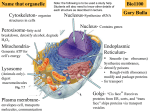
Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Gene expression wikipedia , lookup
Expression vector wikipedia , lookup
Drug design wikipedia , lookup
Epitranscriptome wikipedia , lookup
Biosynthesis wikipedia , lookup
Photosynthetic reaction centre wikipedia , lookup
Interactome wikipedia , lookup
Enzyme inhibitor wikipedia , lookup
Magnesium transporter wikipedia , lookup
Signal transduction wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Structural alignment wikipedia , lookup
Catalytic triad wikipedia , lookup
Western blot wikipedia , lookup
Biochemistry wikipedia , lookup
Protein–protein interaction wikipedia , lookup
Homology modeling wikipedia , lookup
Protein structure prediction wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Proteolysis wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Two-hybrid screening wikipedia , lookup
STRUCTURAL INSIGHTS INTO NOVEL MICROBIAL METALLOENZYMES by Laura Margaret van Staalduinen A thesis submitted to the Biochemistry graduate program in the Department of Biomedical and Molecular Sciences in conformity with the requirements for the degree of Doctor of Philosophy Queen’s University Kingston, Ontario, Canada January, 2013 Copyright © Laura Margaret van Staalduinen, 2013 Abstract Metalloproteins represent a large portion of the total proteome. When bound to a protein a metal ion influences both protein stability and function through structural, catalytic or regulatory roles. Discovery of a metal ion cofactor presents new insight into both the structural and functional aspects of a protein and can be essential for the elucidation of the functional and mechanistic details of a protein of interest. The cupin, 2-oxoglutarate/Fe2+-dependent oxygenases (2OG oxygenases) and the di-iron oxygenase families of metalloproteins exemplify the diversity and catalytic potential of a metal ion cofactor, as well as the conservation of 3-dimensional fold and structural features in proteins with similar functions. The structural and biochemical characterization of three novel microbial metalloenzymes are presented; two Escherichia coli hypothetical proteins of previously unknown function, E. coli cupin sugar isomerase (EcSI) and a 2OG oxygenase, YcfD, and the novel microbial carbon-phosphorus (C-P) bond cleavage enzyme, PhnZ, are presented. In each case the identification of a metal ion cofactor and structure determination led to important functional insights. EcSI is encoded by a gene that is highly conserved among pathogenic bacteria. It has been identified as a sugar isomerase with specificity for the rare sugar D-lyxose as well as D-mannose based on structural homology to the cupin phosphoglucose isomerases, suggesting a role for EcSI in metabolism of alternative carbon sources. Structural homology of YcfD, the second metalloenzyme, to the 2OG oxygenase family, particularily human proteins involved in ribosome assembly, combined with evidence that YcfD interacts with the essential ribosomal protein L-16 provides the first evidence of translational regulation by a 2OG oxygenase in E. coli. The third metalloenzyme, PhnZ, was previously identified as an iron dependent oxygenase. Structural characterization revealed that PhnZ possesses a di-iron cofactor and shows significant structural homology to a di-iron oxygenase, providing structural evidence for its novel mechanism of C-P bond cleavage. Combined, these three structures also highlight several features of metal ion-enzyme interaction and regulation ii mechanisms employed by metalloenzymes as well as the importance of structure in the elucidation of functional and mechanistic characteristics of a protein. iii Co-Authorship A description of the contributions of each author to the experimental work and manuscript preparation for each chapter is presented here. Chapter 2: Structure Based Annotation of a Novel Sugar Isomerase from the Pathogenic E. coli O157:H7 This chapter was published in the Journal of Molecular Biology in August of 2010 (J. Mol. Biol.(2010), 401(5):866-81). This project was initiated by Laura van Staalduinen, Melanie Adams-Cioaba and Zongchao Jia as part of the Montreal-Kingston Bacterial Structural Genomics Project. Laura van Staalduinen and Zongchao Jia were responsible for overall experimental design. Laura van Staalduinen performed native protein expression, purification, crystallization, structure solution and refinement, structure analysis, phosphoglucose isomerase activity detection and cell growth experiments. Melanie Adams-Cioaba was responsible for initial expression and purification. Chang-Su Park, Soo-Jin Yeom and Dr. Deok-Kun Oh were responsible for mutant generation, specific activity and kinetic analysis. The manuscript was written by Laura van Staalduinen with editorial input from Melanie Adams-Cioaba and Dr. Zongchao Jia. Chapter 3: Crystal Structure and Preliminary Functional Analysis of YcfD, a Novel 2oxogluterate/ Fe2+-Dependent Oxygenase Involved in Translational Regulation in E. coli This chapter is currently in preparation for publication. Laura van Staalduinen and Zongchao Jia initiated the project and designed all experimental work. Laura van Staalduinen was responsible for cloning, generation of surface entropy reduction mutants, expression, purification, crystallization, X-ray data collection, structure determination and analysis. Stefanie Novakowski performed site-directed mutagenesis on 2-oxoglutarate binding residues and prepared samples of native protein for isothermal titration experiments. The manuscript was prepared by Laura van Staalduinen with editorial input from Dr. Zongchao Jia. iv Chapter 4: Identification of a Novel Di-iron Oxygenase, PhnZ, with a Unique Mechanism for C-P bond Cleavage This chapter represents unpublished work that is in preparation for publication. The project was initiated as a result of collaboration between Laura van Staalduinen, Zongchao Jia, Fern R. McSorley and David L. Zechel, who were all involved in experimental design. Laura van Staalduinen was responsible for expression, purification, crystallization, X-ray