Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Elias James Corey wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Woodward–Hoffmann rules wikipedia , lookup
Metal carbonyl wikipedia , lookup
George S. Hammond wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Ene reaction wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
Aldol reaction wikipedia , lookup
Wolff rearrangement wikipedia , lookup
Tiffeneau–Demjanov rearrangement wikipedia , lookup
1,3-Dipolar cycloaddition wikipedia , lookup
Strychnine total synthesis wikipedia , lookup
Petasis reaction wikipedia , lookup
Hydroformylation wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
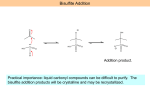
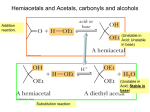
Aldehydes and Ketones Chapter 16 Structure Aldehydes Ketone Carbonyl group O R O R H R sp2 2-pentanone O pentanal O Examples of Naming OH O O O O pentan-2-one 3-oxopentanal O O 2-(1-hydroxyethyl)pentanal CHO CH3 H CHO H CO2H CH3 (E)-3-(but-1-enyl)hexane-2,4-dione 2,3-dimethyl-4-oxobutanoic acid benzaldehyde Resonance O O result O Extension of resonance O O Boiling points For compounds of comparable molecular weight… Alkanes, ethers < aldehydes, ketones < alcohols < carboxylic acids Dispersion Forces Dipole-dipole Hydrogen Bonding Water Solubility Ketones and Aldehydes, like ethers, can function as hydrogen bond acceptors and smaller compounds have significant water solubility. Recall Preparation from Alcohols OH OH Can also be done using KMnO4 in base with heat or bleach in acid solution (HOCl). Na2Cr2O7 acid, 35 deg. HO CH2OH Be sure you can balance this kind of reaction. O CO2H Use PCC to limit oxidation of primary alcohol to the aldehyde. Secondary are oxidized to ketone. Primary alcohol PCC RCH2OH RCH=O Secondary PCC R2CHOH R2C=O Preparations, con’d • Reaction of acid chloride and Gilman O R OH SOCl2 O Na2Cr2O7 O RCH2OH R Cl R R' 2CuLi 1. Li R'X 2. CuX But where do we get these?? R' Note that we have two possible disconnects available HOCH2R R'OH O + R'X HO O R' R R O + RX R' OH ROH HOCH2R' Example: Prepare 2-butanone from ethyl alcohol Requirement to start with ethanol suggests a disconnect into two carbon fragments. CH3CH2OH O Done! CH3CO2H + CH3CH2X via Gilman and acid chloride Aldehydes from carboxylic acids Reduction O R O SOCl2 OH LiAlH(OC(CH3)3)3 R Cl O R H And from alcohols, as before: PCC RCH2OH Oxidation RCHO A Common Sequence PCC RCH2OH Mg PX3 R'OH OH RCHO RX R R' R'MgX Na2Cr2O7 O Observe these parts at this moment. R R' Reactions Addition of a nucleophile: Nucleophilic Addition Good nucleophile, usually basic O O O + Nu: - Nu Nu tetrahedral intermediate OH OH + Nu Attack of nucleophile occurs on both sides of carbonyl group. Produces both configurations. Nu Overall: H – Nu was added to carbonyl group double bond. Notice that the CO bond order was reduced from 2 to 1. The addition reduced the bond order. We will use this idea later. Reaction can also be done in acid environment. Nucleophiles not expected to be as strong (why?) but the oxygen may become protonated making the carbonyl a better electrophile (why?). O OH acid OH OH + Nu: Nu Nu Very electronegative, protonated oxygen. Pulls the pi electrons into itself strongly. Problem: If there is too much acid present the nucleophile may become protonated, deactivating it Addition of Grignard (Trumpets Please) Recall the formation of a Grignard and its addition to an oxirane O Mg R-X R-Mg-X OMgX ether Carbonyls may be added to in same way… Mg R-X O R OMgX R-Mg-X ether If a new chiral center is created both configurations will be produced. OMgX + R R mild acid OH OH R R Common Reactions of Grignards R2R'COH a tertiary alcohol R-H Examine reaction with ester further. R'CO2Et an ester ROHRXRH(D) acid, weak acid O R primary alcohol O ROH + R’CH2OHRX + R’CO2HR2C(OH)R’ O R-Mg-X H R' OH R'' H RCH2OH O CO2 R' Both of these reactions extend carbon chain & keep -OH functionality at end of