Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Ridge (biology) wikipedia , lookup
Quantitative trait locus wikipedia , lookup
Epigenetics in learning and memory wikipedia , lookup
Genetic testing wikipedia , lookup
Long non-coding RNA wikipedia , lookup
Gene desert wikipedia , lookup
Gene nomenclature wikipedia , lookup
Genomic imprinting wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Heritability of IQ wikipedia , lookup
Polymorphism (biology) wikipedia , lookup
Human genetic variation wikipedia , lookup
Gene therapy wikipedia , lookup
Gene therapy of the human retina wikipedia , lookup
Genome evolution wikipedia , lookup
Genetic engineering wikipedia , lookup
History of genetic engineering wikipedia , lookup
Pharmacogenomics wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Genome-wide association study wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Gene expression programming wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Gene expression profiling wikipedia , lookup
Designer baby wikipedia , lookup
Microevolution wikipedia , lookup
Genome (book) wikipedia , lookup
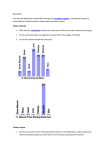
From www.bloodjournal.org by guest on June 15, 2017. For personal use only. Review Article Genetic polymorphisms of vein wall remodeling in chronic venous disease: a narrative and systematic review Vighnesh Bharath,1 Susan R. Kahn,2 and Alejandro Lazo-Langner3,4 1 Department of Medicine, University of Western Ontario, London, ON, Canada; 2Division of Internal Medicine and Department of Medicine, McGill University, Montreal, QC, Canada; 3Department of Medicine, Division of Hematology, University of Western Ontario, London, ON, Canada; and 4 Department of Epidemiology and Biostatistics, Western University, London, ON, Canada Chronic venous disease encompasses a spectrum of disorders caused by an abnormal venous system. They include chronic venous insufficiency, varicose veins, lipodermatosclerosis, postthrombotic syndrome, and venous ulceration. Some evidence suggests a genetic predisposition to chronic venous disease from gene polymorphisms associated mainly with vein wall remodeling. The literature exploring these polymorphisms has not been reviewed and compiled thus far. In this narrative and systematic review, we present the current evidence available on the role of polymorphisms in genes involved in vein wall remodeling and other pathways as contributors to chronic venous disease. We searched the EMBASE, Medline, and PubMed databases from inception to 2013 for basic science or clinical studies relating to genetic associations in chronic venous disease and obtained 38 relevant studies for this review. Important candidate genes/ proteins include the matrix metalloproteinases (extracellular matrix degradation), vascular endothelial growth factors (angiogenesis and vessel wall integrity), FOXC2 (vascular development), hemochromatosis (involved in venous ulceration and iron absorption), and various types of collagen (contributors to vein wall strength). The data on associations between these genes/proteins and the postthrombotic syndrome are limited and additional studies are required. These associations might have future prognostic and therapeutic implications. (Blood. 2014;124(8):1242-1250) Introduction Chronic venous disease (CVD) refers to a spectrum of overlapping diseases involving abnormalities of the venous system, both structural and functional. Chronic venous insufficiency (CVI) is a term that describes functional abnormalities of the venous system, but is often used to describe the full range of CVD manifestations such as varicose veins, venous ulceration, lipodermatosclerosis (LDS), and postthrombotic syndrome (PTS) (essentially a secondary form of CVI).1-3 Almost a quarter of the adult population in the Western world has some form of CVD, but treatment is often delayed or deferred because of an underestimation of the prevalence and burden of the condition, leading to significant disability. Treatment is multifactorial, and often includes mechanical and/or surgical measures such as compression devices, leg elevation, ablations, and vein stripping.1 CVI is a multifactorial disease, and although most commonly caused by valvular incompetence and venous hypertension, the exact pathogenesis remains unclear, although various studies have suggested a potential genetic contribution.4 This condition may be primary (abnormalities of vein walls or valves) or secondary (after venous thrombosis; ie, PTS). Risk factors include age, female gender, pregnancy, family history, obesity, and prolonged orthostasis. Clinical manifestations include leg pain, lower extremity edema, skin changes, varicose veins, and venous ulceration.5 Venous ulcers are a severe complication of CVI, although the underlying mechanisms are not fully known. Varicose veins, a form of CVD and the most common manifestation of CVI, are caused by a loss of vessel wall homeostasis; venous hypertension leads to vein dilation, distortion, leakage, and inflammation, causing valve and wall disease and reflux.6-8 Although sometimes caused by PTS, varicose veins are usually primary in nature. They are often hereditary and result from extensive extracellular matrix (ECM) remodeling, leading to vein wall weakening or dysfunction.9-11 PTS, a type of CVD, is an important and frequent chronic complication of deep venous thrombosis (DVT) that develops in 20% to 50% of patients (severe in 5% to 10%) after DVT despite appropriate anticoagulation.12 Risk factors include incomplete DVT symptom resolution, proximal or previous ipsilateral DVT, obesity, and increased age.12 Although the pathophysiology