Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Nucleic acid analogue wikipedia , lookup
Designer baby wikipedia , lookup
Genomic library wikipedia , lookup
Transposable element wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Cre-Lox recombination wikipedia , lookup
Microevolution wikipedia , lookup
Genome evolution wikipedia , lookup
Transcription factor wikipedia , lookup
Pathogenomics wikipedia , lookup
Non-coding DNA wikipedia , lookup
Primary transcript wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Point mutation wikipedia , lookup
Human genome wikipedia , lookup
Nucleic acid tertiary structure wikipedia , lookup
Metagenomics wikipedia , lookup
Microsatellite wikipedia , lookup
Computational phylogenetics wikipedia , lookup
Smith–Waterman algorithm wikipedia , lookup
Genome editing wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Helitron (biology) wikipedia , lookup
Local Multiple Sequence Alignment
Sequence Motifs
Motifs
• Motifs represent a short common sequence
– Regulatory motifs (TF binding sites)
– Functional site in proteins (DNA binding motif)
Regulatory Motifs
• DNA in every cell is identical
– Different cells have different functions
• Transcription is crucial aspect of regulation
– Transcription factors (TFs) affect transcription
rates
• TFs bind to regulatory motifs
MCM1
– Motifs are 6 – 20 nucleotides long
MCM1
– Activators and repressors
motif
– Usually located near target gene, mostly
upstream
Transcription
Start Site
SBF
SBF
motif
Gene X
E. Coli promoter sequences
Challenges
• How to recognize a regulatory motif?
• Can we identify new occurrences of known
motifs in genome sequences?
• Can we discover new motifs within
upstream sequences of genes?
1. Motif Representation
• Exact motif: CGGATATA
• Consensus: represent only
deterministic nucleotides.
– Example: HAP1 binding sites in
5 sequences.
• consensus motif: CGGNNNTANCGG
• N stands for any nucleotide.
• Representing only consensus
loses information. How can this
be avoided?
CGGATATACCGG
CGGTGATAGCGG
CGGTACTAACGG
CGGCGGTAACGG
CGGCCCTAACGG
-----------CGGNNNTANCGG
Transcription
start site
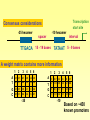
Consensus considerations
-35 hexamer
-10 hexamer
spacer
interval
TTGACA 15 - 19 bases TATAAT 5 - 9 bases
A weight matrix contains more information
3
4
5 6
0.1 0.1
0.7 0.7
0.1
0.2
0.5
0.2
0.2 0.5
0.2 0.2
0.1 0.1
0.5
0.1
0.1 0.2
0.1 0.1
0.2
0.2 0.5
1
A
T
G
C
2
-35
0.1
A
T
G
C
1
2
3
4
0.1
0.7 0.2
0.6
0.5
0.1
0.7
0.1 0.5
0.2
0.2
0.8
0.1
0.1 0.1
0.1
0.1 0.0
0.1
0.1 0.2
0.1
0.1 0.1
-10
5 6
Based on ~450
known promoters
PSPM – Position Specific
Probability Matrix
• Represents a motif of length k
• Defines Pi{A,C,G,T} for i={1,..,k}.
– Pi (A) – frequency of nucleotide A in position i.
1
2
3
4
5
A
0.1
0.25
0.05
0.7
0.6
C
0.3
0.25
0.8
0.1
0.15
T
0.5
0.25
0.05
0.1
0.05
G
0.1
0.25
0.1
0.1
0.2
PSPM – Position Specific
Probability Matrix
• Represents a motif of length k
• Defines Pi{A,C,G,T} for i={1,..,k}.
– Pi (A) – frequency of nucleotide A in position i.
• Each k-mer is assigned a probability.
– Example: P(TCCAG)=0.5*0.25*0.8*0.7*0.2
1
2
3
4
5
A
0.1
0.25
0.05
0.7
0.6
C
0.3
0.25
0.8
0.1
0.15
T
0.5
0.25
0.05
0.1
0.05
G
0.1
0.25
0.1
0.1
0.2
Graphical Representation –
Sequence Logo
• Horizontal axis: position
of the base in the
sequence.
• Vertical axis: amount of
information.
• Letter stack: order
indicates importance.
• Letter height: indicates
frequency.
• Consensus can be read
across the top of the letter
columns.
2. Identification of Known Motifs
within Genomic Sequences
• Motivation:
– identification of new genes controlled by the same
TF.
– Infer the function of these genes.
– enable better understanding of the regulation
mechanism.
Detecting a Known Motif within a
Sequence using PSPM
• The PSPM is moved along the query sequence.
• At each position the sub-sequence is scored for a
match to the PSPM.
1
2
3
• Example:
A
0.1
0.25
0.05
sequence = ATGCAAGTCT…
4
5
0.7
0.6
C
0.3
0.25
0.8
0.1
0.15
T
0.5
0.25
0.05
0.1
0.05
G
0.1
0.25
0.1
0.1
0.2
Detecting a Known Motif within a
Sequence using PSPM
• The PSPM is moved along the query sequence.
• At each position the sub-sequence is scored for a
match to the PSPM.