data collection, structural determination and analysis. Site-directed mutagenesis of active site residues was performed by Laura van Staalduinen and Fern McSorley. Product formation was analyzed by Fern McSorley. The manuscript was prepared by Laura van Staalduinen with editorial input from Fern McSorley, David L. Zechel and Zongchao Jia. v Acknowledgements I wish to express my deepest gratitude to my supervisor, Dr. Zongchao Jia, for his guidance and support throughout my graduate studies. His encouragement has helped me to develop confidence and independence in my work. His keen ability to ask the right questions and provide unique insights into a problem has been pivotal to my success. I am grateful to all of the wonderful people I have had the opportunity to work with throughout my time in the Jia Lab. In particular I would like to thank Natalie Roy for her excellent technical assistance, Drs. Qilu Ye and Susan Yates for their support and friendship, and Dr. Frédérick Faucher for sharing his immense knowledge of X-ray crystallography. I would like to express my sincerest gratitude to Drs. Brent Wathen and Mona Rahman for critical reading of this thesis as well as their friendship and support throughout my graduate career. I would like to thank Dr. Vinay Singh, who was my first supervisor in the Jia Lab and has since become a valued mentor, confidant and cherished friend. I also wish to express my appreciation to Dr. Melanie Adams-Cioaba for guiding me through the fog of functional annotation and for her contagious enthusiasm, which contributed greatly to my choice to study structural biology. I would like to thank my committee members, Drs. Steven Smith and Bruce Hill for countless helpful discussions. I am grateful to my collaborators from the laboratory of Dr. DeokKun Oh for their contributions to the work on EcSI, and also Dr. David Zechel and Fern McSorley for their collaboration on the PhnZ project. I would like to thank Kim Munro and David McLeod from the Protein Function Discovery for their assistance with biophysical experiments. I would like to acknowledge the beamline operators and staff at the NSLS, CHESS, SSRL and APS for making data collection possible. I would like to acknowledge NSERC and Queen’s University for funding my graduate research. I am forever grateful to all of my family, including my amazing in-laws, for their encouragement and support. I am so fortunate to have so many wonderful people in my life. In vi particular I wish to thank my parents, Duane and Caroline Dryden. Their love and support provided the foundation for every success in my life, they gave me the courage to reach further, knowing they would always be there to for me, and countless times they have gone above and beyond to help me attain my personal and academic goals. To my loving husband Dan I am eternally grateful. He has supported me in every way possible and his unwavering faith and love are a limitless source of strength without which this thesis would not have come to fruition. His determination, strength and generosity are an inspiration to me. I wish to thank him for sharing his love and his life with me. Finally I wish to thank my son, Owen, for being a constant source of love, laughter and joy and for teaching me the important things in life. I thank my lucky stars for him each and every day. vii Table of Contents Abstract ............................................................................................................................................ ii Co-Authorship ................................................................................................................................ iv Acknowledgements ......................................................................................................................... vi List of Figures. ................................................................................................................................ xi List of Tables ................................................................................................................................ xiii List of Abbreviations .................................................................................................................... xiv Chapter 1- General Introduction ...................................................................................................... 1 1.1 Metalloproteins ...................................................................................................................... 1 1.2 Cupin superfamily .................................................................................................................. 5 1.2.1 The cupin DSBH: a versatile and stable scaffold............................................................ 7 1.2.2 Functional classes of the cupin superfamily ................................................................... 9 1.2.3 Metal binding and cupins .............................................................................................. 11 1.3 2OG/FeII-dependent dioxygenases ....................................................................................... 13 1.3.1 Biological functions of 2OG oxygenases...................................................................... 14 1.3.2 Structure ........................................................................................................................ 16 1.3.3 2OG and Fe2+ binding ................................................................................................... 18 1.3.4 General reaction mechanism ......................................................................................... 20 1.4 Di-iron oxygenases .............................................................................................................. 22 1.4.1 Mechanistic insights...................................................................................................... 22 1.4.2 Structure of di-iron oxygenases and the di-iron site ..................................................... 