of PTS is not well understood, it involves venous hypertension caused by persistent venous obstruction and/or valvular reflux from valve destruction, and recent evidence also supports a role for inflammation.13 Symptoms are similar to CVI and can be debilitating, often including constant or intermittent limb swelling, aching, cramps, or numbness/tingling (improved with rest or recumbency). Treatment involves symptomatic relief using graduated elastic compression stockings or compression devices, leg elevation, or a trial of horse chestnut seed extract as a last resort.12,14,15 Recently, there has been some research into the genetic contributors and risk factors of CVD, such as varicose veins and venous ulceration, mainly relating to certain noted polymorphisms in genes associated with vein wall remodeling. In fact, genetic risk factors are already known to affect wound progression and healing, and screening in this regard may aid in the planning of appropriate individualized treatment and prophylaxis.16 Also, various thrombophilic single nucleotide polymorphisms (SNPs) may contribute to CVI and ulcers by increasing the risk of DVT. More recently, however, other SNPs have been studied in genes relating specifically to vein wall remodeling and proinflammatory or angiogenesis-regulating factors and receptors. Although studies in this area do support a moderate to strong genetic predisposition Submitted March 3, 2014; accepted July 3, 2014. Prepublished online as Blood First Edition paper, July 8, 2014; DOI 10.1182/blood-2014-03-558478. © 2014 by The American Society of Hematology 1242 BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 From www.bloodjournal.org by guest on June 15, 2017. For personal use only. BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 GENETIC POLYMORPHISMS IN CHRONIC VENOUS DISEASE 1243 toward CVI and varicose veins,16 the exact genes are still unknown— though they likely involve quantitative or qualitative defects in proteins associated with the vein wall, ECM, and cell organization/ regulation.16 These polymorphisms, in turn, could plausibly increase the likelihood of CVD, including PTS after DVT, but evidence in this area is lacking. Though various candidate genes have been identified as contributors to CVD, the evidence has not previously been compiled and critically reviewed. In this review, we use a systematic approach to explore the current evidence regarding genetic polymorphisms in vein wall structure and healing/remodeling as potential contributors to CVD, including PTS. Understanding these genetic associations may help clarify the underlying pathophysiology, identify patients at risk, and suggest novel therapeutic options. Methods To identify potential genes associated with CVD, we conducted a broad computerized literature search. We considered for inclusion any studies, either basic or clinical, describing or potentially describing an association between any gene, gene mutation, or genetic polymorphism and any of the following conditions: varicose veins, chronic leg ulcers, CVI, lipodermatosclerosis, PTS, or any combination of these. The search was conducted in September 2013 from the inception of each of the following databases: EMBASE, Medline (through the OVID interface), and PubMed. The search strategy used was: [“varicose veins” OR “chronic venous insufficiency” OR “leg ulcer” OR “post-thrombotic syndrome” OR “post thrombotic syndrome” OR “post-phlebitic syndrome” OR “post phlebitic syndrome”] AND [“genetics” OR “gene” OR “genes” OR “mutation” OR “polymorphism”]. We restricted the search to studies published in English. The retrieved references were initially screened for eligibility based on titles by 1 author and confirmed by another author. A preliminary list of potentially relevant studies was then independently screened by 2 authors based on titles and abstracts and a final list of studies to review in full was generated by consensus. Any studies whose relevance was unclear were reviewed in full. Quality of the studies was not assessed because of the lack of validated scales for the type of studies included. We did not plan a meta-analysis because we anticipated substantial heterogeneity among studies. Our results are presented descriptively by CVI subtype. Finally, although no standards exist for reporting systematic reviews of basic science studies, whenever possible we attempted to adhere to the available reporting standards.17,18 Results Search results The search process is summarized in Figure 1. The initial search generated 682 results. The initial screening by title alone resulted in 142 potentially relevant studies. The final selection for review in full included 41 studies, of which 3 were excluded and 38 articles were included in the final review. Genetic polymorphisms and CVD Our literature search yielded various candidate genes that have evidence-based associations with a spectrum of CVD including CVI, varicose veins, LDS, venous ulceration, and PTS (Table 1). There was significant overlap in that these genes were often associated with more than 1 CVD manifestation (Figure 2), but they are presented here according to their most significant associations with a given subtype. The matrix metalloproteinases (MMPs) are discussed Figure 1. Flow diagram of the systematic review separately, given their consistent strong associations with various forms of CVD. Varicose veins The ECM is a complex and dynamic framework of collagen, proteoglycans, elastin, glycoproteins, and cellular components. Degradation or destruction of the ECM disrupts the homeostasis of the vein (much of which is maintained by the MMPs, discussed