family (DP1 and DP2) [36]. E2F-7 is unique in that it binds to DNA independent of a DP partner [37]. The basic structure of E2F proteins includes an N-terminal DNA-binding domain, a central dimerization domain, and a C-terminal transactivation domain. The C-terminal transactivation domain contains the docking site for the pocket proteins. E2F-1, E2F-2, and E2F-3 also contain an Nterminal cyclin/Cdk binding site as well as a nuclear localization sequence, whereas E2F-6 lacks the transactivation domain. E2F-7 is comprised only of two independent DNA binding domains, setting it apart from the other E2Fs. While the majority of E2F family members share significant structural homology, they differ with respect to their abilities to promote cell growth and cell death, respectively. E2F-1, E2F-2, and E2F-3 can override G1-mediated growth arrest and reactivate DNA synthesis in quiescent cells. In contrast, E2F-4 and E2F-5 marginally stimulate S phase entry [38,39]. The mechanism by which E2F factors influence cell growth may be related to their ability to interact with specific pocket protein members, basal transcription factors such as TFIIH and TBP, as well as recruit the CBP/p300 histone acetylase [40,41]. 3. The tumor suppressor proteins The retinoblastoma gene product pRb is a tumor suppressor protein and member of the bpocket proteinQ family consisting of pRb, p107, and p130 (reviewed in Ref. [28]). pRb was first identified as a putative negative regulator of cell growth by the observation that functional 4. Regulation of Cdk activity during the cell cycle The major transitions of the cell cycle are controlled by the cyclin/Cdk complexes and as such, numerous mechanisms have evolved to regulate this important function 398 K.M. Regula et al. / Cardiovascular Research 64 (2004) 395–401 (reviewed in Ref. [42]). Regulation begins with assembly of the cyclin/Cdk complex. Cyclins display a periodic pattern of expression. They accumulate during different phases of interphase and then rapidly degrade before the next round of the cell cycle. Once the Cdk has associated with its cyclin partner, it is not catalytically active until it is phosphorylated on a threonine around position 160 by Cdk-activating kinase (CAK), an enzyme comprised of Cdk7 and cyclin H. The cyclin/Cdk complexes are also subject to negative regulation. Proteins known as Cdk inhibitors (CKIs) bind to and inhibit the activity of cyclin/Cdk complexes. CKIs are categorized into two families: the Cip/Kip family and the Ink4 family. Members of the Cip/Kip family which include p21Cip1, p27Kip1, and p57Kip2 bind to and inhibit cyclin D-, cyclin E-, and cyclin A-dependent kinases, whereas members of the Ink4 family (p16INK4a, p15INK4b, p18INK4c, and p19INK4d) bind directly to Cdk4 and Cdk6, and thus inhibit only D-type cyclin/Cdk4/6 complexes. In addition, Cdk2 and Cdc2 are regulated by inhibitory phosphorylation of N-terminal tyrosine14 and tyrosine15 [43]. Members of the Cdc25 family of protein phosphatases reverse this effect, thus enabling Cdk activity. 5. Promoting cell cycle re-entry in cardiac myocytes Cell cycle withdrawal is believed to be the primary mechanism for the dramatic reduction in proliferative capacity of cardiomyocytes. In the human heart, cardiomyocytes cease dividing between 3–6 months after birth, whereas in the postnatal rat heart, this occurs between days 3 and 4, with evidence of Go/G1cell cycle arrest [44–47]. These and other studies have supported the longstanding dogma that cardiomyocytes do not regenerate. Challenging this dogma, however, are two recent reports of cardiomyocyte cell division in the failing and infarcted human heart [48,49]. If adult myocytes do in fact undergo cell division, it does not appear to be sufficient to balance the loss of working myocytes that compromises heart function leading to heart failure [50,51]. Therefore, among the therapeutic strategies