23 1.5 Significance and Implications .............................................................................................. 26 1.5.1 Specific Objectives ....................................................................................................... 27 Chapter 2 - Structure-Based Annotation of a Novel Sugar Isomerase from the Pathogenic E. coli O157:H7......................................................................................................................................... 28 2.1 Abstract ................................................................................................................................ 28 2.2 Introduction .......................................................................................................................... 29 2.3 Materials and Methods ......................................................................................................... 31 2.3.1 Protein expression, purification, crystallization, and X-ray diffraction data collection 31 2.3.2 Structure solution and refinement ................................................................................. 32 2.3.3 Inductively coupled plasma-mass spectrometry ........................................................... 33 2.3.4 Generation of EcSI mutants .......................................................................................... 33 2.3.5 Enzymatic assays .......................................................................................................... 33 viii 2.3.6 Determination of specific activity and kinetic parameters ............................................ 34 2.3.7 Metal ions, pH, and temperature effects ....................................................................... 35 2.3.8 Isomerization product identification ............................................................................. 35 2.3.9 Growth complementation experiments ......................................................................... 35 2.3.10 PDB accession numbers .............................................................................................. 36 2.4 Results .................................................................................................................................. 36 2.4.1 Crystal structure of EcSI ............................................................................................... 36 2.4.2 EcSI is structurally similar to cupin phosphoglucose isomerases ................................. 39 2.4.3 Identification of substrate preference for EcSI ............................................................. 40 2.4.4 Effects of metal ions, pH, and temperature on the activity of EcSI .............................. 43 2.4.5 Complex structure of EcSI with D–fructose bound....................................................... 47 2.4.6 Active site mutations..................................................................................................... 49 2.4.7 Substrate binding and product release .......................................................................... 51 2.4.8 Growth complementation assay .................................................................................... 53 2.5 Discussion ............................................................................................................................ 53 Chapter 3 - Crystal Structure and Preliminary Functional Analysis of YcfD, a Novel 2oxogluterate/Fe2+-Dependent Oxygenase Involved in Translation Reculation in E. coli .............. 61 3.1 Abstract ................................................................................................................................ 61 3.2 Introduction .......................................................................................................................... 62 3.3 Materials and Methods ......................................................................................................... 64 3.3.1 Cloning and generation of YcfD variants ..................................................................... 64 3.3.2 Protein expression and purification............................................................................... 64 3.3.3 Crystallization and X-ray data collection...................................................................... 65 3.3.4 Structure solution and refinement ................................................................................. 66 3.3.5 Metal identification ....................................................................................................... 67 3.3.6 Isothermal titration calorimetry .................................................................................... 67 3.3.7 Pull-down assay and mass spectrometry ....................................................................... 67 3.3.8 Colony forming assay ................................................................................................... 68 3.4 Results and Discussion ........................................................................................................ 69 3.4.1 Crystal structure of YcfD .............................................................................................. 69 3.4.2 YcfD binds 2OG ........................................................................................................... 73 3.4.3 YcfD is structurally similar to human ribosome assembly proteins ............................. 75 3.4.4 YcfD Pulls-Down Ribosomal Protein RL-16 of the 50S subunit ................................. 76 3.4.5 Growth effects of YcfD overexpression ....................................................................... 80 ix 3.4.6 Summary ....................................................................................................................... 82 Chapter 4 - Identification of a Novel Di-iron Oxygenase, PhnZ, with a Unique Mechanism for C-P Bond Cleavage ........................................................................................................................ 83 4.1 Abstract ................................................................................................................................ 