later), thereby leading to varicosity. Vein wall weakness is a key player in the pathophysiology of varicose veins, and collagen is an important matrix component that provides strength; however, collagen dysregulation leads to vein wall abnormalities.4 In varicose veins, the total elastin content is decreased, type I collagen is upregulated, and type III collagen is downregulated.7,19 Jin et al4 conducted a recent study that examined a 7-base pair insertion/ deletion polymorphism (rs3917) in the COL1A2 (a-2 type I collagen) gene. They found that this polymorphism upregulated COL1A2 expression and produced a 1.6-fold increase in CVI risk and speculated that genetic variations in this gene alter transcriptional activity, affect messenger RNA (mRNA) structure, and ultimately allow expressional upregulation.4 Kowalewski et al20 demonstrated increased expression of the cytokine vascular endothelial growth factor A (VEGF-A) and receptor VEGF R2 in the walls of varicose veins as compared with normal saphenous veins, particularly if complicated by thrombophlebitis. Similarly, Hollingsworth et al21 observed increased transcription of VEGF and its receptors in varicose veins, reflecting a potential early role in varicogenesis. Of note, VEGF-A is involved in the maintenance of vessel wall integrity. Increased expression of VEGF-A or its receptor R2 leads to increased activation of nitric oxide synthase, which then leads to vessel wall damage mediated by oxygen free radicals as well as decreased vessel tone that predisposes to venous stasis.20 VEGF also plays a key role in angiogenesis, so SNPs in the VEGF gene (C936T and 21780 T/C have been described) can be considered risk factors for impaired wound healing and venous ulceration.22 Chang et al6 conducted a study using microarray bioinformatics that systematically explored various genes involved in biological expression Basic science/gene ECM integrity case-control study Basic science BAT1 TNF-a Oct-1 MGP with TNFa Apoptosis; linkage Inflammation Intron 10 [–/C] patients vs 16.3% in controls Presence of allele: 28.8% in ulcer patients vs 22.6% in controls Presence of allele: 43.1% in ulcer veins vs normal vein tissue Significantly higher mRNA levels and (1.74 vs 1.1) varicose vs nonvaricose vein tissue Higher relative MGP expression in collagen 2.33, type III collagen 1.70 vs nonvaricose vein tissue: type I Relative mRNA expression in varicose type I collagen and mRNA expression muscle cells, n 5 8); no difference in (control vs varicose vein smooth gene expression 2308G → A –SNP 3: 1/24 (4.2%) Type III collagen dpm/106: ;290 vs ;40 protein expression of Oct-1 in varicose Upregulation Overexpression Overexpression deposition decreased Type III collagen stress/regulation of Cellular response to Vein wall remodelling ER, estrogen receptor; HFE, hemochromatosis; NA, not available; NS, not significant; VV, varicose veins. 28 Venous ulcers/wound healing 27 expression Basic science/gene immunostaining Basic science/vein –SNP 2: 2/24 (8%) 3. 241G → T (not in controls) –SNP 1: 5/24 (20.8%) 2. 241G → A P 5 .012 OR: 2.00 P 5 .000155 OR: 2.48 P , .001 P , .0001 Type III: P , .01 Type I: P , .001 P , .005 NA P , .0001 P 5 2.2E-6 increase in intensity VV:CV (control veins) $2-fold P , .001 Strength of association BHARATH et al 8 19 vein reflux: 14/18) vs 1/12 in referents SNPs in proximal upstream region of sequencing 1. 291C → G with various FOXC2 mutations (deep Great saphenous vein reflux in 18/18 (VV: 67% vs 45%) monozygotic vs dizygotic Concordance rates significantly higher for with control 32/74 genes upregulated in VV compared VEGF-R2: 57.47 vs 28.92 ng/g VEGF-A: 41.76 vs 25.79 ng/g in VV vs normal wall tissue Increased VEGF-A and VEGF-R2 content Results FOXC2 in VV/hemorrhoid patients Basic science/genomic 25 (varied) FOXC2 mutations Functional variants Upregulated expression Increased mRNA/protein Polymorphisms/abnormality analysis and Basic science Type I/III collagen development D16S520 using marker gene Lymphatic and apoptosis Cell proliferation and apoptosis Cell signaling, Protein degradation angiogenesis Vessel wall integrity, Proposed function vascular/venous FOXC2 TGF-b1 ILK HSP-90 VEGF-A/VEGF-R2 Gene linkage analysis Basic science/twin 26 24 Basic science/genetic 6 microarrays Basic science Study details 20 VV Reference 1244 Table 1. Candidate gene polymorphisms/abnormalities and their proposed evidence-based associations in chronic venous disease From www.bloodjournal.org by guest on June 15, 2017. For personal use only. BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 Study details Gene genotyping gene expression (COL1A1) Proposed function Results ECM integrity ECM integrity metabolism Homocysteine Iron export absorption Regulation of iron 3. 13% vs 7% 4. 47% vs 38% 3. 210808G → T 4. 21569C → A Overexpression in 39UTR) indel polymorphism rs3917 (7-base pair 677C → T 28C → G 282G → A 2. 34% vs 25% 50 000 mm2) (LDS vs control: 180 vs 97 per mesenchymal cells/fibroblasts groups from increased (COL1A1) in LDS vs other patient Increased procollagen type I mRNA of CVI cases vs 8% controls Indel polymorphism present in 12.2% vs 5% in healthy population Overall, 15% homozygous for C677T C4-6: 20% C2-3: 10% homozygous for C677T Based on CEAP classification: Homozygotes: 8.6% vs 2.3% vs 30.8% in controls Heterozygotes: 34% in ulcer patients Homozygotes: 0.6% vs 0% vs 1.8% in controls Heterozygotes: 9.9% in ulcer patients associated with venous ulceration T-T-T-A haplotype significantly 1. 58% vs 49% 2. 213850T → C exon and promoter repair processes 1. 