1
2
3
• Example:
A
0.1
0.25
0.05
sequence = ATGCAAGTCT…
C
0.3
0.25
0.8
• Position 1: ATGCA
0.1*0.25*0.1*0.1*0.6=1.5*10-4
4
5
0.7
0.6
0.1
0.15
T
0.5
0.25
0.05
0.1
0.05
G
0.1
0.25
0.1
0.1
0.2
Detecting a Known Motif within a
Sequence using PSPM
• The PSPM is moved along the query sequence.
• At each position the sub-sequence is scored for a
match to the PSPM.
1
2
3
• Example:
A
0.1
0.25
0.05
sequence = ATGCAAGTCT…
C
0.3
0.25
0.8
• Position 1: ATGCA
0.1*0.25*0.1*0.1*0.6=1.5*10-4
• Position 2: TGCAA
0.5*0.25*0.8*0.7*0.6=0.042
4
5
0.7
0.6
0.1
0.15
T
0.5
0.25
0.05
0.1
0.05
G
0.1
0.25
0.1
0.1
0.2
Detecting a Known Motif within a
Sequence using PSSM
Is it a random match, or is it indeed an
occurrence of the motif?
PSPM -> PSSM (Probability Specific Scoring Matrix)
– odds score matrix: Oi(n) where n {A,C,G,T} for i={1,..,k}
– defined as Pi(n)/P(n), where P(n) is background frequency.
Oi(n) increases => higher odds that n at position i is part of
a real motif.
PSSM as Odds Score Matrix
•
Assumption: the background frequency of each
nucleotide is 0.25.
1
2
3
4
1. Original PSPM (Pi): A 0.1
0.25
0.05 0.7
2. Odds Matrix (Oi):
A
5
0.6
1
2
3
4
5
0.4
1
0.2
2.8
2.4
3. Going to log scale we get an additive score,
Log odds Matrix (log2Oi):
A
1
2
3
4
5
-1.322
0
-2.322
1.485
1.263
Calculating using Log Odds Matrix
• Odds 0 implies random match;
Odds > 0 implies real match (?).
• Example: sequence = ATGCAAGTCT…
1
2
• Position 1: ATGCA
-1.32+0-1.32-1.32+1.26=-2.7
odds= 2-2.7=0.15
• Position 2: TGCAA
1+0+1.68+1.48+1.26 =5.42
odds=25.42=42.8
3
4
5
A
-1.32
0
-2.32
1.48
1.26
C
0.26
0
1.68
-1.32
-0.74
T
1
0
-2.32
-1.32
-2.32
G
-1.32
0
-1.32
-1.32
-0.32
Calculating the probability of a Match
ATGCAAG
• Position 1 ATGCA = 0.15
Calculating the probability of a Match
ATGCAAG
• Position 1 ATGCA = 0.15
• Position 2 TGCAA = 42.3
Calculating the probability of a Match
ATGCAAG
• Position 1 ATGCA = 0.15
• Position 2 TGCAA = 42.3
• Position 3 GCAAG = 0.18
Calculating the probability of a match
ATGCAAG
• Position 1 ATGCA = 0.15
• Position 2 TGCAA = 42.3
• Position 3 GCAAG = 0.18
P (i) = S / (∑ S)
Example 0.15 /(.15+42.8+.18)=0.003
P (1)= 0.003
P (2)= 0.993
P (3) =0.004
Building a PSSM
• Collect all known sequences that bind a
certain TF.
• Align all sequences (using multiple
sequence alignment).
• Compute the frequency of each nucleotide
in each position (PSPM).
• Incorporate background frequency for each
nucleotide (PSSM).
PROBLEMS…
• When searching for a motif in a genome using PSSM or
other methods – the motif is usually found all over the place
->The motif is considered real if found in the vicinity of a
gene.
• Checking experimentally for the binding sites of a specific
TF (location analysis) – the sites that bind the motif are in
some cases similar to the PSSM and sometimes not!
3. Finding new Motifs
• We are given a group of genes, which
presumably contain a common regulatory
motif.
• We know nothing of the TF that binds to the
putative motif.
• The problem: discover the motif.
Difficulties in Computational
Identification
• Each motif can appear in any of m-k columns;
there are (m-k)n possibilities.
• Noise:
Mismatches are allowed, the motif is not exact.
Not all sequences contain the motif.
• Statistical significance:
k is short (6-20 nucleotides).
m ranges from 10s (prokaryotes) to 1000s (eukaryotes) of
nucleotides.
=> a random motif can appear by chance in sequences.
Computational Methods
• This problem has received a lot of attention from
CS people.
• Methods include:
– Probabilistic methods – hidden Markov models
(HMMs), expectation maximization (EM), Gibbs
sampling, etc.
– Enumeration methods – problematic for inexact motifs
of length k>10. …
• Current status: Problem is still open.
Tools on the Web
• MEME – Multiple EM for Motif Elicitation.
http://meme.sdsc.edu/meme/website/
• metaMEME- Uses HMM method
http://meme.sdsc.edu/meme
• MAST-Motif Alignment and Search Tool
http://meme.sdsc.edu/meme
• TRANSFAC - database of eukaryotic cis-acting regulatory DNA
elements and trans-acting factors.
http://transfac.gbf.de/TRANSFAC/
• eMotif - allows to scan, make and search for motifs in the protein
level.
http://motif.stanford.edu/emotif/