to reduce the morbidity and mortality of patients with heart failure, reactivating cardiomyocyte proliferation through the regulation of cell cycle regulators is particularly appealing. Proliferating cells express high levels and activity of cell cycle promoting factors such as cyclins D1, E, A, and B, Cdk2, Cdk4/6, and Cdc2 as well as E2F family members and low levels of the Cdk inhibitors p21Cip1and p27Kip1 [44,45,52]. In contrast, a marked down regulation of cell cycle promoting factors and increased activity and/or expression of cyclin/Cdk inhibitors is observed in the postmitotic adult heart [44,45,53]. Immunodepletion of p21Cip1 has been shown to increase Cdk2 activity in adult cardiac cell lysate [45]. Strategies to induce cardiomyocyte proliferation have therefore been directed towards the genetic manipulation of cell cycle regulatory factors to promote cell cycle progression. In recent years, an increasing number of reports have suggested that cardiomyocyte cell cycle reentry can be achieved through the manipulation of specific cell cycle regulatory proteins. For example, constitutive expression of c-myc mRNA in the heart resulted in enhanced hyperplastic growth during fetal development, presumably through the activation of cyclin E/Cdk2 [54,55]. In another study, transgenic hearts that expressed high levels of Cdk2 mRNA showed significantly increased levels of Cdk4 and cyclins A, D3, and E and augmented DNA synthesis in the adult hearts but lacked significant differences in heart weight to body weight ratios compared to wild-type mice [56]. Transgenic mice overexpressing cyclin D1 showed elevated levels of Cdk2 and Cdk4 as well as increased DNA synthesis and multinucleated cardiomyocytes in the mature animal [57]. While the manipulation of these cellular factors to promote cardiomyocyte cell division in the postmitotic adult heart has been mostly unsuccessful, p27Kip1 knockout mice have been shown to have larger hearts with more cardiomyocytes compared to p27Kip1+/+ mice. Analysis of myocytes demonstrated a greater proportion of cells in S phase and fewer in Go/G1. The authors suggest that loss of p27Kip1 prolongs the duration of cardiomyocyte division after birth [58]. Interestingly, postnatal cardiomyocyte hyperplasia has also been recently demonstrated in cyclin A2 overexpressing mice [59]. Cyclin A2, which is silenced in the postnatal heart, is an important mediator of S phase in cells. Chaudhry et al. report cardiac enlargement and evidence of mitosis during postnatal development of cyclin A2 transgenic hearts that is absent in wild-type hearts. Independently, these studies support the possibility of modifying a cell’s internal cell cycle clock to extend its proliferative potential [60]. The ability of Rb and related family members to sequester E2F proteins also plays an important role in cell cycle control. Viral proteins such as adenovirus E1A, human papilloma virus E6, and SV40 large T antigen (SV40) that bind to and inactivate Rb and p107, promote G1-exit of nonproliferating cells. In postnatal ventricular myocytes, E1A expression results in partial cell cycle reactivation including DNA synthesis with cells arresting in G2/M [21]. Moreover, expression of E2F-1 in cardiac muscle alone is sufficient to reactivate DNA synthesis in adult ventricular muscles in vitro and in vivo [22,61], supporting the role of Rb and related family members as key regulators of cell cycle control. However, a compounding effect of reactivating DNA synthesis in cardiac muscle by E1A or E2F-1 proteins is an increased incidence of apoptosis that can be abrogated by Bcl-2, adenovirus E1B proteins, or insulin-like growth factor (IGF-1) [22,62]. In contrast, delivery of SV40 to cardiomyocytes has been shown to promote G1 exit and augment growth of atrial and ventricular myocytes without provoking apoptosis [63–65]. These differences may reflect differential effects