216849C → T regions, including 0N Upstream regulatory Cases vs controls Homozygotes: 23.17% vs 13.42% (rs7895676) (rs2981578) patients vs 45.12% in controls inflammatory and Estrogen receptor; expression Heterozygotes: 53.66% in leg ulcer (Grzela et al) FGF-R2: 2451A → G normal veins No significant difference in FGF-R aFGF mRNA wound healing, ECM metabolism protein in VV vs 24.68 pg/g in Vein wall aFGF content: 32.21 pg/g of aFGF and increased Overexpression of Polymorphisms/abnormality regeneration, Connective tissue ER, estrogen receptor; HFE, hemochromatosis; NA, not available; NS, not significant; VV, varicose veins. 33 Procollagen (COL1A2) extraction and Basic science/pathology 1 collagen Basic science/DNA 4 LDS a-2 Type I Basic science/genetics MTHFR FPN1 HFE ERb a-FGF/FGF-R2 32 array study Basic science/DNA control study Basic science case analysis Basic science/genetic expression Basic science/gene P , .001 P 5 .008 OR: 1.60 P , .001 P 5 .005 OR: 5.2 P 5 .001 OR: 4.5 T-T-T-A haplotype OR: 2.9 4. P 5 .013 3. P 5 .002 2. P 5 .010 1. P 5 .025 P 5 .0103 P , .001 Strength of association BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 CVI 31 30 29 9 Reference Table 1. (continued) From www.bloodjournal.org by guest on June 15, 2017. For personal use only. GENETIC POLYMORPHISMS IN CHRONIC VENOUS DISEASE 1245 Study details Basic science/gene 23 Basic science (mice) IL-6 TIMPs Inflammation Inhibitors of MMP ECM degradation ER, estrogen receptor; HFE, hemochromatosis; NA, not available; NS, not significant; VV, varicose veins. 37 PTS Basic science/gene 34 expression Basic science/genotyping 40 expression Basic science/gene expression (mice) Basic science/gene expression Basic science/gene expression Basic science/gene MMPs Proposed function IL-6 as therapeutic target Diminished TIMP-1,2 TIMP-2: 2418G -→ C of TIMP-1 Overexpression generated) vs controls; MMP-14 mRNA (MT1-MMP) rat IgG 44%) vs those treated with control fibrosis (intimal thickness reduced by anti-IL-6 had decreased vein wall Mice with IVC thrombus treated with dermatitis vs controls immunoreactivity of TIMP-1,2 in stasis Low gene expression and 14.2% in controls Allele frequency: 23.3% in VV group vs (2.06 6 0.26) in varicose vs nonvaricose veins Higher relative TIMP-1 mRNA expression controls upregulated 2.5-fold compared with MMP-2 activity in ligated cavae (DVT During resolution of DVT: 71% increase in least an 80-fold increase controls; MMP-1, -9, -8, and -13 had at levels elevated in ulcer tissue vs MMP-1, -2, -3, -8, -9, -12, and -13 protein dermatitis) vs controls -13 mRNA in lesional skin (stasis Increased expression of MMP-1, -2, and and TIMP-1 class 4-5 patients relative to MMP-1 Active component of MMP-2 increased in TIMP-1: 2.56 (control) vs 23.45 (class 6) (class 4) vs 31.2 (class 6) MMP-1 mRNA: 0.002 (control) vs 4.15 CVI patients, class 1-6 injury at 8 and 21 d after stasis thrombosis less collagen content/fibrosis vs controls Vein walls of MMP92/2 mice had 45% group vs 48.3% in controls 21562C allele frequency: 81.7% in VV 282AA 1 AG: 11.1 6 17.1 cm2 282GG: 5.4 6 2 cm2 282GG genotype Smaller ulcer size in patients with Results MMP-2 and MMP-14 Overexpression of MMP-1,2,3,8,9,12,13 Overexpression of of MMP-1,2,13 Overexpression protein mRNA and MMP-2 Overexpression of MMP-1 MMP-92/2 MMP-9: 21562C/C MMP-12: 282A → G Polymorphisms/abnormality P , .05 NS Unclear significance OR: 2.213 P 5 .038 P , .01 P , .05 P , .005 (majority) P , .05 TIMP-1: P , .05 MMP-1 mRNA: P , .01 P , .01 OR: 6.102 P 5 .000 P 5 .001 Strength of association BHARATH et al 8 36 42 41 Basic science (mice) 38 expression Basic science/genotyping array study Basic science/DNA 40 31 Gene 1246 MMPs: VV, CVI, venous ulcers Reference Table 1. (continued) From www.bloodjournal.org by guest on June 15, 2017. For personal use only. BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 From www.bloodjournal.org by guest on June 15, 2017. For personal use only. BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 pathways that may contribute to varicosity. The results showed that 32 genes were upregulated and 74 genes were downregulated in varicose veins, most of which were related to apoptosis and angiogenesis. Important examples of upregulated genes included: HSP90, ILK, and TGF-b1.6 In fact, Saito et al23 earlier noted that TGF-b1 stimulates collagen synthesis and alters levels of MMPs (discussed later). Mutations in the FOXC2 gene have also been associated with varicose veins. FOXC2 encodes a transcription factor involved in lymphatic and vascular development, and mutations in FOXC2 are seen in lymphoedema distichiasis, which is characterized by lymphoedema and varicose veins.7,24 Ng et al24 conducted an early twin linkage study that strongly implicated FOXC2 in the development of varicose veins as a heritable condition. Similarly, knowing that FOXC2 had previously been implicated in primary venous valve failure, Al-Batayneh et al25 identified 3 specific SNPs in the FOXC2 gene that may contribute to varicose vein and hemorrhoid development. More recently, Mellor et al26 conducted a small study of 18 patients and noted a strong association between FOXC2 and primary venous valve failure in both superficial and deep veins of the lower limb. Other, lesser-known genes have also been examined in preliminary studies and must be mentioned. Jeong et al27 performed large scale mRNA screens among normal varicose veins and found the greatest differential expression in the octamer-binding transcription factor-1 gene (Oct-1), which was upregulated in primary varicose veins. Cario-Toumaniantz et al8 found overexpressed vitamin K–dependent matrix gla protein (MGP) in varicose veins, and speculated that its role in wall remodeling