chain. Can extend further. primary alcohol H RR'R''C-OH tertiary alcohol ROH + R’R”CHOH RX + R’R”CO RR’C (OH)R” RCHR' secondary alcohol OH RCO2H carboxylic acid ROHRXRCO2H ROH + R’CH2OHRX + R’CHORCH(OH)R’ Grignard Reacting with an Ester. Look for two kinds of reactions. O OMgX O O R-Mg-X R-Mg-X R' R' Any alcohol will do here. OEt OEt R' R R' R R R Substitution EtOH R'COCl But where does an ester come from? OH Acid chloride R' SOCl2 R'CO2H R R Perhaps this carboxylic acid comes from the oxidation of a primary alcohol or reaction of a Grignard with CO2. Addition Synthetic Planning… Use of epoxides and carbonyls offer different disconnect sites. Pattern 1. RMgX HO-C-C-R HO O 2. H2O, HCl epoxide R New bond. Disconnect site. O OH R-Mg-X carbonyl R New bond. Disconnect site Nucleophile Pattern HO-C-R Want this to be the nucleophile (Grignard). Patterns to recognize: carbonyl vs oxirane We can create the following fragments of target molecules by using an organometallic (carbon nucleophile) reaction with a carbonyl O RMgX dil. acid OH The difference is the extra CH2 when using an oxirane. R reaction with a oxirane O RMgX dil. acid OH R Synthetic Planning… Three different disconnects possible OH Give synthetic routes to O + R3-Mg-X C R1 R1 R3 R2 R2 O + R2-Mg-X O R1 R3 + R1-Mg-X R2 R3 If none of the Rs are H then these three synthetic routes may be available. Example: Synthesize from ethanol CH3CH2OH CH3CH2X O OH Done OH 2 MgX CH3CO2Et O CH3CH2MgX CH3CO2H CH3CH2OH CH3CH2OH Preliminary Analysis •Hmmm, even number of carbons, at least that is good; ethanol is a two carbon molecule. •Now the problem is to divide it up into smaller fragments. •Ether linkage is easily constructed. Williamson. •Two butyl groups attached to the central 2 carbon fragment. Grignard + ester. Bisulfite Addition O OH O S HO S HO O O S O O O O O Addition product. Practical importance: liquid carbonyl compounds can be difficult to purify. The bisulfite addition products will be crystalline and may be recrystallized. Addition of Organolithium Compounds to Carbonyls Generally the reactions are the same as for Grignards but the lithium compounds are more reactive (and more difficult to handle). O O Br OH Li Li mild acid bromocyclohexane Decreased reactivity of electrophile due to steric hindrance to attack. So we used the alkyl lithium instead of a Grignard. Nucleophiles derived from terminal alkynes For example, once formed, the new alkynyl alcohol can be hydrated in two ways, Markovnikov and anti Markovnikov. Carefully observe the structure of the products, the relationship of the OH and the carbonyl. Can do all the reactions of an alkyne and an alcohol. But remember that we have two acidic groups: the more acidic OH and the less acidic terminal alkyne. We discussed this problem earlier. Note that the regioselectivity used here is only effective if this alkyne is terminal. Otherwise get a mixture. Addition of hydrogen cyanide basic Think of what the mechanism should be…. Followed by protonation of the alkoxide ion (perhaps by unionized HCN). pH issue. Slightly basic media so that HCN has partially ionized to cyanide ion, the actual nucleophile. Follow-up reactions on the cyanohydrins… H2O R N RHC acid catalysis dehydration O NaCN dil acid OH We saw this hydrogenation before. OH H2 R CH2R R NH2 N CH2R catalyst R CH2R Hydrolysis. Acid, heat or base, heat OH R CO2H CH2R Let’s see what we can do with the mechanism of the hydrolysis of the nitrile group to a carboxylic acid. Overall aq. acid, heat + NH3 R R CO2H N The action is at the nitrile group, CN --> CO2H. But how does a nitrile group behave? What could be happening? We are breaking the CN bond; bond order goes from 3 to 0. Probably stepwise. Chemically speaking: the nitrogen of the nitrile is basic (lone pair) and can be protonated. This makes it a better electrophile (cf. carbonyl). Multiple bond can undergo addition (cf. carbonyl) reducing bond order. Goal: Break