Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Artificial gene synthesis wikipedia , lookup
Genome (book) wikipedia , lookup
DNA barcoding wikipedia , lookup
Gene expression programming wikipedia , lookup
Non-coding DNA wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Genomic library wikipedia , lookup
Human genome wikipedia , lookup
Metagenomics wikipedia , lookup
Human Genome Project wikipedia , lookup
Quantitative comparative linguistics wikipedia , lookup
Microevolution wikipedia , lookup
Genome editing wikipedia , lookup
Minimal genome wikipedia , lookup
Pathogenomics wikipedia , lookup
Maximum parsimony (phylogenetics) wikipedia , lookup
Smith–Waterman algorithm wikipedia , lookup
Koinophilia wikipedia , lookup
Sequence alignment wikipedia , lookup
Genome evolution wikipedia , lookup
Phylogenetic Tree Construction
and Related Problems
Bioinformatics
Problems
Multiple Sequence Alignment
Construction of Phylogenetic Trees
Determining Genomic Distance through
Rearrangements
Alignment of Multiple
Sequences
We can extend the notion of alignment to
multiple strings:
-att-ct-ttactat-a-t
An alignment of strings S1…Sn is described
by strings S1’…Sn’
Each Si’ contains the characters of Si in order
interspersed with spaces (-)
No position exists that contain spaces for all Si’
Scoring a Multiple Alignment
Sum of pairs
Consider each pair in the set of sequences,
determine the similarity score (using gap, match,
and mismatch weights) for each pair, and add all
pair-wise scores
Distance from consensus
Consider all columns and count the total number
of differences from the consensus character
Variations: just count characters that differ from
consensus or have a difference score for each
differing character
Multiple Alignment Problem
Formulation: Given sequences S1…Sn,
obtain an optimal multiple alignment of
the sequences
“Optimal” depends on multiple
alignment scoring method
No known (correct) efficient algorithms
for this problem
Strategies
Brute force algorithm: consider all possible
alignments, then determine the one that
results in a best score (time complexity?)
Common Heuristic: Use regular (two-string)
alignment and then repeatedly add a string to
a growing alignment
O(nm2) – where m is the the max string length
Does not always produce an optimal alignment
Phylogenetic Tree Construction
Multiple alignments are often performed on
similar species
Next step: Construct an evolutionary tree of
these species
Input to problem: evolutionary distances
between each pair
Not the same as similarity score
Example: edit distance--count number of indels
needed to transform one string to the other
Problem Formulation
Given a set of species and evolutionary
distance between each pair (2d matrix
of numbers), construct a phylogenetic
tree consistent with the distances
Details needed:
Characteristics/constraints of a tree
Notion of “consistent”
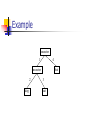
Phylogenetic Tree
Rooted
Leaves are species
Internal nodes are speculated ancestors
Distances associated with each edge
Example
ancestor
1
4
ancestor
2
dog
bat
3
cat
Minimizing Distance Deviation
Phylogenetic tree implies pairwise
distances (sum all edge distances in
path)
Compare with input distances to assess
consistency
Sum of Differences
Alternative computations for deviation
(least squares)
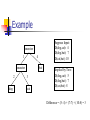
Example
Suppose Input:
D(dog,cat): 4
D(dog,bat): 7
D(cat,bat): 10
ancestor
1
4
ancestor
2
dog
bat
3
cat
Implied by Tree:
D(dog,cat): 5
D(dog,bat): 7
D(cat,bat): 8
Difference = (5- 4) + (7-7) + (10-8) = 3
Character-based Tree
Construction
Input:
a set C of m characters
possible values for each character
a set S of n species where each element of S is associated
with a value for each character in C
Output: a phylogenetic tree T such that
Elements in S are the leaves of T
Each internal node of T has an assigned value per character
Each character value induces a connected subtree of T
Mutations on Genomes
Mutations can occur at the gene level
(indels of nucleotides) or at the genome
level (operations on genes)
At the genome level, for similar species,
the operations are rearrangements
Reversals
Transpositions
Translocations
Genomes, Chromosomes, and
Permutations
Genome: set of chromosomes
Chromosome: sequence of genes
Genes: unique across entire genome
Can simplify the representation of a
genome by arbitrarily concatenating the
chromosomes
Result: a permutation of a set of unique
genes
Determining Genomic Distance
Recall: Phylogenetic tree construction
need pair-wise distances
For similar species with the same set of
genes:
Can use number of rearrangement
operations for distance
Common distance model: obtain the
fewest (optimal) rearrangements necessary
to transform genome X to genome Y
Some Assumptions Made
Can limit allowed rearrangements to
some reasonable subset of possible
arrangements
e.g., reversals apparently most common for
plant species
Sometimes, gene-blocks instead of
individual genes are the elements that
are permuted
Problem Formulations
Given permutations (species) p and q of
the set {1…n}, find the shortest
sequence of rearrangements
(mutations) that transform p to q
p.r1. r2 … rs = q
Given a permutation p, find the shortest
sequence of rearrangements that sort p
p.r1. r2 … rs =
123…n