involved smooth muscle proliferation and mineralization processes. GENETIC POLYMORPHISMS IN CHRONIC VENOUS DISEASE 1247 ulcers; as mentioned previously, this likely led to abnormal reepithelialization and angiogenesis. Estrogen is a known contributor to ECM metabolism; in fact, hormone replacement therapy prevents CVI and topical estrogen promotes wound healing in the elderly by decreasing the inflammatory response.30 Although convincing associations have not been elucidated, several SNPs in the ER-b receptor have been associated with venous ulcers in the elderly.22,30 Ashworth et al30 found that polymorphisms in the upstream regulatory regions of the ER-b gene were significantly associated with venous ulceration, but did not conduct further functional studies to determine the precise mechanism. Hemochromatosis is an inherited iron-overload disease caused by mutations in the HFE gene (most commonly allele C282Y), a major histocompatibility complex class 1-type membrane protein associated with b2-microglobulin. The mutated gene results in abnormalities in the regulation of iron absorption related to interactions between transferrin and its receptor. In patients with CVI, the HFE C282Y allele increases the risk of ulceration by almost 7-fold, likely because of accumulation of iron in tissues surrounding blood vessels, increased free radicals and oxidative stress, and upregulation of the inflammatory response that finally leads to tissue destruction.22 Moreover, Gemmati et al31 found that the 28CG polymorphism in the FPN1 gene (ferroportin; involved in exporting iron out of the cell) increases susceptibility to leg ulcers. Finally, Sam et al32 suggested from preliminary data that mild to moderate hyperhomocysteinemia is common in patients with CVI (approximately 65%, especially with ulceration), and is associated in one-third of patients with an underlying methylenetetrahydrofolate reductase C677T homozygous polymorphism. LDS Venous ulceration 22 Grzela et al reviewed 4 genes that may play a role in venous ulceration: tumor necrosis factor (TNF), fibroblast growth factor-R (FGF-R), estrogen receptor, and HFE. Levels of TNF-a and interleukin-1 (IL-1) are higher in venous leg ulcers compared with normal acute wounds, with certain SNPs (such as the 2308A variant in TNF-a) conferring a higher risk of venous ulceration compared with wild-type.5,22,28 Along with demonstrating the previously mentioned association with TNF-a, Wallace et al28 also identified a polymorphism in intron 10 of the BAT1 gene (human leukocyte antigen B–associated transcript-1) as being a significant risk factor for venous ulceration. Similarly, FGFs and their receptors (FGF-R) are cytokines that are imperative for the control of connective tissue regeneration in wound healing. Kowalewski et al9 found increased acidic fibroblast growth factor (a-FGF) expression in the walls of varicose veins. They noted that these walls contained extensive ECM remodeling and altered collagen and glycosaminoglycan differentiation; a-FGF likely influences the expression of certain enzymes through various pathways (eg, mitogen-activated protein kinase pathway) involved in ECM metabolism and varicosity. Furthermore, a-FGF synthesis is enhanced by hypoxia, which may be a consequence of venous stasis.9 Previous studies indicate that certain SNPs in FGF-R2, most frequently the polymorphism A2451G, are more commonly observed in CVI or ulceration. Though the mechanism is unclear, these SNPs likely result in lower expression of FGF-R2, causing impaired regeneration of connective tissue, longer vein wall reepithelialization, and eventually CVI or ulceration. 22 Nagy et al29 conducted an association study and identified an SNP in the FGF-R2 gene that was more prevalent CVI patients with nonhealing LDS is a consequence of CVI and is characterized by severe skin changes including hardening, atrophy, dark pigmentation, and edema. It often progresses to skin breakdown, venous ulceration, and delayed healing.33,34 deGiorgio-Miller et al33 analyzed leg skin biopsies from patients with LDS and found enhanced cell proliferation and procollagen gene expression as well as significant fibrotic changes which correlated directly with ulcer formation and healing time. Furthermore, imbalances in the MMPs and their inhibitors (discussed later) have also been implicated in LDS through the generation of epidermal and dermal skin defects.34 PTS PTS is a frequent yet poorly understood complication of DVT. Previous studies have explored genes involved in vein wall remodeling in relation to CVI and its manifestations (eg, varicose veins, venous ulcers). These are also manifestations of PTS, and it is logical to infer that polymorphisms in these very genes may potentially increase the risk of PTS as well. In fact, Deatrick et al35 noted ongoing vein wall remodeling 6 months after an acute DVT that was associated with biomarkers such as MMP-9, which directly correlate with resolution and predict PTS. There is, however, a dearth of literature that directly associates the above genes with PTS. An early study by Dahi et al36 demonstrated increased MMP-2 and MT1-MMP activity (potentially mediated by thrombin) during DVT resolution, which, in turn, increased the risk of PTS. Wojcik et al37 used a mouse model to conclude that IL-6 is associated with reduced monocyte recruitment, leading to reduced vein wall thickness and fibrosis. Given that PTS involves extensive perivenous and mural fibrosis, IL-6 may serve as a therapeutic target to prevent these fibrotic complications. Another recent mouse model study by From www.bloodjournal.org by guest on June 15, 2017. For personal