Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Linear motifs and
phosphorylation sites
What is a linear motif?
(in molecular biology)
…a first taste
Short sequence of amino acids encoding
a particular molecular function
Linear Motifs
Functional sites
We need a more accurate definition!
What are you going to learn about Linear Motifs?
Where can we find them?
Why are they important?
Can we classify them?
How can we represent them?
How can we discover them?
When and how can we use them?
What are tools and resources to handle them?
What are you going to learn about Linear Motifs?
Where can we find them?
Why are they important?
Can we classify them?
How can we represent them?
How can we discover them?
When and how can we use them?
What are tools and resources to handle them?
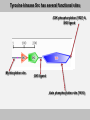
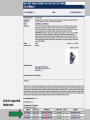
Tyrosine kinsase Src has several functional sites
CSK phosphorylation (Y527) &
SH2 ligand
Myristoylation site
SH3 ligand
Auto phosphorylation site (Y416)
p53 is full of functional sites
CYCLIN
MDM2
NES
TAFII31
P300
P300
Pin1 P-Ser-Pro isomerisation
Acetylation
SUMO
Ubiquitinylation
phosphorylation
NLS
CBP
S100B
SIR2
The sequences of many proteins contain short,
conserved motifs that are involved in recognition
and targeting activities, often separate from other
functional properties of the molecule in which they
occur.
These motifs are linear, in the sense that threedimensional organization is not required to bring
distant segments of the molecule together to make
the recognizable unit.
Tim Hunt (TIBS 1990)
The conservation of these motifs varies: some are
highly conserved while others, for example, allow
substitutions that retain only a certain pattern of
charge across the motif.
Tim Hunt (TIBS 1990)
A more accurate definition
• short, common stretches of polypeptide chains (~ 3-10 amino acid
residues long)
• embody a distinct molecular function independent of a larger
sequence/structure context.
• bind with low affinity (1.0-150 M). Mediate transient interactions.
• are nearly always involved in regulation
• are involved in protein/domain-protein/domain interactions
• often reside in disordered or low-complexity regions
• often become ordered upon binding to another protein or domain
• occurrences of LMs seem to arise or disappear as a result of point mutations
What are you going to learn about Linear Motifs?
Where can we find them?
Why are they important?
Can we classify them?
How can we represent them?
How can we discover them?
When and how can we use them?
What are tools and resources to handle them?
Why are they important?
Evolutionary unrelated protein sharing a functional feature are likely to
contain similar linear motifs
This may be the result of
- convergent evolution
- evolutionary conservation in a divergent evolution process
In any case, linear motifs are indicative of functions
In other words…
They are made up of the amino acid residues encoding
a functional site
With the appropriate tools, they can be used to identify:
•protein functions
•functional regions (in a protein sequence and on its threedimensional structure, if available)
What are you going to learn about Linear Motifs?
Where can we find them?
Why are they important?
Can we classify them?
How can we represent them?
How can we discover them?
When and how can we use them?
What are tools and resources to handle them?
Can we classify LMs? How?
Can we classify LMs? How?
Functional group
Functional site (Linear Motif)
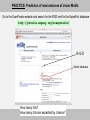
PRACTICE: Let’s find linear motifs in human p53…
Go to the UniProt website: http://www.uniprot.org/
Type p53 in the Query text box and select P04637
or
Type directly either P04637 or P53_HUMAN in the Query text box
Work in groups and analyse the p53 entry record:
- how many LMs can you identify?
- which function(s) are they indicative of?
- are they always annotated as “motif”?
- can you classify them according to the 4 categories?
What are you going to learn about Linear Motifs?
Where can we find them?
Why are they important?
Can we classify them?
How can we represent them?
How can we discover them?
When and how can we use them?
What are tools and resources to handle them?
How can we represent LMs?
Alignment of cyclin ligands
inhibitors
Regular expression: [RK].L.{0,1}[FLIV]
How can we represent LMs?