conferred by binding partners. Indeed, SV40 has been found to have two unique binding partners, the proapoptotic proteins p193 and p53. Interestingly, K.M. Regula et al. / Cardiovascular Research 64 (2004) 395–401 inactivation of both p53 and p193 pathways block E1Ainduced apoptosis, allowing an unimpeded proliferative response similar to that seen with SV40. Moreover, these results suggest that p53 and p193 coordinate the signals which control cell cycle and cell death pathways mediated by viral proteins. Manipulation of one or more tumor suppressor proteins in combination with increased expression of cyclin and CdK proteins may together be sufficient to overcome the G2-M checkpoint without provoking apoptosis. 399 Acknowledgements This work was supported by grants to L.A.K. from the Canadian Institutes for Health Research, an Interdisciplinary Heart Research Program grant from CHIR (CHFNET). K.M.R. holds a studentship from the CIHR, L.A.K. is a Canada Research Chair in Molecular Cardiology. References 6. Cell transplantation An alternative approach for regenerating cardiac muscle involves repopulating the damaged or diseased myocardium with pluripotent stem cells. Stem cells are characterized by their unique ability for prolonged self-renewal through cell division and their ability to differentiate into one or more mature cell types. Several recent studies have evaluated the efficacy and feasibility of using pluripotent cells for as transplantation therapy [66,67]. Embryonic stem cells (ESC) derived from the blastocyst are pluripotent and capable of developing into different cell types of the body. In contrast, however, adult stem cells derived from bone marrow exhibit a more limited ability to be phenotypically reprogrammed. Restrictions placed on the use of embryonic stem cells for obvious ethical considerations have limited their immediate experimental and clinical use, which has been the driving impetus for developing alternative strategies using adult stem cells for cell therapy. Advancements in the area of stem cell biology raise the interesting possibility for de novo generation or phenotypic conversion of endothelial progenitor cells, bone marrow derived, or hematopoietic stem cells into cells of the cardiac lineage [68–71]. Moreover, several reports now purport the exciting prospects of using these cells to improve cardiac function after injury [70]. Perhaps even more compelling is the recent discovery of cardiac progenitor cells within the adult myocardium itself [72,73]. These cells likely hold the most promise as the bholy grailQ for regenerating adult myocardium after injury. 7. Conclusions Despite many unresolved issues, new innovative gene therapy strategies to genetically reactivate DNA synthesis and cell progression in the adult myocardium together with cell transplantation therapies to repopulate damaged or diseased myocardium with pluripotent progenitors each suggest the exciting possibility for regenerating cardiac muscle postmyocardial infarction and/or enhancing cardiac performance in patients with heart failure or diminished cardiac performance. [1] Schwartz K, Chassagne C, Boheler KR. The molecular biology of heart failure. J Am Coll Cardiol 1993;22:30A – 3A. [2] Mercadier JJ, Lompre AM, Duc P, Boheler KR, Fraysse JB, Wisnewsky C, et al. Altered sarcoplasmic reticulum Ca2(+)-ATPase gene expression in the human ventricle during end-stage heart failure. J Clin Invest 1990;85:305 – 9. [3] Afzal N, Dhalla NS. Differential changes in left and right ventricular SR calcium transport in congestive heart failure. Am J Physiol 1992;262:H868–74. [4] Hill MF, Singal PK. Antioxidant and oxidative stress changes during heart failure subsequent to myocardial infarction in rats. Am J Pathol 1996;148:291 – 300. [5] Singal PK, Kirshenbaum LA. A relative deficit in antioxidant reserve may contribute in cardiac failure. Can J Cardiol 1990;6:47 – 