use only. 1248 BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 BHARATH et al development and severity of PTS. However, their preliminary data did show increased MMP-9 and decreased Toll-like receptor 9 expression in acute DVT. MMP-9 levels have been shown to be increased in varicose veins and venous ulcers, and some data suggest a potential role in DVT resolution, with gene deletions being associated with less collagen deposition. In fact, Beidler et al42 used multiplexed protein analysis to show that all MMPs except MMP-7 were highly expressed in venous leg ulcers, especially MMP-8 and MMP-9. In addition, Singh et al43 analyzed various SNPs associated with venous leg ulcers and found that MMP-12 gene polymorphisms may have a role in ulcer progression. Interestingly, Kurzawski et al44 demonstrated that polymorphisms in MMP-1 and MMP-3 did not predict susceptibility to varicose veins, but this study likely did not have the power to uncover the milder effects of these genes on an otherwise multifactorial disorder. Discussion Figure 2. Candidate genes and their overlapping associations with the spectrum of chronic venous disease. Deatrick et al38 reveals that MMP-9 modulates vein wall collagen content and contributes to inflammation and fibrosis, thus implicating it as a potential target to reduce the fibrotic complications of PTS. To our knowledge, there has been no study to date that has analyzed polymorphisms in the previously mentioned candidate genes to uncover a direct association with PTS. Some of the studies presented in Table 1 involve tissue blocks from veins complicated by thrombophlebitis, and some of the patients were noted to have a history of venous thrombosis (however, scant patient data were provided), but none of the studies specifically included PTS as a subset. MMPs Perhaps the most studied genes in venous disease are the MMPs and tissue inhibitors of metalloproteinases (TIMPs). A systematic review by Lim et al7 revealed that MMP-1, MMP-2, MMP-3, MMP-7, MMP-9, TIMP-1, and TIMP-3 were all upregulated in varicose veins. Similarly, Raffetto et al39 noted that MMPs are involved in ECM degradation, which subsequently leads to venous remodeling, structural wall changes, and ultimately venous dilation and valve dysfunction; they are thus heavily involved in the pathogenesis of CVD. Though the molecular mechanisms of these genetic anomalies have not been fully elucidated, it appears that MMPs regulate or degrade the ECM through hydrolysis, whereas TIMPs are tissue inhibitors of MMPs that influence vascular remodeling. Hence, an imbalance between these proteins may lead to abnormalities in the vessel wall and, ultimately, vascular disease such as varicosity and ulceration.7,35,40 Moreover, misregulation of MMP activity and TIMP counterregulation contributes to impaired ulcer healing.34 An early study by Saito et al23 suggested that increased MMP-2 levels affect tissue remodeling and contribute to a proulcer environment. Subsequently, Herouy et al41 examined stasis dermatitis, a common consequence of impaired venous drainage characterized by dermal neovascularization, and noted elevated MMP-1, 22, and 213 and diminished TIMP-1 and 22 in the skin lesions. More recently, Xu et al40 showed that specific polymorphisms in the MMP-9 and TIMP2 genes potentially put patients at a higher risk for developing varicose veins. Deatrick et al35 examined vein remodeling and associated gene expressions, but did not correlate results with the In this review, we examined different candidate genes and their polymorphisms (mostly SNPs) that have been linked to various forms of CVD. In assessing these studies, a few candidate genes should be highlighted as having the strongest link to the development of CVD (because we could not perform a meta-analysis, these assessments are qualitative and based on the available evidence). Three studies agree that SNPs in FOXC2, a transcription factor involved in lymphatic and vascular development, provide a strong link toward venous varicosity, valve failure, and hemorrhoids. On the other hand, a polymorphism in the HFE gene increases the risk of venous ulceration by almost 7-fold, while also causing the known dysregulation in iron absorption. The MMPs (and, to a lesser extent, their inhibitors, TIMPs) are perhaps the best studied in venous disease. MMPs are involved in ECM degradation and structural vein wall changes, ultimately contributing to venous remodeling, dilation, and valve dysfunction. They are thus heavily involved in the pathogenesis of CVD. The exact mechanisms have yet to be elucidated and further research is required in this area. Although genetic markers in CVD have been examined in some detail, there is a lack of evidence correlating these genes with the development and severity of PTS. If these polymorphisms are potentially associated with CVI, LDS, varicose veins, venous ulcers, and DVT resolution, it is plausible that they could also provide an underlying substrate for the development of PTS. The MMPs have once again been implicated in this respect, but data are scarce and further research is warranted. In general, a limitation of all of the studies included in this review relates to the fact that they were conducted either in animal models, in very limited numbers of patients, or using surgical samples (eg, saphenectomy). It is difficult to assess the quality of each study because no standardized tools exist in this regard—in contrast to the quality assessment scales used for clinical studies. We believe that the included studies were methodologically sound; however, we cannot totally rule out the possibility of biased results. Clearly, the genetic predisposition to CVD, particularly PTS, is controversial and unclear at best, and more studies are needed to clarify these associations. Earlier studies have promoted the use of a genome-wide approach; this allows for the identification of previously unknown markers, and may be a consideration for the future. Clinically, this would be a novel way to reexamine a patient’s propensity toward developing CVD, predict those who go on to From www.bloodjournal.org by guest on June 15, 2017. For personal use only. BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 GENETIC POLYMORPHISMS IN CHRONIC VENOUS DISEASE develop it, and provide a better understanding of the underlying mechanisms to ultimately improve treatment options. 