the C to N bonding and create C-O bonds. Considerations: neither the electrophile (RCN) nor the nucleophile (water) is very reactive. Since we are in acid protonate the CN group to make it a better electrophile. Then attack it with the water nucleophile to add water. This results in reduction of C-N bond order and creation of C to O bonds . Again, we are in acid environment. Let’s protonate something…. Protonate the multiple bonded N atom to make better electrophile and attack with the nucleophile, water. Note the bonding pattern here. We have seen it before. acid R R N NH HO similar to NH HO CH2 which tauermerizes in acid or base, keto-enol HO R R CH2 O R R + H+ + H+ NH H2O NH HO NH O NH2 HOH What have done so far? Reduced the CN bond order from 3 to 2 and added one O to the C. Moving in the right direction! Want to reduce the CN bond order to zero and introduce more O on the C. Keep going! To induce the water to attack again (adds another O) need to increase the reactivity of the electrophile. Protonate again!! On the O. R HOH acid R NH2 O O R NH2 OH2 H Initial equilibrium with acid NH2 HO Now want to get rid of the NH2. We have all the O’s we need. We know what we have to do. Have to get the N protonated to make it a good leaving group. OH2 H R HO NH2 reposition the H+ + NH3 O R R HO NH3 HO Done. O Wittig Reaction O H I PPh3 Bu-Li R R or NaH Substitution Elimination Example, synthesize R R CHCH3 or combine them the other way… Wittig Reaction Mechanism Ph3P: Acidic hydrogen H H I Ph3P + Nucleophilic substitution Nucleophilic center Phosphonium ylide H strong base, BuLi Ph3P Ph3P R O O R O R R + Ph3P R Ph3P Ph3P R oxaphosphetane betaine R R O R Ph3P oxaphosphetane Ph3PO R Friedel Crafts Acylation R O O R Cl AlCl3 And then all the reactions of ketones… Formation of Hydrates, carbonyls and water. Carbonyl side of equilibrium is usually favored. Hemiacetals and Acetals, carbonyls and alcohols Addition reaction. (Unstable in Acid; Unstable in base) (Unstable in Acid; Stable in base) Substitution reaction Formation of Hemiacetals, catalyzed by either acid or base. Let’s do it in Base first. But first let’s take stock. We have an addition reaction. Just mixing a carbonyl and an alcohol do not cause a Use Base to reaction. set-up good necleophile. One of them must be made a better reactant. Carbonyl can be made into a Poor nucleophile better electrophile by protonating in acid. Good nucleophile Alcohol can become a better nucleophile in base by ionization. An addition of the alcohol to the carbonyl has taken place. Same mechanism as discussed earlier. hemiacetal Alternatively, hemiacetal formation in Acid Protonation of carbonyl (making the oxygen more electronegative) Attack of the (poor) nucleophile on (good) electrophile. Deprotonation Overall, we have added the alcohol to the carbonyl. Hemiacetal to Acetal, Acid Only Protonate the hemiacetal, setting up leaving group. Departure of leaving group. Attack of nucleophile Deprotonation Substitution reaction, cf S 1. Equilibria Generally, the hemiacetals and acetals are only a minor component of an equilibrium mixture. In order to favor formation of acetals the carbonyl compound and alcohol is reacted with acid in the absence of water. Dry HCl) The acetals or hemiacetals maybe converted back to the carbonyl compound by treatment with water and acid. An exception is when a cyclic hemiacetal can be formed (5 or 6 membered rings). Hemiacetal of D-Glucose The alcohol The carbonyl Try following the stereochemistry here for yourself The hemiacetal can form with two different configurations at the carbon of the carbonyl group. The carbon is called the anomeric carbon and the two configurations are called the two anomers. The two anomers are interconverted via the open chain form. Stabilities of the Anomers… Here note the alternating up-down relationships. More stable b form, with the OH of the anomeric carbon is equatorial Less stable a