Alignment of cyclin ligands
inhibitors
Regular expression: [RK].L.{0,1}[FLIV]
Regular Expression (regexp)
L: single amino acid “L” = Leucine
[KR]: different amino acids allowed at this position
x or .: wildcard
{0,1}: variable length
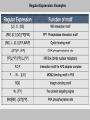
Regular Expression: Examples
Before we describe what regexp are useful for, let’s briefly see how
to discover de novo motifs
In some cases, the structure and function of
an unknown protein which is too distantly
related to any protein of known structure to
detect its affinity by overall sequence
alignment may be identified by its possession
of a particular cluster of residues types
classified as a motifs. The motifs, or
templates, or fingerprints, arise because of
particular requirements of binding sites that
impose very tight constraint on the evolution
of portions of a protein sequence
Arthur Lesk, 1988
What are you going to learn about Linear Motifs?
Where can we find them?
Why are they important?
Can we classify them?
How can we represent them?
How can we discover them?
When and how can we use them?
What are tools and resources to handle them?
In contrast to domains, which are readily
detectable by sequence comparison, linear
motifs are difficult to discover due to
their short length, a tendency to reside in
disordered regions in proteins, and limited
conservation outside of closely related
species.
Neduva et al. PLoS Biology 2005
De novo Linear Motif discovery
Study literature paper(s)/review(s) on a group of unrelated proteins
sharing a function
Build an alignment of these proteins
Add to the alignment other sequences relevant to the subject under
consideration
Pay attention to the residues and regions thought or proved to be
important to the biological function of that group of proteins:
• enzyme catalytic sites
• PTM sites
• regions involved in binding
Try to find a short conserved sequence which includes functionally
important residues
Discovery of de novo Linear Motif
There are algorithms that do it automatically
Neduva et al. PLoS Biology 2005
Discovery of de novo Linear Motif
Our central hypothesis is that proteins with
a common interaction partner will share a
feature that mediates binding, either a
domain or a linear motif. In the absence of a
shared domain, a linear motif could well be
the only common sequence feature and might
thus be detectable simply by virtue of overrepresentation, which is the basis of our
approach.
Neduva et al. PLoS Biology 2005
A probabilistic method for identifying over-represented, convergently
evolved, short linear motifs in proteins.
Edwards et al. PLoS ONE 2007
PRACTICE: Discovery of de novo Linear Motifs
Dilimot
http://dilimot.russelllab.org/
SLIMFinder
http://www.southampton.ac.uk/~re1u06/software/slimfinder/
What are you going to learn about Linear Motifs?
Where can we find them?
Why are they important?
Can we classify them?
How can we represent them?
How can we discover them?
When and how can we use them?
What are tools and resources to handle them?
Linear Motif Databases
ELM
PROSITE
R.[RK]{1,2}.R
R-x-[RK]-x(1,2)-R
1632 documentation entries
(domains and functional sites)
174 manually annotated motifs
16-03-2012
What regular expressions are useful for?
How can we use regular expressions?
Regular expressions can be used to search for motif occurrences in
(uncharacterised) protein sequences
There are algorithms that do this for us
We call the occurrence of a motif in a sequence an INSTANCE of that motif
A motif (a regexp) can have many instances
SH3 ligand motif
[RKY]..P..P
KKVAVVRTPPKSPSSAKSRL
ISPPTPKPRPPRPLPVAPGS
EDQILKKPLPPEPAAAPVST
SHRKTKKPLPPTPEEDQILK
TRICKIYDSPCLPEAEAMFA
TAU_HUMAN
P85A_HUMAN
BTK_HUMAN
BTK_HUMAN
RAD51_HUMAN
Prediction of new instances of Linear Motifs
ScanProsite
INPUT: a protein sequence
OUTPUT: PROSITE or user-defined motif matches in the input sequence
Allows the search for user-defined regular expressions
Scansite
ELM
MiniMotifMiner
INPUT: a protein sequence
OUTPUT: scansite motif matches in the input sequence
INPUT: a protein sequence
OUTPUT: ELM motif matches in the input sequence
INPUT: a protein sequence
OUTPUT: MiniMotifMiner motif matches in the input sequence
PRACTICE: Prediction of new instances of Linear Motifs
Go to the ScanProsite website and search for the RGD motif in the SwissProt database
http://prosite.expasy.org/scanprosite/
R-G-D
Select database
How many hits?
How many hits are expected by chance?
Regular expression pros and cons
Unfortunately matches to these motifs are not significant, providing
a signal-to-noise problem for bioinformatics tools
Advantages
Disadvantages
Memorable to humans
Over determined
Computationally fast
Motif may vary in other lineages
Standardised in scripting
languages (Python, Perl)
Do not capture weaker
preferences
Often, they can descrive a motif
very well
Easy to make a poor
representation
Overprediction and context information
Functional sites only work in proper context
The cell knows how to discriminate TP from FP !!!