1249 Acknowledgments The researching, writing, and editing were performed solely by the authors and no other individuals or organizations were involved. Conclusions Herein, we examine the role of various candidate genes and their polymorphisms in the development of CVD, including the lesser studied PTS. Our observations support a genetic predisposition to CVD related to vein wall remodeling. Genes of significance include FOXC2, HFE, and the MMPs, all of which show strong associations with varicose veins, CVI, or venous ulceration. The data surrounding PTS are more limited, but given that it is a type of CVD with similar manifestations, we may infer that some of the above genes may be implicated. Thus, further studies are needed to examine these associations more directly. Most importantly, given the burden of this disease worldwide and the paucity of treatment options currently available, studying these polymorphisms could potentially allow us to better identify patients at higher risk of developing CVD, and also provide novel therapeutic targets. Authorship Contribution: A.L.L. and S.K. conceived the idea; V.B. and A.L.L. performed the systematic review; V.B. wrote an initial draft of the article; V.B., S.K., and A.L.L. reviewed and edited the manuscript; and all authors reviewed and approved the final manuscript. Conflict-of-interest disclosure: The authors declare no competing financial interests. Correspondence: Alejandro Lazo-Langner, Hematology Division, London Health Sciences Centre, 800 Commissioners Rd E, Rm E6-216A, London, ON, N6A 5W9, Canada; e-mail: [email protected]. References 1. Lattimer CR. CVD: a condition of underestimated severity. Int Angiol. 2014;33(3):222-228. 2. Partsch H. Varicose veins and chronic venous insufficiency. Vasa. 2009;38(4):293-301. 3. Eberhardt RT, Raffetto JD. Chronic venous insufficiency. Circulation. 2005;111(18): 2398-2409. 4. Jin Y, Xu G, Huang J, Zhou D, Huang X, Shen L. Analysis of the association between an insertion/ deletion polymorphism within the 39 untranslated region of COL1A2 and chronic venous insufficiency. Ann Vasc Surg. 2013;27(7): 959-963. 5. Hot‚oleanui C, Jurj C. The involvement of genetic factors in chronic venous insufficiency. Rom J Intern Med. 2008;46(2):119-123. 6. Chang M-Y, Chiang P-T, Chung Y-C, et al. Apoptosis and angiogenesis in varicose veins using gene expression profiling. Fooyin J Health Sci. 2009;1(2):85-91. 7. Lim CS, Davies AH. Pathogenesis of primary varicose veins. Br J Surg. 2009;96(11): 1231-1242. 8. Cario-Toumaniantz C, Boularan C, Schurgers LJ, et al. Identification of differentially expressed genes in human varicose veins: involvement of matrix gla protein in extracellular matrix remodeling. J Vasc Res. 2007;44(6):444-459. 9. Kowalewski R, Malkowski A, Sobolewski K, Gacko M. Evaluation of aFGF/bFGF and FGF signaling pathway in the wall of varicose veins. J Surg Res. 2009;155(1):165-172. 10. Labropoulos N. Why do varicose veins develop? Phlebolymphology. 2010;17(1):25. 11. Naoum JJ, Hunter GC. Pathogenesis of varicose veins and implications for clinical management. Vascular. 2007;15(5):242-249. 12. Kahn SR. How I treat postthrombotic syndrome. Blood. 2009;114(21):4624-4631. 13. Rabinovich A, Cohen JM, Cushman M, et al. Inflammation markers and the risk of post thrombotic syndrome: results from the Bio-Sox study. Blood. 2013;122(21):36. 14. Tick LW, Kramer MHH, Rosendaal FR, Faber WR, Doggen CJ. Risk factors for post-thrombotic syndrome in patients with a first deep venous thrombosis. J Thromb Haemost. 2008;6(12): 2075-2081. 15. Kahn SR, Kearon C, Julian JA, et al; Extended Low-intensity Anticoagulation for Thromboembolism (ELATE) Investigators. Predictors of the post-thrombotic syndrome during long-term treatment of proximal deep vein thrombosis. J Thromb Haemost. 2005;3(4):718-723. FOXC2 region of chromosome 16 for varicose veins in otherwise healthy, unselected sibling pairs. J Med Genet. 2005;42(3):235-239. 25. Al-Batayneh KM, Al Battah RM. Genetic variation in the proximal 59 UTR of FOXC2 gene in varicose veins and hemorrhoids patients. Int J Integ Biol. 2008;4(2):78-80. 16. Krysa J, Jones GT, van Rij AM. Evidence for a genetic role in varicose veins and chronic venous insufficiency. Phlebology. 2012;27(7): 329-335. 26. Mellor RH, Brice G, Stanton AWB, et al; Lymphoedema Research Consortium. Mutations in FOXC2 are strongly associated with primary valve failure in veins of the lower limb. Circulation. 2007;115(14):1912-1920. 17. Stroup DF, Berlin JA, Morton SC, et al. Metaanalysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA. 2000;283(15):2008-2012. 27. Jeong G-A, Choi ET, Chang J-H. Octamerbinding transcription factor-1 gene is upregulated in primary varicose veins. Ann Vasc Surg. 2008; 22(1):115-120. 18. Moher D, Liberati A, Tetzlaff J, Altman DG; PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. PLoS Med. 2009;6(7): e1000097. 