form. Here see the cis relationship of these two OH groups, one must be axial. Acetals as Protecting Groups Synthetic Problem, do a retrosynthetic analysis E Target molecule N Form this bond by reacting a nucleophile with an electrophile. Choose Nucleophile and Electrophile centers. The nucleophile could take the form of an organolithium or a Grignard reagent. The electrophile would be a carbonyl. Grignard would react with this carbonyl. Br-Mg Do you see the problem with the approach?? Use Protecting Group for the carbonyl… Acetals are stable (unreactive) in neutral and basic solutions. Create acetal as protecting group. Now create Grignard and then react Grignard with the aldehyde to create desired bond. protect react Remove protecting group. Same overall steps as when we used silyl ethers: protect, react, deprotect. deprotect Tetrahydropyranyl ethers (acetals) as protecting groups for alcohols. Recall that the key step in forming the acetal was creating the carbocation as shown… There are other ways to create carbocations…… Recall that we can create carbocations in several ways: 1. As shown above by a group leaving. This resonance stabilized carbocation then reacts with an alcohol molecule to yield the acetal. An acid 2. By addition of H+ to a C=C double bond as shown next. This cation can now react with an alcohol to yield an acetal. The alcohol becomes part of an acetal and is protected. Sample Problem Provide a mechanism for the following conversion O HCl/H2O HO OH O O OH First examination: have acid present and will probably protonate Forming an acetal. Keep those mechanistic steps in mind. Ok, what to protonate? Several oxygens and the double bond. Protonation of an alcohol can set-up a better leaving group. Protonation of a carbonyl can create a better electrophile. We do not have a carbonyl but can get a similar species as before. Strongly electrophilic center, now can do addition to the C=O The protonation of the C=C O O H+ O H Now do addition, join the molecules HO HO OH O Product O O Now must open 5 membered ring here. Need to set-up leaving group. HO HO H+ O O H O O Leaving group leaves…. HO HO HO H H H O O O O O O Followed by new ring closure. Done. Wow! Sulfur Analogs Consider formation of acetal O OH OH O dry HCl acetaldehyde ethanal Sulfur Analog O SH SH S dry HCl acetaldehyde ethanal dithiane S O The aldehyde hydrogen has been made acidic Bu Li + BuH S S S S H Why acidic? Sulfur, like phosphorus, has 3d orbitals capable of accepting electrons: violating octet rule. S S S S Recall early steps from the Wittig reaction discussed earlier H Ph3P: H I Ph3P + H strong base, BuLi Ph3P Ph3P This hydrogen is acidic. Why acidic? The P is positive and can accept charge from the negative carbon into the 3d orbitals PH Some Synthetic Applications Umpolung – reversed polarity What we have done in these synthetic schemes is to reverse the polarity of the carbonyl group; change it from an electrophile into a nucleophile. O CN O CN electrophile O O OH O S S nucleophilic Can you think of two other examples of Umpolung we have seen? Nitrogen Nucleophiles Mechanism of Schiff Base formation Attack of nucleophile on the carbonyl Followed by transfer of proton from weak acid to strong base. Protonation of –OH to establish leaving group. Leaving group departs, double bond forms. Hydrazine derivatives Note which nitrogen is nucleophilic O O H2N N H NH2 H2N N H Nucleophilic nitrogen Favored by resonance Less steric hinderance NH2 Reductive Amination Pattern: R2C=O + H2N-R’ R2CH-NH-R’ Enamines Recall primary amines react with carbonyl compounds to give Schiff bases (imines), RN=CR2. Primary amine But secondary amines react to give enamines See if you can write the mechanism for the reaction. Secondary Amine Acidity of a Hydrogens a hydrogens are weakly acidic Weaker acid than alcohols but stronger than terminal alkynes. Learn this table…. Keto-Enol Tautomerism (Note: we saw tautomerism