9. [6] Liu J, Wang C, Murakami Y, Gong G, Ishibashi Y, Prody C, et al. Mitochondrial ATPase and high-energy phosphates in 5 failing hearts. Am J Physiol Heart Circ Physiol 2001;281:H1319–26. [7] Sorescu D, Griendling KK. Reactive oxygen species, mitochondria, and NAD(P)H oxidases in the development and progression of heart failure. Congest Heart Fail 2002;8:132 – 40. [8] Schwartz K, Carrier L, Lompre AM, Mercadier JJ, Boheler KR. Contractile proteins and sarcoplasmic reticulum calcium-ATPase gene expression in the hypertrophied and failing heart. Basic Res Cardiol 1992;87(Suppl 1):285 – 290. [9] Brilla CG, Rupp H. Myocardial collagen matrix remodeling and congestive heart failure. Cardiologia 1994;39:389 – 93. [10] Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, et al. Apoptosis in myocytes in end-stage heart failure. N Engl J Med 1996;335:1182 – 9. [11] Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, et al. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 2003;111:1497 – 504. [12] Anversa P, Kajstura J. Ventricular myocytes are not terminally differentiated in the adult mammalian heart. Circ Res 1998;83: 1 – 14. [13] Nadal-Ginard B, Kajstura J, Leri A, Anversa P. Myocyte death, growth, and regeneration in cardiac hypertrophy and failure. Circ Res 2003;92:139 – 50. [14] Orlic D. Stem cell repair in ischemic heart disease: an experimental model. Int J Hematol 2002;76(Suppl 1):144 – 5. [15] Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 2002;91:226 – 31. [16] Kirshenbaum LA, De Moissac D. The bcl-2 gene product prevents programmed cell death of ventricular myocytes. Circulation 1997;96: 1580 – 5. [17] Yeh ET, Zhang S, Wu HD, Korbling M, Willerson JT, Estrov Z. Transdifferentiation of human peripheral blood CD34+-enriched cell population into cardiomyocytes, endothelial cells, and smooth muscle cells in vivo. Circulation 2003;108:2070 – 3. [18] Murry CE, Wiseman RW, Schwartz SM, Hauschka SD. Skeletal myoblast transplantation for repair of myocardial necrosis. J Clin Invest 1996;98:2512 – 23. 400 K.M. Regula et al. / Cardiovascular Research 64 (2004) 395–401 [19] Dowell JD, Rubart M, Pasumarthi KB, Soonpaa MH, Field LJ. Myocyte and myogenic stem cell transplantation in the heart. Cardiovasc Res 2003;58:336 – 50. [20] Orlic D, Kajstura J, Chimenti S, Bodine DM, Leri A, Anversa P. Transplanted adult bone marrow cells repair myocardial infarcts in mice. Ann N Y Acad Sci 2001;938:221 – 9. [21] Kirshenbaum LA, Schneider MD. Adenovirus E1A represses cardiac gene transcription and reactivates DNA synthesis in ventricular myocytes, via alternative pocket protein- and p300-binding domains. J Biol Chem 1995;270:7791 – 4. [22] Kirshenbaum LA, Abdellatif M, Chakraborty S, Schneider MD. Human E2F-1 reactivates cell cycle progression in ventricular myocytes and represses cardiac gene transcription. Dev Biol 1996;179:402 – 11. [23] Cluzeaut F, Maurer-Schultze B. Proliferation of cardiomyocytes and interstitial cells in the cardiac muscle of the mouse during pre- and postnatal development. Cell Tissue Kinet 1986;19:267 – 74. [24] Zak R. Development and proliferative capacity of cardiac muscle cells. Circ Res 1974;35(suppl II):17 – 26. [25] Holtzer H, Schultheiss T, Dilullo C, Choi J, Costa M, Lu M, et al. Autonomous expression of the differentiation programs of cells in the cardiac and skeletal myogenic lineages. Ann N Y Acad Sci 1990;599:158 – 69. [26] Kobayashi H, Stewart E, Poon R, Adamczewski JP, Gannon J, Hunt T. Identification of the domains in cyclin A required for binding to, and activation of, p34cdc2 and p32cdk2 protein kinase subunits. Mol Biol Cell 1992;3:1279 – 94. [27] Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 1998;12:3499 – 511. [28] Mulligan G, Jacks T. The retinoblastoma gene family: cousins with overlapping interests. Trends Genet 1998;14:223 – 9. [29] Benedict WF, Murphree AL, Banerjee A, Spina CA, Sparkes MC, Sparkes RS. Patient with 13 chromosome deletion: evidence that the retinoblastoma gene is a recessive cancer gene. Science 1983;219:973 – 5. [30] Lee EY, To H, Shew JY, Bookstein R, Scully P, Lee WH. Inactivation of the retinoblastoma susceptibility gene in human breast cancers. Science 1988;241:218 – 21. [31] Horowitz JM, Park SH, Bogenmann E, Cheng JC, Yandell DW, Kaye FJ, et al. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc Natl Acad Sci U S A 1990;87:2775 – 9. [32] Dynlacht BD, Flores O, Lees JA, Harlow E. Differential regulation of E2F transactivation by cyclin/cdk2 complexes. Genes Dev 1994;8:1772 – 86. [33] Classon M, Dyson N. p107 and p130: versatile proteins with interesting pockets. Exp Cell Res 2001;264:135 – 47. [34] Dyson N. pRB, p107 and the regulation of the E2F transcription factor. J Cell Sci, Suppl 1994;18:81 – 7. [35] Ogawa H, Ishiguro K, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Mycresponsive genes in G0 cells. Science 2002;296:1132 – 6. [36] Stevens C, La Thangue NB. E2F and cell cycle control: a doubleedged sword. Arch Biochem Biophys 2003;412:157 – 69. [37] Logan N, Delavaine L, Graham A, Reilly C, Wilson J, Brummelkamp TR, et al. E2F-7: a distinctive E2F family member with an unusual organization of DNA-binding domains. Oncogene 2004. [38] Nevins JR, Leone G, DeGregori J, Jakoi L. Role of the Rb/ E2F pathway in cell growth control. J Cell Physiol 1997;173: 233 – 236. [39] Lukas J, Petersen BO, Holm K, Bartek J, Helin K. Deregulated expression of E2F family members induces S-phase entry and overcomes p16INK4A-mediated growth suppression. Mol Cell Biol 1996;16:1047 – 57. [40] Fry CJ, Pearson A, Malinowski E, Bartley SM, Greenblatt J, Farnham PJ. Activation of the murine dihydrofolate reductase promoter by [41] [42] [43] [44] [45] [46] [47] [48] [49] [50] [51] [52] [53] [54] [55] [56] [57] [58] [59] [60] [61] E2F1. A requirement for CBP recruitment. J Biol Chem 1999;274:15883 – 91. Trouche D, Kouzarides T. E2F1 and E1A(12S) have a homologous activation domain regulated by RB and CBP. Proc Natl Acad Sci U S A 1996;93:1439 – 42. Obaya AJ, Sedivy JM. Regulation of cyclin-Cdk activity in mammalian cells. Cell Mol Life Sci 2002;59:126 – 42. Chow JP, Siu WY, Ho HT, Ma KH, Ho CC, Poon RY. Differential contribution of inhibitory phosphorylation of CDC2 and CDK2 for unperturbed cell cycle control and DNA integrity checkpoints. J Biol Chem 2003;278:40815 – 28. Brooks G, Poolman RA, McGill CJ, Li JM. Expression and activities of cyclins and cyclin-dependent kinases in developing rat ventricular myocytes. J Mol Cell Cardiol 1997;29:2261 – 71. Poolman RA, Gilchrist R, Brooks G. Cell cycle profiles and expressions of p21CIP1 and P27KIP1 during myocyte development. Int J Cardiol 1998;67:133 – 42. Brooks G, Poolman RA, Li JM. Arresting developments in the cardiac myocyte cell cycle: role of cyclin-dependent kinase inhibitors. Cardiovasc Res 1998;39:301 – 11. Li JM, Poolman RA, Brooks G. Role of G1 phase cyclins and cyclindependent kinases during cardiomyocyte hypertrophic growth in rats. Am J Physiol 1998;275:H814–22. Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci U S A 1998;95:8801 – 5. Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, et al. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med 2001;344:1750 – 7. Colucci WS. Molecular and cellular mechanisms of myocardial failure. Am J Cardiol 1997;80:15L – 25L. Sabbah HN, Sharov VG, Goldstein S. Programmed cell death in the progression of heart failure. Ann Med 1998;30(Suppl 1):33 – 8. Li JM, Brooks G. Cell cycle regulatory molecules (cyclins, cyclindependent kinases and cyclin-dependent kinase inhibitors) and the cardiovascular system; potential targets for therapy? Eur Heart J 1999;20:406 – 20. Yoshizumi M, Lee WS, Hsieh CM, Tsai JC, Li J, Perella MA, et al. Disappearance of cyclin A correlates with permanent withdrawal of cardiomyocytes from the cell cycle in human and rat hearts. J Clin Invest 1995;95:2275 – 80. Jackson T, Allard MF, Sreenan CM, Doss LK, Bishop SP, Swain JL. The c-myc proto-oncogene regulates cardiac development in transgenic mice. Mol Cell Biol 1990;10:3709 – 16. Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H. Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J 1999;18: 5310 – 5320. Liao HS, Kang PM, Nagashima H, Yamasaki N, Usheva A, Ding B, et al. Cardiac-specific overexpression of cyclin-dependent kinase 2 increases smaller mononuclear cardiomyocytes. Circ Res 2001;88: 443 – 450. Soonpaa MH, Koh GY, Pajak L, Jing S, Wang H, Franklin MT, et al. Cyclin D1 overexpression promotes cardiomyocyte DNA synthesis and multinucleation in transgenic mice. J Clin Invest 1997;99:2644 – 54. Poolman RA, Li JM, Durand B, Brooks G. Altered expression of cell cycle proteins and prolonged duration of cardiac myocyte hyperplasia in p27KIP1 knockout mice. Circ Res 1999;85:117 – 27. Chaudhry HW, Dashoush NH, Tang H, Zhang L, Wang X, Wu EX, et al. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem 2004;279:35858 – 66. Bicknell KA, Surry EL, Brooks G. Targeting the cell cycle machinery for the treatment of cardiovascular disease. J Pharm Pharmacol 2003;55:571 – 91. Agah R, Kirshenbaum LA, Abdellatif M, Truong LD, Chakraborty S, Michael LH, et al. Adenoviral delivery of E2F-1 directs cell K.M. Regula et al. / Cardiovascular Research 64 (2004) 395–401 [62] [63] [64] [65] [66] cycle reentry and p53-independent apoptosis in postmitotic adult myocardium in vivo. J Clin Invest 1997;100:2722 – 8. von Harsdorf R, Hauck L, Mehrhof F, Wegenka U, Cardoso MC, Dietz R. E2F-1 overexpression in cardiomyocytes induces downregulation of p21CIP1 and p27KIP1 and release of active cyclindependent kinases in the presence of insulin-like growth factor I. Circ Res 1999;85:128 – 36. Steinhelper ME, Lanson Jr NA, Dresdner KP, Delcarpio JB, Wit AL, Claycomb WC, et al. Proliferation in vivo and in culture of differentiated adult atrial cardiomyocytes from transgenic mice. Am J Physiol 1990;259:H1826–34. Katz EB, Steinhelper ME, Delcarpio JB, Daud AI, Claycomb WC, Field LJ. Cardiomyocyte proliferation in mice expressing alphacardiac myosin heavy chain-SV40 T-antigen transgenes. Am J Physiol 1992;262:H1867–76. Sen A, Dunnmon P, Henderson SA, Gerard RD, Chien KR. Terminally differentiated neonatal rat myocardial cells proliferate and maintain specific differentiated functions following expression of SV40 large T antigen. J Biol Chem 1988;263:19132 – 6. Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, et al. Cardiac stem cell and myocyte aging, heart failure, and insulin-like growth factor-1 overexpression. Circ Res 2004;94: 514 – 524. 401 [67] Lanza R, Moore MA, Wakayama T, Perry AC, Shieh JH, Hendrikx J, et al. Regeneration of the infarcted heart with stem cells derived by nuclear transplantation. Circ Res 2004;94:820 – 7. [68] Makino S, Fukuda K, Miyoshi S, Konishi F, Kodama H, Pan J, et al. Cardiomyocytes can be generated from marrow stromal cells in vitro. J Clin Invest 1999;103:697 – 705. [69] Jackson KA, Majka SM, Wang H, Pocius J, Hartley CJ, Majesky MW, et al. Regeneration of ischemic cardiac muscle and vascular endothelium by adult stem cells. J Clin Invest 2001;107:1395 – 402. [70] Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, et al. Bone marrow cells regenerate infarcted myocardium. Nature 2001;410:701 – 5. [71] Badorff C, Brandes RP, Popp R, Rupp S, Urbich C, Aicher A, et al. Transdifferentiation of blood-derived human adult endothelial progenitor cells into functionally active cardiomyocytes. Circulation 2003;107:1024 – 32. [72] Oh H, Bradfute SB, Gallardo TD, Nakamura T, Gaussin V, Mishina Y, et al. Cardiac progenitor cells from adult myocardium: homing, differentiation, and fusion after infarction. Proc Natl Acad Sci U S A 2003;100:12313 – 8. [73] Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell 2003;114:763 – 76.