Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Magnesium transporter wikipedia , lookup
Western blot wikipedia , lookup
Catalytic triad wikipedia , lookup
Nucleic acid analogue wikipedia , lookup
Plant nutrition wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Fatty acid synthesis wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
Fatty acid metabolism wikipedia , lookup
Point mutation wikipedia , lookup
Peptide synthesis wikipedia , lookup
Metalloprotein wikipedia , lookup
Nitrogen cycle wikipedia , lookup
Protein structure prediction wikipedia , lookup
Glyceroneogenesis wikipedia , lookup
Proteolysis wikipedia , lookup
Genetic code wikipedia , lookup
Citric acid cycle wikipedia , lookup
Biochemistry wikipedia , lookup
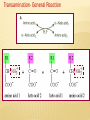
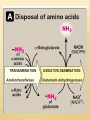
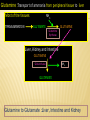
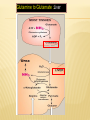
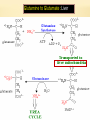
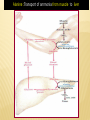
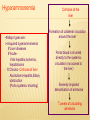
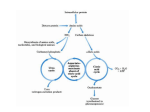
AMINO ACIDS: DISPOSAL OF NITROGEN & UREA CYCLE Dr Vivek Joshi, MD CONTENTS Amino acid pool Protein Degradation Amino acid degradation Disposal of Body Nitrogen Transamination Deamination Transport of ammonia OVERALL NITROGEN METABOLISM Amino Acid pool Amino acids-Not stored in the body. Maintain a supply of amino acids-Amino acid pool PROTEIN TURNOVER-PURPOSE Affords metabolic flexibility Protects cells from the accumulation of abnormal proteins Plays an important role in numerous physiological processes e.g. Eukaryotic cell cycle control/antigen presentation Measured in half-lives Structural proteins (Collagen)typically have long half-lives Regulatory enzymes have half-lives that are typically measured in minutes Proteins with Ser as N-terminal aa -Half life >20 h Proteins with shorter half life: Asp as N-terminal aa Rich in proline (P), glutamate (E),serine (S), and threonine (T) PROTEIN DEGRADATION Enzyme systems for degradation of proteins Energy dependent ubiquitinproteasome pathway Cytosolic pathway for degradation of damaged /unneeded proteins Important in degrading proteins of intracellular origin Non energy dependent degrading enzymes of lysosmes. Way by which extracellular and some intracellular proteins are degraded Ubiquitin - Proteasomes pathway Present in all eukaryotic cells Cytosolic proteins destined for degradation are enzymatically tagged with activated ubiquitin Ubiquitin-tagged proteins are then attacked by cytosolic ATPdependent proteases Amino acid Degradation Regardless of the source ,aa not immediately incorporated into a new protein are rapidly degraded. AA catabolism involves: Removal of the a amino group as ammonia Conversion of the ammonia into urea Conversion of remaining AA carbon skeleton ( a-keto acid) into TCA cycle intermediates Disposal of Body Nitrogen Transamination Deamination Transport of Ammonia TRANSAMINATION- INTRODUCTION Most common reaction involving free amino acids Reversible reaction Transfer of amino group from an amino acid to a keto acid to form a newer keto acid and a newer amino acid respectively. No release of ammonia Major process for removing nitrogen from amino acids with EXCEPTION- Lysine,Threonine,proline Require Pyridoxal phosphate as an essential coenzyme Transamination- Introduction Transamination- General Reaction Amino transferase/ Transaminase Catalyses Transamination reaction Located in the Cytosol and mitochondria of liver, muscle Each Aminotransaminases is specific for 1 pair of substrate but non-specific for the other pair Aminotransaminases are named after the specific amino group donor Major Aminotransaminases #Aminotransaminases family Conserve amino group of most of the amino acids into Glutamate #Alanine Aminotransaminases(SGPT)Alanine /Pyruvate conversion #Aspartate Aminotransaminases(SGOT)Aspartate/Oxaloacetate conversion Amino Transaminase Family Family Glutamate All the amino nitrogen from amino acids that undergo transamination can be concentrated in glutamate The acceptor of the amino group is almost always a-keto glutarate Alanine Amino Transaminase Alanine-The principal amino acid released from muscle during starvation Pyruvate -Form an important substrate for hepatic Gluconeogenesis. Aspartate Aminotransaminases Aspartate which is a source of nitrogen in the UREA cycle. Oxaloacetate can form Glucose via Gluconeogenesis DIAGNOSTIC VALUE OF PLASMA AMINOTRANSAMINASES •Aminotransaminases- Intracellular enzymes with low levels in plasma •Elevated plasma amino transaminase- Cell Damage Diagnostic purpose •Non hepatic diseases Myocardial infarction and muscle disorders Serum AST more specific than ALT •Hepatic diseases Serum ALT more specific than serum AST Serum AST more sensitive