28. Wallace HJ, Vandongen YK, Stacey MC. Tumor necrosis factor-alpha gene polymorphism associated with increased susceptibility to venous leg ulceration. J Invest Dermatol. 2006;126(4): 921-925. 19. Sansilvestri-Morel P, Rupin A, Badier-Commander C, et al. Imbalance in the synthesis of collagen type I and collagen type III in smooth muscle cells derived from human varicose veins. J Vasc Res. 2001;38(6):560-568. 29. Nagy N, Szolnoky G, Szabad G, et al. Single nucleotide polymorphisms of the fibroblast growth factor receptor 2 gene in patients with chronic venous insufficiency with leg ulcer. J Invest Dermatol. 2005;124(5):1085-1088. 20. Kowalewski R, Malkowski A, Sobolewski K, et al. Vascular endothelial growth factor and its receptors in the varicose vein wall. Acta Angiol. 2011;17(2):141-149. 30. Ashworth JJ, Smyth JV, Pendleton N, et al. Polymorphisms spanning the 0N exon and promoter of the estrogen receptor-beta (ERbeta) gene ESR2 are associated with venous ulceration. Clin Genet. 2008;73(1): 55-61. 21. Hollingsworth SJ, Powell G, Barker SGE, Cooper DG. Primary varicose veins: altered transcription of VEGF and its receptors (KDR, flt-1, soluble flt1) with sapheno-femoral junction incompetence. Eur J Vasc Endovasc Surg. 2004;27(3):259-268. 22. Grzela T, Bialoszewska A. Genetic risk factors of chronic venous leg ulceration: Can molecular screening aid in the prevention of chronic venous insufficiency complications? Mol Med Rep. 2010; 3(2):205-211. 23. Saito S, Trovato MJ, You R, et al. Role of matrix metalloproteinases 1, 2, and 9 and tissue inhibitor of matrix metalloproteinase-1 in chronic venous insufficiency. J Vasc Surg. 2001;34(5):930-938. 24. Ng MYM, Andrew T, Spector TD, Jeffery S; Lymphoedema Consortium. Linkage to the 31. Gemmati D, Federici F, Catozzi L, et al. DNA-array of gene variants in venous leg ulcers: detection of prognostic indicators. J Vasc Surg. 2009;50(6):1444-1451. 32. Sam RC, Burns PJ, Hobbs SD, et al. The prevalence of hyperhomocysteinemia, methylene tetrahydrofolate reductase C677T mutation, and vitamin B12 and folate deficiency in patients with chronic venous insufficiency. J Vasc Surg. 2003; 38(5):904-908. 33. Degiorgio-Miller AM, Treharne LJ, McAnulty RJ, Coleridge Smith PD, Laurent GJ, Herrick SE. Procollagen type I gene expression and cell proliferation are increased in lipodermatosclerosis. Br J Dermatol. 2005;152(2):242-249. From www.bloodjournal.org by guest on June 15, 2017. For personal use only. 1250 BLOOD, 21 AUGUST 2014 x VOLUME 124, NUMBER 8 BHARATH et al 34. Herouy Y. The role of matrix metalloproteinases (MMPs) and their inhibitors in venous leg ulcer healing. Phlebolymphology. 2004;44:231-243. 35. Deatrick KB, Elfline M, Baker N, et al. Postthrombotic vein wall remodeling: preliminary observations. J Vasc Surg. 2011;53(1):139-146. 36. Dahi S, Lee JG, Lovett DH, Sarkar R. Differential transcriptional activation of matrix metalloproteinase-2 and membrane type-1 matrix metalloproteinase by experimental deep venous thrombosis and thrombin. J Vasc Surg. 2005; 42(3):539-545. 37. Wojcik BM, Wrobleski SK, Hawley AE, Wakefield TW, Myers DD Jr, Diaz JA. Interleukin-6: a potential target for post-thrombotic syndrome. Ann Vasc Surg. 2011;25(2):229-239. 38. Deatrick KB, Obi A, Luke CE, et al. Matrix metalloproteinase-9 deletion is associated with decreased mid-term vein wall fibrosis in experimental stasis DVT. Thromb Res. 2013; 132(3):360-366. 39. Raffetto JD, Khalil RA. Matrix metalloproteinases in venous tissue remodeling and varicose vein formation. Curr Vasc Pharmacol. 2008;6(3): 158-172. 40. Xu HM, Zhao Y, Zhang XM, Zhu T, Fu WG. Polymorphisms in MMP-9 and TIMP-2 in Chinese patients with varicose veins. J Surg Res. 2011; 168(1):e143-e148. 41. Herouy Y, Mellios P, Bandemir E, et al. Inflammation in stasis dermatitis upregulates MMP-1, MMP-2 and MMP-13 expression. J Dermatol Sci. 2001;25(3):198-205. 42. Beidler SK, Douillet CD, Berndt DF, Keagy BA, Rich PB, Marston WA. Multiplexed analysis of matrix metalloproteinases in leg ulcer tissue of patients with chronic venous insufficiency before and after compression therapy. Wound Repair Regen. 2008;16(5):642-648. 43. Singh AV, Subhashree L, Milani P, Gemmati D, Zamboni P. Interplay of iron metallobiology, metalloproteinases, and FXIII, and role of their gene variants in venous leg ulcer. Int J Low Extrem Wounds. 2010;9(4): 166-179. 44. Kurzawski M, Modrzejewski A, Pawlik A, Droździk M. Polymorphism of matrix metalloproteinase genes (MMP1 and MMP3) in patients with varicose veins. Clin Exp Dermatol. 2009;34(5): 613-617. From www.bloodjournal.org by guest on June 15, 2017. For personal use only. 2014 124: 1242-1250 doi:10.1182/blood-2014-03-558478 originally published online July 8, 2014 Genetic polymorphisms of vein wall remodeling in chronic venous disease: a narrative and systematic review Vighnesh Bharath, Susan R. Kahn and Alejandro Lazo-Langner Updated information and services can be found at: http://www.bloodjournal.org/content/124/8/1242.full.html Articles on similar topics can be found in the following Blood collections Review Articles (710 articles) Vascular Biology (499 articles) Information about reproducing this article in parts or in its entirety may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requests Information about ordering reprints may be found online at: http://www.bloodjournal.org/site/misc/rights.xhtml#reprints Information about subscriptions and ASH membership may be found online at: http://www.bloodjournal.org/site/subscriptions/index.xhtml Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036. Copyright 2011 by The American Society of Hematology; all rights reserved.