83 4.2 Introduction .......................................................................................................................... 84 4.3 Materials and Methods ......................................................................................................... 87 4.3.1 Expression and purification .......................................................................................... 87 4.3.2 Crystallization and X-ray data collection...................................................................... 88 4.3.3 Structure solution and refinement ................................................................................. 88 4.3.4 Generation of PhnZ variants ......................................................................................... 89 4.3.5 Detection of product formation ..................................................................................... 89 4.4 Results .................................................................................................................................. 90 4.4.1 Structure determination ................................................................................................. 90 4.4.2 Overall structure of PhnZ.............................................................................................. 93 4.4.3 PhnZ is a di-iron oxygenase .......................................................................................... 95 4.4.4 The di-iron site .............................................................................................................. 97 4.4.5 Co-Crystal Structure of PhnZ with substrate reveals conformational change upon substrate binding .................................................................................................................. 100 4.4.6 Investigation of key active site residues ..................................................................... 102 4.5 Discussion .......................................................................................................................... 105 4.5.1 Di-iron oxygenases ..................................................................................................... 105 4.5.2 Di-iron binding site in PhnZ ....................................................................................... 106 4.5.3 Active site and catalytic mechanism ........................................................................... 107 4.5.4 Active site substitutions .............................................................................................. 110 4.5.5 Structural insights for substrate specificity ................................................................. 111 Chapter 5 - General Discussion and Summary ............................................................................ 113 5.1 Importance of metals in metalloproteins ............................................................................ 113 5.2 Structure and function ........................................................................................................ 115 5.3 Active site closure upon substrate binding ........................................................................ 117 5.4 Future directions ................................................................................................................ 120 5.4.1 EcSI ............................................................................................................................. 120 5.4.2 YcfD............................................................................................................................ 123 5.4.3 PhnZ ............................................................................................................................ 125 5.5 Conclusions ........................................................................................................................ 127 x List of Figures. Figure 1-1 Amino acids involved in metal ion coordination ........................................................... 4 Figure 1-2 Consensus sequence conservation in representative cupin proteins............................... 6 Figure 1-3 Representative cupin structures ...................................................................................... 8 Figure 1-4 Coordination of metal-ions by cupin proteins. ............................................................. 12 Figure 1-5 Examples of reactions catalyzed by 2OG oxygenases. ................................................ 15 Figure 1-6 Crystal structure of DAOCS, a representative 2OG oxygenase ................................... 17 Figure 1-7 Binding of Fe2+ and 2OG by 2OG oxygenases ............................................................ 19 Figure 1-8 General mechanism for 2OG oxygenase formation of a reactive ferryl intermediate.. 21 Figure 1-9 Representative di-iron oxygenase structures. ............................................................... 24 Figure 1-10 Representative di-iron binding sites ........................................................................... 25 Figure 2-1 The crystal structure of EcSI from E. coli O157:H7 .................................................... 37 Figure 2-2 Structural comparison of EcSI to cupin phosphoglucose isomerases .......................... 41 Figure 2-3 Schematic representation of aldose–ketose isomerization reactions catalyzed by E. coli D-lyxose isomerase ........................................................................................................................ 44 Figure 2-4 Michaelis-Menten Plot for EcSI ................................................................................... 45 Figure 2-5 Complex structure of EcSI with fructose. .................................................................... 48 Figure 2-6 Substrate stabilization and product release mechanism.. ............................................. 52 Figure 2-7 The z5688 gene from E. coli O157:H7 confers the ability for E. coli BL21 (DE3) to grow on D-lyxose.. ......................................................................................................................... 