than serum ALT ALANINE TRANSAMINASE :INDICATOR OF HEPATIC DAMAGE Transamination-Pair Amino Acid Keto Acid Glutamate a ketoglutarate Alanine Pyruvate Aspartate Oxaloacetate Transamination- Pyridoxal phosphate (PLP) AMINOTRANSAMINASES REQUIRE PYRIDOXAL PHOSPHATE (PLP) AS AN ESSENTIAL COFACTOR Deamination Removal of amino group as AMMONIA Types Oxidative Deamination Oxidase- L . AA oxidase / D-AA oxidase Dehydrogenase –Glutamate Dehydrogenase Non Oxidative Deamination Hydroxy and Sulfur containing AA Catalysed by Dehydratase and Desulfurase PLP dependent OXIDATIVE DEAMINATION BY GLUTAMATE DEHYDROGENASE In mammals: almost entirely confined to the liver mitochondrial matrix / Readily reversible Glutamate –The only amino acid that undergoes rapid oxidative deamination Oxidative Deamination by Glutamate Dehydrogenase Amino Transaminase ADP and GDP Indicative of low cellular energy level Stimulates glutamate degradation ATP and GTP Indicative of ample energy supply Allosteric activator in the direction of glutamate synthesis Transamination is followed by Deamination: Transdeamination Glutamate Dehydrogenase TRANSPORT OF AMMONIA Nitrogen travels in blood mainly in amino acids, particularly alanine and glutamine. Alanine and glutamine are synthesized in the peripheral tissues to act as nitrogen carrier Glutamine Transport of ammonia from peripheral tissue to liver Alanine Transport of ammonia from the muscle to the liver Glutamine :Transport of ammonia from peripheral tissue to liver Glutamine Provides a non-toxic transport form of ammonia Transport form of Ammonia from peripheral tissue to the liver Synthesized by Glutamine synthase -Liver, Muscle and Brain Most common free amino acid in human blood plasma. Major mechanism for detoxification of ammonia in the brain. 50% of circulating amino acid molecules are glutamine, an ammonia transporter Glutamine :Transport of ammonia from peripheral tissue to liver Most of the tissues TRANSAMINATION NH3 GLUTAMATE Glutamine Synthase GLUTAMINE Liver ,Kidney and Intestine GLUTAMINE Glutaminase NH3 GLUTAMATE Glutamine to Glutamate :Liver, Intestine and Kidney Glutamine to Glutamate :Liver Glutamine to Glutamate :Liver Most of the tissues TRANSAMINATION NH3 GLUTAMATE Glutamine Synthase GLUTAMINE Intestine GLUTAMINE Glutaminase NH3 Portal Blood GLUTAMATE Another Source of NH3 in the intestine :Bacterial flora Glutamine to Glutamate :Intestine LIVER Glutamine to Glutamate :Kidney Glutamine is removed from circulation by the kidneys Glutamine converted into Glutamate by Glutaminase releasing ammonia Most of the ammonia is excreted in the urine as NH4+ An important mechanism for maintaining the body’s acid-base balance Glutaminase Glutamate Dehydrogenase Alanine :Transport of ammonia from muscle to liver Alanine :Transport of ammonia from muscle to liver Glucose –Alanine cycle UREA CYCLE UREA Major end-product of nitrogen catabolism in humans Principal Non protein nitrogenous waste products (Uric acid and Creatinine ) Synthesis- liver (Cytosol and mitochondria) Released in the blood-Cleared by the kidneys Urea Clearance-Measure of Glomerular filtration rate(GFR) UREA CYCLE First metabolic pathway to be discovered Transdeamination of the AA results in release of NH4 + in the liver Ammonia is toxic and is converted into non toxic ,water soluble product –UREA. Mammals are primarily Ureotelic Birds and reptiles are Uricotelic UREA CYCLE Urea Cycle Urea synthesis begins with reaction of ammonia with C02 (Bicarbonate) and ATP to give Carbamoyl phosphate Reaction catalyzed by Carbamoyl phosphate synthetase I Reaction requires Mg2+ Carbamoyl phosphate synthetase II-Pyrimidine synthesis Carbamoyl phosphate synthetase-I Catalyzes the rate-limiting step in urea cycle Active only in the presence of the allosteric activator Nacetylglutamate(NAG) N-Acetyl Glutamate is synthesized from Acetyl CoA and Glutamate by N-Acetyl Glutamate synthase Binding of NAG to CPS I induces a conformational change that enhances the affinity of the synthase for ATP Intrahepatic concentration of NAG increases after a protein rich meal –Stimulates UREA synthesis. Know CPS –I ……. UREA CYCLE Carbamoyl phosphate combines with ornithine to form Citrulline –Ornithine trans Carbomylase(OTC) Citrulline combines with Aspartate to form Arginosuccinate-Arginosuccinate synthase Arginosuccinase splits Arginosuccinate into Arginine and Fumarate Arginase split Arginine into Urea and Ornithine Fate of UREA Renal failure patients- Urease acts on urea-Important source of NH3 Oral neomycin administration-Reduces intestinal Bacteria-Decreased NH3 SUMMARY-UREA CYCLE Overall Reaction: Aspartate+NH3 +CO2 +3 ATP Urea+Fumarate +2 ADP +AMP +2Pi +2 PPi +3 H2O 4 High energy phosphates –Synthesis of each molecule of urea Source of one nitrogen of