The site must be in the correct
cellular context
(subcellular localisation)
The site must be in correct molecular
context
- accessible
- usually not in globular domains,
- often together with certain types of co-domains
The site is only relevant in a specific
taxonomy range
Knowledge of context can provide the basis
for filters for improved prediction of
functional sites
For example…
Globular domain filter
Motifs are mostly found in disordered regions
The disordered regions are proving to be rich in Linear Motifs
Src kinase
We can exploit this observation and filter out motif matches inside
domains
Structural Filter
Motif matches are not ALWAYS outside domains
Inside domains they are unlikely unless in surface loops
When inside a domain, a motif match is more likely to be a True
Positive (TP) if it occurs in a flexible (i.e. loop, turn or linker) and
accessible region of the domain
The RGD motif is recognized by different members of the integrin family
An exposed instance of
the RGD motif in a domain
An instance of the RGD
motif in a region outside
a domain
MOD_N-GLC_1 (.(N)[^P][ST]..) is a motif for N-glycosilation site
Two MOD_N-GLC_1
motifs in a domain
Structural Filter
We can think to implement a filter that is based on the three-dimensional
features of motifs (i.e. their accessibility and secondary structure types)
If the match is not accessible
low score
If the match is in -helix
low score
If the match is in -strand
low score
Other features that can be used to filter out FPs:
•Taxonomy
•Cellular compartment
•Evolutionary conservation
Davey NE et al. Mol Biosyst 2011
Why is a Conservation Score useful for linear motif prediction?
Improve the prediction of LM instances by discarding those
matches that are unlikely to be functional because they
have not been conserved during the evolution of the protein
sequences
There is a resource which implements these filters
It associates a score to occurrences of motifs based on
•Cellular context
•Molecular context
•Domain context
•Disorder
•Taxonomy
•Evolutionary conservation
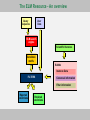
The Eukaryotic Linear Motif (ELM) Resource implements
a logical filtering system to reduce false matches
The Eukaryotic Linear Motif (ELM) Resource
• Repository of information about functional sites
(including experimentally reported instances)
• A motif-based query tool to find possible new functional
sites
• A logical filtering system to reduce false matches
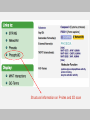
The ELM Resource - An overview
Query
Sequence
User
Data
ELM search
engine
Scientific literature
Candidate
motifs
ELMdb
Instance Data
FILTERS
Conextual information
Filter information
Rejected
predictions
Retained
predictions
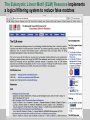
PRACTICE: The ELM server (http://elm.eu.org/)
Go to the ELM server
Search for motif matches in the EH domain-binding mitotic phosphoprotein
Output 1
instance in
structurally
unfavourable
context
annotated
instance
Instance in
unfavourable
context
highly
conserved
instance
Output 2
Output 2
Browse the ELMs page for the Clathrin Box motif in Endocytosis cargo adaptor
proteins (ELM: LIG_AP2alpha_2)
Link to reported
instances
Exploring unknown
protein sequences
Phosphorylation sites
Phosphorylation is the addition of a phosphate group (PO4) to a protein
molecule or small molecule.
The hydroxyl groups (-OH) of SER, THR or TYR residues side chain are
the most common targets
Reversible protein phosphorylation
A protein
kinase moves
a phosphate
group from
ATP to the
protein
ATP (adenosine triphosphate) is
the energy currency of the living
world. Every cellular process that
requires energy gets it from ATP
A protein phosphatase
removes the
phosphate and the
protein reverts to
its original state.
•It is rapid (few seconds)
•It is easily reversible
Reversible protein phosphorylation regulates most aspects of cell life
~ one third of cellular proteins could undergo phosphorylation
It is involved in regulation of
metabolism, motility, growth,
division, differentiation,
trafficking, membrane transport,
learning, memory
Even subtle changes in the activity
of protein kinases can lead to a
variety of diseases (cancer)
Phosphorylation is a Post Translational Modification (PTM)
A kinase recognises its substrate and adds a phosphate
group (PO4) to one of its residues, typically a Serine (Ser, S),
Threonine (Thr, T), or Tyrosine (Tyr, Y)
Amino acid phosphorylation is probably the most
abundant of the intracellular PTMs used to regulate the
state of eukaryotic cells, with estimates ranging up to
500,000 phosphorylation sites in the human proteome
Nevertheless…
Substrate recognition is specific
In other words…
Each kinase is capable of recognising its substrate(s) in the cell
In fact, the enzymes must be specific and act only on a defined subset of
cellular targets to ensure signal fidelity.