54 Figure 2-8 Schematic representation of the two potential mechanisms for enzyme catalyzed aldose-ketose isomerization.. ......................................................................................................... 60 Figure 3-1 Crystal structure of YcfD ............................................................................................. 70 Figure 3-2 Isothermal titration calorimetry of YcfD binding to 2-oxoglutarate ............................ 74 Figure 3-3 Structural comparisons of YcfD ................................................................................... 77 Figure 3-4 Structure based multiple sequence alignment of YcfD, Mina53 and NO66 ................ 78 Figure 3-5 YcfD interacts with a ribosomal protein. ..................................................................... 79 Figure 3-6 Overexpression of YcfD inhibits cell growth .............................................................. 81 Figure 4-1 A general reaction scheme for PhnY/PhnZ catalyzed C-P bond cleavage ................... 86 Figure 4-2 Crystals for apo PhnZ and PhnZ in the presence of (±)-2-amino-1hydroxyethylphosphonic acid ........................................................................................................ 91 Figure 4-3 Crystal structure of PhnZ ............................................................................................. 94 Figure 4-4 Structural homology to MIOX reveals PhnZ is a di-iron oxygenase.. ......................... 96 xi Figure 4-5 The di-iron site of tartrate bound and 1-OH-AEP bound PhnZ. .................................. 98 Figure 4-6 Conformational changes observed upon substrate binding. ....................................... 101 Figure 4-7 Comparison of tartrate and substrate binding............................................................. 103 Figure 4-8 Key steps in the PhnZ catalyzed C-P bond cleavage of 1-OH-AEP .......................... 109 Figure 5-1 Active site closure upon substrate binding ................................................................. 118 Figure 5-2 A schematic of the genes surrounding the gene for EcSI in the E. coli O157:H7 genome. ........................................................................................................................................ 122 xii List of Tables Table 2-1 EcSI data collection and refinement statistics ............................................................... 38 Table 2-2 Specific activity of EcSI ................................................................................................ 42 Table 2-3 Kinetic parameters of EcSI ............................................................................................ 46 Table 2-4 Specific activities of the wild-type and mutant EcSI..................................................... 50 Table 3-1 Data collection and refinement statistics for MAD data ............................................... 71 Table 4-1 PhnZ data collection and refinement statistics. ............................................................. 92 Table 4-2 Fe ion-ligand inter-atomic distances.............................................................................. 99 Table 4-3 Activity of active site mutants of PhnZ as measured by 31P-NMR ............................. 104 xiii List of Abbreviations The abbreviations used are: 1-OH-AEP 2-amino-1-hydroxyethylphosphonic acid 2OG 2-oxoglutarate AEP 2-Aminoethylphosponic acid AurF p-amino benzoate N-oxygenase BOG n-octyl-β-D-glucoside CFU Colony forming unit C-P Carbon-phosphorus cPGI Cupin phosphoglucose isomerase DACS Deacetlycephalosporin C synthase DAOCS Deacetoxycephalosporin C synthase DSBH Double stranded beta-helix EcSI Escherichia coli sugar isomerase F-6-P Fructose-6-phosphate FIH Factor inhibiting hypoxia inducible factor-1α G-6-P Glucose-6-phosphate GLP Germin-like protein GST Glutathione S-transferase HIF Hypoxia inducible factor-1α ICP Inductively coupled plasma ICP-OES Inductively coupled plasma optical emission spectrometry IPNS Isopenicillin N synthase IPTG Isopropyl β-D-1-thiogalactopyanoside ITC Isothermal titration calorimetry xiv JmjC Jumonji C Kd Dissociation constant LB Luria Broth LI Lyxose isomerase MAD Multiple-wavelength Anomalous Dispersion MESG 2-amino-6-mercapto-7-methylpurine riboside MI Mannose isomerase MIOX myo-inositol oxygenase MS/MS Tandem sequencing mass spectrometry PBS Phosphate buffered saline PDB Protein data bank PEG Polyethyleneglycol PGI Phosphoglucose isomerase Pi Inorganic phosphate PIPES Piperazine-N,N’-bis(2-ethanesulfonic acid) PMI Phosphomannose isomerase 31 Phosphorus 31 nuclear magnetic resonance P-NMR PNP Purine nucleoside phophorylase PTS Phosphotransfer system RL-16 Ribosomal protein L16 RMSD Root mean squared deviation RNR-R2 Ribonucleotide reductase-R2 subunit SAD Single-wavelength Anomalous Dispersion SBP Sucrose binding protein SERp Surface entropy reduction prediction xv sMMO Soluble methane monooxygenase SSM Secondary Structure Matching TLS Translation/Liberation/Screw ToMO Toluene/o-xylene monooxygenase XAS X-ray absorption spectroscopy xvi Chapter 1 General Introduction 1.1 Metalloproteins Metalloproteins, defined as proteins that require one or more metal-ions for their biological function, comprise at least 1/3 of the proteome based the composition of structures in the Protein Data Bank (PDB) (1). The largest subset of these are the metalloenzymes which utilize the metal ion cofactor for catalysis (2,3). The metal ion cofactor in such proteins can function in a structural, catalytic or regulatory role by influencing both protein stability and catalytic potential. Several fundamental properties of metal ions make them ideal cofactors. First, they typically hold a positive charge, where this electrophilic property allows them to act as Lewis acids for acid-base catalysis and substrate binding. Second, they are able to participate in redox reactions due to the fact that many metals can exist in multiple