urea-Free ammonia Source of second nitrogen of urea-Aspartate In effect, both nitrogen atoms of urea come from glutamate, which in turn gathers nitrogen from other amino acids. Summary-UREA cycle Two nitrogen of the Urea comes from # Ammonia # Aspartate AMMONIA Constantly produced in the tissues Toxic to the central nervous system even in trace amounts Amino acids are quantitatively the most important source of ammonia Most of the ammonia generated in amino acid degradation is produced by the oxidative deamination of glutamate DISPOSAL OF AMMONIA Amines/Monoamines GLUTAMATE DEHYDROGENASE Asparagine L –AA Oxidase Non –oxidative Deamination Ammonia pool UREA Glutaminase Glutamine Purines/Pyrimidines Bacterial Urease(25%) Urea – formation is quantitatively the most important disposal route for ammonia HYPERAMMONEMIA Normal Ammonia level -5–50 mol/L Ammonia is normally detoxified into Urea in the liver Liver functions compromisedHyperammonemia (1000mol/L) Hyperammonemia-Medical emergency Hyperammonemia Ammonia-Directly Neurotoxic Tremors,slurring of speech and blurring of vision Coma and death at high levels Brain is particularly vulnerable -Depends on the CAC to maintain its high rate of energy production Shift in the equilibrium of the glutamate dehydrogenase reaction toward the direction of glutamate formation Depletes a-ketoglutarate, an essential intermediate in CAC Results in a decrease in cellular ATP production Hyperammonemia Major types are: Acquired hyperammonemia # Liver diseases # AcuteViral hepatitis,ischemia, hepatotoxins # Chronic- Cirrhosis of liver Alcoholism,Hepatitis,Biliary obstruction (Porto systemic shunting) Cirrhosis of the liver Formation of collateral circulation around the liver Portal blood is shunted directly to the systemic circulation (no access to the liver) Severely impaired detoxification of ammonia Levels of circulating ammonia HYPERAMMONEMIA Hereditary Hyperammonemia # Inherited deficiency of the enzymes of Urea cycle # Failure to synthesize urea leads to hyperammonemia during the first week following birth # Hyperammonemia Type I- Deficiency of CPS-I # Hyperammonemia Type II -Deficiency of OTC - 1;30,000 live births - Most common of the inherited urea cycle disorders - X-linked (Predominantly affecting males) - All others urea cycle disorders are AR - Presents typically with mental retardation ,few weeks after birth #Treatment - Limiting protein in diet - Orally Phenyl butyrate -Converted to phenyl acetatephenylacetylglutamine -Excreted - Gene Therapy HYPERAMMONEMIA Hyperammonemia I Hyperammonemia II CPS –I Deficiency OTC Deficiency Blood Glutamine increased Blood Glutamine increased BUN Decreased BUN Decreased No increase in Orotic acid and uracil increased in blood Orotic acid and uracil increased in blood Cerebral edema Cerebral edema Lethargy ,convulsions, coma ,death Lethargy ,convulsions, coma ,death UCD Enzyme Deficiency Type I Hyperammonemia Carbamoylphosphate synthetase I N-acetylglutamate synthetase Deficiency N-acetylglutamate synthetase Type 2 Hyperammonemia Ornithine transcarbamoylase Classic Citrullinemia Argininosuccinate synthetase Argininosuccinic aciduria Argininosuccinate lyase (Argininosuccinase) Hyperargininemia Arginase Symptoms/Comments With 24h - 72h after birth infant becomes lethargic, needs stimulation to feed, vomiting, increasing lethargy, hypothermia and hyperventilation; without measurement of serum ammonia levels and appropriate intervention infant will die: treament with arginine which activates N-acetylglutamate synthetase severe hyperammonemia, mild hyperammonemia associated with deep coma, acidosis, recurrent diarrhea, ataxia, hypoglycemia, hyperornithinemia: treatment includes administration of carbamoyl glutamate to activate CPS I Most commonly occurring UCD, only X-linked UCD, ammonia and amino acids elevated in serum, increased serum orotic acid due to mitochondrial carbamoylphosphate entering cytosol and being incorporated into pyrimidine nucleotides which leads to excess production and consequently excess catabolic products: treat with high carbohydrate, low protein diet, ammonia detoxification with sodium phenylacetate or sodium benzoate episodic hyperammonemia, vomiting, lethargy, ataxia, seizures, eventual coma: treat with arginine administration to enhance citrulline excretion, also with sodium benzoate for ammonia detoxification episodic symptoms similar to classic citrullinemia, elevated plasma and cerebral spinal fluid argininosuccinate: treat with arginine and sodium benzoate rare UCD, progressive spastic quadriplegia and mental retardation, ammonia and arginine high in cerebral spinal fluid and serum, arginine, lysine and ornithine high in urine: treatment includes diet of essential amino acids excluding arginine, low protein diet