Even though the determinants of specificity are still unclear
Substrate recruitment is one of the known specificity mechanisms
The protein composition around the phosphorylatable site is another factor
Kinases are capable
of recognising the
region surrounding
the phosphoacceptor
residue (in sequence
and/or in structure)
In fact, kinases do not phosphorylate every Ser, Thr, Tyr they encounter
Kreegipuu et al, NAR 1998
in the cell
A phosphorylation site can be represented by a phosphorylation motif
Experimentally verified phosphorylation motifs can
be used to predict new phosphorylation sites and
characterise kinase substrates
There are many resources collecting P-sites and many tools to
predict P-sites in user-defined protein sequences
Collection of instances of P-sites
Prediction of new instances of P-sites
Phospho.ELM
phospho.elm.eu.org/
Phospho.ELM
phospho.elm.eu.org/
PhosphoSitePlus
www.phosphositePlus.org/
Scansite
scansite.mit.edu/
PHOSIDA
www.phosida.com/
NetPhos
www.cbs.dtu.dk/services/NetPhos/
PHOSPHORYLATION SITE DATABASE
www.phosphorylation.biochem.vt.edu/
NetPhosK
www.cbs.dtu.dk/services/NetPhos/
Phospho.3D
www.phospho3d.org/
NetworKIN
networkin.info/search.php
KinasePhos
KinasePhos.mbc.nctu.edu.tw/
Predikin
predikin.biosci.uq.edu.au/
Phospho.ELM
phospho.elm.eu.org
Database of experimentally verified phosphorylation sites
in eukaryotic proteins
Current release contains:
•42,914 instances (fully linked to literature references)
• 299 kinases
• 11,224 sequences
• 8,698 substrates
PRACTICE
Go to the Phospho.ELM website
and search P-sites for p53
ELM and Phospho.ELM are interconnected
PhosphoBlast
Structural information on P-sites and 3D scan
Phospho.3D
http://www.phospho3d.org/
PRACTICE
Go to the Phospho.3D
website and search all the
substrates of the Src
kinase
Suggestions to predict P-sites in unknown sequences
MEESQSDISLELPLSQETFSGLWKLLPPEDILPSPH
CMDDLLLPQDVEEFFEGPSEALRVSGAPAAQDPVTE
TPGPVAPAPATPWPLSSFVPSQKTYQGNYGFHLGFL
QSGTAKSVMCTYSPPLNKLFCQLAKTCPVQLWVSAT
PPAGSRVRAMAIYKKSQHMTEVVRRCPHHERCSDGD
GLAPPQHLIRVEGNLYPEYLEDRQTFRHSVVVPYEP
PEAGSEYTTIHYKYMCNSSCMGGMNRRPILTIITLE
DSSGNLLGRDSFEVRVCACPGRDRRTEEENFRKKEV
LCPELPPGSAKRALPTCTSASPPQKKKPLDGEYFTL
KIRGRKRFEMFRELNEALELKDAHATEESGDSRAHS
SYLKTKKGQSTSRHKKTMVKKVGPDSD
?
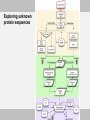
Exploring unknown protein sequences
• Go to UniProt (or Blast your sequence against the UniProt database)
and explore the sequence annotation
• Go to Phospho.ELM and scan the sequence
• Go to PHOSIDA and PhosphoSitePlus and do the same
• Use different predictors and select only high scoring sites
• Use evolutionary information:
- is the site conserved?
• Use domain (SMART and Pfam) databases:
- is the site inside a domain?
• Use structural information if available:
- is the site exposed?
- is it in a flexible region?
Exploring unknown protein sequences
When all information is collected, only retain sites predicted by more
than one tool
Amongst these, for further experimental tests, preferably choose sites
that are:
•Not inside domain(s)
•Not in secondary structure elements (helices and strands)
•Accessible to the solvent
•Evolutionary conserved