oxidation states. Finally, the ability of metal ions to be coordinated by four or more protein side chains allows them to act as cross-linking reagents bringing together distant parts of proteins to stabilize protein folds and/or form active sites. The prevalence and versatility of metalloproteins result in their involvement in many biological processes, including but not limited to cellular respiration, photosynthesis, reduction of molecular oxygen, nitrogen fixation, and water oxidation. The relative amount of individual metal-ions found in metalloproteins has been shown to vary between Archaea, Bacteria and Eukaryotes, and to be dependent on the total proteome size within a given species (4). However, there are ten metals which are commonly found as cations associated with proteins: Zn, Fe, Co, Ni, Cu, Mg, Ca, Mn, Na, and K (5). Less commonly observed metals include W, Mo, Cd, Se and Hg, which can be toxic at high concentrations, but 1 are essential in some species. Metal ions can be coordinated through direct contact with multiple side chains from the protein, or as part of larger prosthetic groups. For example, Mg2+-bound chlorophyll, Fe2+-bound heme or Co2+-bound corrin groups with the metal making minimal contact to the protein. A survey of structures in the PDB revealed that Zn2+ was the most abundant, followed closely by Fe2+/3+, Mg2+ and Ca2+ (6). The function of a metalloprotein is dictated by the interaction between the metal ion and the protein. This interaction is largely defined by the electronic properties of the metal and its coordination chemistry. The replacement or removal of a metal cofactor is often found to result in a reduction or loss of biological activity of a protein. Each metal plays specialized roles within a metalloprotein, and depending on the protein environment, metals can assume multiple roles. For example, Zn2+, the most abundant divalent cation, exists in a single oxidation state and therefore does not participate in redox reactions; instead it acts as a Lewis acid to drive catalysis when associated with three protein based ligands (7). When associated with four protein based ligands Zn2+ is found to stabilize and organize protein structures. For example, the well known zinc finger proteins require the Zn2+ ion to fold. Fe2+/3+ ions are found both directly bound to proteins or as part of prosthetic groups, such as heme or iron-sulfur clusters. Due to the stability of Fe in multiple oxidations states it is involved in both electron transfer and oxygen metabolism reactions. Though the ionic radii of the previously mentioned divalent cations varies only by approximately 0.3 Å (~0.65 Å for Mg2+ - 0.99 Å for Ca2+), these seemingly small differences greatly affect their interactions with proteins (5). As a result, despite the overall inherent flexibility of proteins, there is a high degree of metal selectivity observed in proteins. The propensity for proteins to bind a particular divalent cation obeys a universal order of preference 2 that is defined by the Irving-Williams series (Mg2+ and Ca2+ < Mn2+ < Fe2+ < Co2+ < Ni2+ < Cu2+ > Zn2+) (8). Based on this affinity profile, it would be expected that Cu2+ would be bound to the majority of proteins. In order to prevent all metalloproteins from incorporating Cu2+, cells have developed a number of mechanisms to assure the right metal is bound, which have been extensively studied in bacterial cells (9). These mechanisms include metal selection by cellular location of folding, metallochaperone assisted metal insertion, and taking advantage of variable protein-metal affinity. The metal binding site within a metalloprotein is typically comprised of amino acid side chains, with occasional contributions from main chain carbonyl oxygens. The functional groups which interact with the metal are the imidazole group of histidine, glutamic or aspartic acid carboxylate oxygens, the oxygen of threonine or serine, and the sulphur of cysteine (Figure 1-1). Based on ligand preference, metals can be separated into three groups: the alkali metal ions (Na+, K+, Mg2+, and Ca2+) which possess the highest preference for coordination with carboxylate and oxygen groups, the imidazole class (Mn2+, Co2+, and Fe2+/3+), and the sulfur class (Ni2+, Zn2+, and Cu2+) (1). Both the imidazole and sulfur classes are found to interact with imidazole and thiol groups, however the sulfur class has a higher affinity for thiol groups. Metalloproteins are a diverse and adaptable class of proteins. A metal ion cofactor adds significant catalytic potential, resulting in some of the most difficult reactions being catalyzed by metalloenzymes. The diversity and catalytic potential of metalloproteins are exemplified by three important families of metal dependent proteins, namely the cupin superfamily, the 2oxoglutarte/Fe2+-dependent oxygenases and the di-iron oxygenases. The latter two classes both belong to a family of non-heme iron oxygenases. 3 Figure 1-1 Amino acids involved in metal ion coordination. (A) Amino acids involved in metal ion coordination adapted from (5). (B) The metals are grouped based on their preferred coordination residues. 4 1.2 Cupin superfamily With numerous members divided in to more than 18 different functional subclasses, the cupin superfamily is one of the largest, most functionally diverse protein superfamilies. Examples of cupin proteins are found in all three domains of life (Archaea, Bacteria and Eukaryotes), and despite low sequence homology the family is thought to have arisen by divergent evolution from a common ancestor due to the fact that they are characterized by a common architecture. The structures are characterized by a conserved double-stranded β-helix (DSBH) fold that is a derivative of the classic β-barrel architecture; indeed, the name cupin is derived from the latin word for small barrel, ‘cupa’. Two sequence motifs are found within the DSBH fold of cupin proteins, and can be used to identify cupin proteins based on primary sequence. The consensus sequences are G(X)5HXH(X)3,4E(X)6G for motif 1 and G(X)5PXG(X)2H(X)3N for motif 2 (Figure 1-2) (10). The two His and the Glu residues of motif 1, together with the His of motif 2, are often found to ligate a metal ion. The robust scaffold of the cupin domain allows for a diverse array of functions to be catalyzed by cupin proteins ranging from non-enzymatic seed storage proteins, to transcription factors, to isomerases and epimerases as well as oxygenases. The cupin superfamily was founded following the discovery of a conserved nine amino acid sequence between wheat germin, a thermostable protein that is produced during the germination of wheat embryos, and spherulin, a stress-related protein that is produced by the slime mould Physarum polycephalum in response to starvation (11,12). This initial conserved sequence (HI/THPRATEI) was termed the ‘germin box’, and sequence analysis combined with structure modeling led to the discovery that this conservation was found not only in germin-like proteins (GLPs) but spread across a wide variety of proteins with diverse function (13). The 5 Figure 1-2 Consensus sequence conservation in representative cupin proteins. The amino acid sequence, identified by PDB ID, for the regions corresponding to motif 1 and 2 are shown in boxes separated by the intermotif region. The A/B suffix after the PDB ID indicates that the structure contained two cupin domains, where A is the N-terminal domain and B is the C-terminal domain. The β-strands are shown above the alignment, with the corresponding residues shaded grey. The predicted metal binding residues are bold, with the consensus motif shown below the alignment. Adapted from Dunwell et al. (2004) (10). 6 ‘germin box’ was found to be part of a larger twenty or twenty-one amino acid sequence that corresponds to motif 1, and a second shorter conserved sequence corresponding to motif 2 was found to be separated from motif 1 by a variable intermotif region (Figure 1-2). Structural analysis revealed that motifs 1 and 2 each comprise two β-strands of the DSBH, and that the variable intermotif region can be anywhere from 11 amino acids to greater than 100 (10). As the family has grown it has become clear that the consensus motif 1 and 2 are much less conserved than initially thought and that the real homology lies in the three dimensional fold. 1.2.1 The cupin DSBH: a versatile and stable scaffold As previously stated, cupin proteins share a conserved DSBH fold, which is responsible for a wide variety of functions. The DSBH fold is typically comprised of six to ten anti-parallel βstrands that make up two β-sheets (Figure 1-3). At the center of the DSBH fold is the ‘active site’ of the cupin protein, including the potential metal ion binding site. The ability of cupins to act in such a diverse array of functions is directly related to the flexibility of the active site provided by the DSBH fold (14). Further diversity is defined by the surrounding secondary structure, including in some cases multiple cupin domains, though only cupins that possess one or two cupin domains, referred to as monocupins and bicupins, respectively, have been characterized. In the case of bicupins the two cupin domains maintain structural similarity, but often differ significantly in primary sequence (15). Cupin proteins also differ in their oligomerization, ranging from monomeric proteins such as phosphomannose isomerase (16) to the hexameric oxalate decarboxylase (17). The cupin fold is an ancient one, explaining its conservation in all species sequenced to date. This can be attributed to the fact that in addition to facilitating the catalysis of a diverse array of reactions, the cupin fold is also highly stable. The archetypal cupin protein is known for 7 Figure 1-3 Representative cupin structures. (A) A monocupin of unknown function (1LKN). (B) The bicupin YhhW, a quercertinase enzyme (1TQ5) (C) The hexamer of germin (1FI2). The β-strands are shown in magenta and α-helices in cyan. The manganese ions of germin are shown as grey spheres. 8 its stability to abiotic stresses such as heat, pH, H2O2 , as well as proteases (18). This stability can be attributed to the compact nature of the DSBH, the increase in subunit contacts upon oligomerization, hydrophobic interactions and hydrogen bonding within the core barrel, covalent linkages between subunits, as well as external factors such as metal ion binding, cofactor binding, and glycosylation (14). The stability of the cupin fold is often employed by organisms in times of abiotic stress. For example, the micororgansim Physarum polycephalum produces spherulin under conditions of starvation (19), and Azotobacter vinelandii and Pseudomonas aeruginosa both utilize phosphomannose isomerases to produce exopolysaccharide alginate for growth under desiccating conditions (20). A class of thermophilic cupin phosphoglucose isomerases (PGI) has also been identified that replace the conventional PGIs in hyperthermophilic archaea (21). 1.2.2 Functional classes of the cupin superfamily Cupin proteins have been found to have both enzymatic and non-enzymatic functions. One of the founding members of the cupin superfamily, germin, along with the GLPs, possess both oxalate oxidase and superoxide dismutase activity (22). The former involves the manganese -dependent oxidative decarboxylation of oxalate to carbon dioxide and hydrogen peroxide, while the latter deactivates superoxide radicals that are formed under stressful conditions. The expression of certain GLPs have been linked to plant defense mechanisms as a means to protect against pathogens (23,24). With the subsequent discovery of each cupin protein the functional diversity has grown to include more than eighteen different functional subclasses, and there remains a number of cupin proteins for which the structure has been determined but have been classified as hypothetical since their function has not yet been determined. The subclasses can be further divided based on whether the protein is a monocupin or a bicupin. Monocupin proteins can be either single domain or multidomain proteins. Most 9 monocupins discovered to date are enzymes, although a class of