before in the hydration of alkynes.) Fundamental process acid or base catalysis O HO CH2 CH3 keto form enol form usually small component Mechanism in base: O :OH- O O HO H-O-H CH3 Negative carbon, a carbanion, basic, nucleophilic carbon. CH2 CH2 Additional resonance form, stabilizing anion, reducing basicity and nucleophilicity. CH2 Protonation to yield enol form. Details… Base strength Alkoxides will not cause appreciable ionization of simple carbonyl compounds to enolate. Strong bases (KH or NaNH2) will cause complete ionization to enolate. O O Double activation (1,3 dicarbonyl compounds) will be much more acidic. For some 1,3 dicarbonyl compounds the enol form may be more stable than the keto form. H H More details… Nucleophilic carbon O nucleophilicity CH2 Some examples: O O base O O O R-X O R :OH- O O O Br-Br CH3 CH2 CH2Br Some reactions related to acidity of a hydrogens Racemization Exchange Oxidation: Aldehyde Carboxylic Recall from the discussion of alcohols. Milder oxidizing reagents can also be used Ag(NH3)2+ RCHO RCO2- + Ag Tollens Reagent test for aldehydes “Drastic Oxidation” of Ketones dichromate, etc CO2H HO2C at high temperature O CO2H HO2C Obtain four different products in this case. Reductions: two electron OH O NaBH4 H Or LiAlH4 OH O H2/Pt H Reductions: Four Electron Clemmenson acid H O H Zn(Hg), HCl Wolf-Kishner base H O H2N-NH2 KOH, heat H Mechanism of Wolf-Kishner, C=O CH2 H Recall reaction of primary amine and carbonyl to give Schiff base. Here is the formation of the Schiff base. We expect this to happen. Weakly acidic hydrogen removed. Resonance occurs. Same as keto/enol tautomerism. N O N - These hydrogens are weakly acidic, just as the hydrogens a to a carbonyl are acidic. OH H N H H N C N N H N C O Here is the resonance for the anion from the keto-enol system N H N H-O-H N Protonation (like forming the enol) Perform an elimination reaction to form N2. N H2N-NH2 H H H C N H C C C O O H - N N OH H N H N N N H H-O-H H H H H Haloform Reaction, overall O O CH3 CX3 CO2- X2 NaOH O CH3 a methyl NaOH + HCX3 The last step which produces the haloform, HCX3 only occurs if there is an a methyl group, a methyl directly attached to the carbonyl. If done with iodine then the formation of iodoform, HCI3, a bright yellow precipitate, is a test for an a methyl group (iodoform test). Steps of Haloform Reaction O The first reaction: O CX3 CH3 X2 NaOH O O CH2X CH3 All three H’s replaced by X. This must happen stepwise, like this: X2 NaOH Pause for a sec: We have had three mechanistic discussions of how elemental halogen, X2, reacts with a hydrocarbon to yield a new C-X bond. Do you recall them? Radical Reaction: R. + X-X R-X + X. (initiation required) Addition to double bond: C=C + X-X + Br- (alkene acts as nucleophile, ions) :OH- O Nucleophilic enolate anion: Br O O Br-Br CH3 CH2 CH2Br Mechanism of Haloform Reaction-1 Using the last of the three possibilities :OH- O O O Br- Br-Br C H2 C H3 R R CH2Br One H has been replaced by halogen. R O Repeat twice again to yield C Br3 R Where are we? The halogens have been introduced. First reaction completed. But now we need a substitution reaction. We have to replace the CBr3 group with OH. Mechanism of Haloform - 2 O O CH3 R CX3 X2 O NaOH O- + HCX3 R NaOH This is a substitution step; OHreplaces the CX3 and then ionizes to become the carboxylate anion. R Here’s how: Attack of hydroxide nucleophile. Formation of tetrahedral intermediate. Anticipate the attack… Reform the carbonyl double bond. CX3- is ejected. The halogens stabilize the negative carbon. Neutralization. O CX3 O - CX3 OH OH R R O CX3 O OH O O- OH R R + -:CX3 R + HCX3 Cannizaro Reaction Overall: conc. KOH RCO2- + RCH2OH 2 RCHO heat Restriction: no a hydrogens in the aldehydes. CHO O CHO H H3C a hydrogens No a hydrogens Why the restriction? The a hydrogens are acidic leading to ionization. Mechanism What can happen? Reactants are the aldehyde and concentrated hydroxide. Hydroxide ion can act both as Base, but remember we have no acidic hydrogens (no a hydrogens). Nucleophile, attacking carbonyl group. O O O O R + R H - HO : Attack of nucleophilic HO- + R R H R OH H O R OH H R H H O H Re-establish C=O and eject H- which is immediately received by second RCHO O Acid-base OH Experimental Evidence KOH, H2O 2 RCDO RD2OH + RCO2- These are the hydrogens introduced by the reaction. They originate in the aldeyde and do not come from the aqueous hydroxide solution. Kinetic vs Thermodynamic Contol of a Reaction Examine Addition of HBr to 1,3 butadiene H H HBr + Br 1,2 product Br 1,4 product Mechanism of reaction. Allylic resonance H H H-Br Br Br H H Br Br 1,2 product But which is the dominant product? 1,4 product Nature of the product mixture depends on the temperature. H H HBr + Br 1,2 product Product mixture at -80 deg Product mixture at + 40 deg 80% 20% Br 1,4 product 20% 80% Goal of discussion: how can temperature control the product mixture? When two or more products may be formed in a reaction A X or A B Thermodynamic Control: Most stable product dominates Kinetic Control: Product formed fastest dominates Thermodynamic control assumes the establishing of equilibrium conditions and the most stable product dominates. Kinetic Control assumes that equilibrium is not established. Once product is made it no longer changes. Equilibrium is more rapidly established at high temperature. Thermodynamic control should prevail at high temperature where equilibrium is established. Kinetic Control may prevail at low temperature where reverse reactions are very slow. Nature of the product mixture depends on the temperature. H H HBr + Br 1,2 product Product mixture at -80 deg Product mixture at + 40 deg 80% 20% Br 1,4 product 20% 80% More stable product Thermodynamic Control Kinetic Control Product formed most quickly, lowest Ea Formation of the allylic carbocation. Can react to yield 1,2 product or 1,4 product. Most of the carbocation reacts to give the 1,2 product because of the smaller Ea leading to the 1,2 product. This is true at all temperatures. At low temperatures the reverse reactions do not occur and the product mixture is determined by the rates of forward reactions. No equilibrium. Most of the carbocation reacts to give the 1,2 product because of the smaller Ea leading to the 1,2 product. This is true at all temperatures. At higher temperatures the reverse reactions occur leading from the 1,2 or 1,4 product to the carbocation. Note that the 1,2 product is more easily converted back to the carbocation than is the 1,4. Now the 1,4 product is dominant. Diels Alder Reaction/Symmetry Controlled Reactions Quick Review of formation of chemical bond. Electro n donor Electron acceptor Note the overlap of the hybrid (donor) and the s orbital which allows bond formation. For this arrangement there is no overlap. No donation of electrons; no bond formation. Diels Alder Reaction of butadiene and ethylene to yield cyclohexene. We will analyze in terms of the pi electrons of the two systems interacting. The pi electrons from the highest occupied pi orbital of one molecule will donate into an lowest energy pi empty of the other. Works in both directions: A donates into B, B donates into A. B HOMO donates into A LUMO LUMO acceptor LUMO acceptor HOMO donor B A Note the overlap leading to bond formation A HOMO donates into B LUMO HOMO Note the donor overlap leading to bond formation Try it in another reaction: ethylene + ethylene cyclobutane LUM O HOMO LUMO HOMO Equal bonding and antibonding interaction, no overlap, no bond formation, no reaction Reaction Problem Br excess sodium methoxide Br Synthesis problem OEt HO using only compounds having two carbons as the source of all carbons in the target molecule Mechanism Problem Give the mechanism for the following reaction. Show all important resonance structures. Use curved arrow notation. O OH aq. acid + EtOH heat OEt