microbial non-enzymatic transcription factors including AraC are known to have a single cupin sugar binding domain fused to a DNA binding domain (10). The largest subclass of the monocupins – and the cupins as a whole – are the dioxygenases, specifically the 2-oxoglutarate/FeII-dependent oxygenases, which will be discussed at length later. A number of monocupins have been identified as sugar isomerases, with the first identified being phosphomannose isomerase (PMI). PMI catalyzes the conversion of D-fructose-6-phosphate to D-mannose-6-phosphate and is essential for the metabolism of D-mannose as well as the production of D-mannose for eukaryotic glycosylation reactions. Three types of PMI have been discovered, with type II being the bifunctional PMI/GDP-d-mannose pyrophosphorylases (25). In addition to the PMIs, a novel class of PGIs from thermophilic archaea that catalyze the conversion of glucose-6-phosphate to fructose-6phosphate, an essential step in energy metabolism, have been identified to belong to the cupin superfamily (26). Auxin binding protein (ABP) is a monocupin that binds auxin and is involved in a number of plant growth responses. In this case, though enzymatic function is predicted based on the structure, the exact function of this protein has yet to be determined (27). The final class of monocupins are nuclear proteins such as pirin, a quercetin dioxygenase involved in programmed cell death (28). The major subclasses of the bicupins include oxalate decarboxylase and a group of nonenzymatic seed storage proteins. There are also examples of bicupin dioxygenases, though only one domain is reported active in all instances reported to date, with the other representing a nonfunctional remnant (10). Oxalate decarboxylase converts oxalate to formate and carbon dioxide in a Mn2+ ion dependent reaction. The crystal structure of oxalate decarboxylase revealed that it is a symmetrical hexamer, where each cupin domain binds to a Mn for a total of twelve 10 bound Mn ions (17). The non-enzymatic storage proteins are represented by the seed storage globulins and the sucrose binding protein (SBP). The globulins, which include vicilins and legumins, are seed storage proteins that are highly similar to germin but have lost the ability to bind metals, and are therefore thought to represent deactivated enzymes (11,14,29). SBP is involved in sucrose uptake in plants, and possesses GTP binding capabilities that have been shown to be independent of its sucrose binding and transport activity, indicating an alternate but thus far undetermined function (30,31). 1.2.3 Metal binding and cupins The conserved sequence motifs that define cupin proteins have been shown to coordinate a metal ion in the majority of cupin proteins that have been characterized. The metal binding site contains the two His residues and the Glu residue from motif 1 and the His residue found in motif 2, though one of the His residues is often found to be substituted by a Glu or Asp residue in motif 1 (Figure 1-4). Additional coordination sites are occupied by substrate ligation, or in the case of apo proteins, water. The most common metal ion found in cupin domains is Fe2+, but examples of Zn2+, Cu2+, Mn2+, Ni2+, Co2+ and Cd2+ ion bound proteins have been identified (10). For the enzymatic cupin proteins, the bound metal ion plays a direct role in catalysis, and the flexibility of metal ion selection allows for different chemical interactions and reactions to be catalyzed. Structure-based phylogenetic analysis of the cupin superfamily revealed that metal ion preference in cupin proteins is independent of the structure homology. That said, in most cases proteins with similar function bound identical metals (15). For example, the PMIs, which are not part of a phylogenetic cluster together based on structure, all utilize Zn2+ ion for their activity. This is not the case for all cupin functional classes; for example quercetin 2,3-dioxygenase from Bacillus subtilis has been shown to bind iron while the homologue from Aspergillus japonicus 11 Figure 1-4 Coordination of metal-ions by cupin proteins. (A) The manganese ion binding site of germin (1FI2), the histidines and glutamic acids of the cupin motifs (purple sphere) are shown coordinating the manganese ion, along with two water molecules (red spheres). (B) The nickel ion binding site of phosphoglucose isomerase (1QXR) bound to an inhibitor. The inhibitor is shown in yellow sticks, the nickel ion a green sphere. 12 binds copper (32). Interestingly, germin and oxalate decarboxylase are both Mn2+-dependent enzymes and both catalyze a reaction involving oxalate as a substrate, however the reactions yield different products (15). In the case of the bicupin acireducton dioxygenase, the activity of the enzyme is determined by the identity of the metal ion bound in the active site: nickel ion binding results in the production of formate, carbon monoxide, and methylthiopropionate, whereas iron binding yields methylthioketobutyrate and formate. Both of these functions involve the reaction of oxygen and acireductone (33,34). 1.3 2OG/FeII-dependent dioxygenases Uncatalyzed oxidation reactions involving organic compounds are thermodynamically favourable but kinetically very slow, which is necessary to prevent spontaneous combustion of organic matter. To get around this kinetic barrier, a class of metalloenzymes has evolved that utilize transition metals that are stable in multiple redox states, such as iron and copper, to activate dioxygen and catalyze the oxidation of organic compounds in a controlled and selective environment. There are two main subgroups of iron-containing oxygenases. The heme-containing enzymes such as cytochrome P450 (35) and the non-heme iron enzymes, which are further subdivided into mono- and di-nuclear iron enzymes (36-38). Here, the largest subclass of the mononuclear iron oxygenases, the 2-oxoglutarate/Fe2+-dependent dioxygenases, will be discussed, as well as an introduction to the potent diiron oxygenases. The 2-oxoglutarate/Fe2+-dependent dioxygenases (2OG oxygenases) represent not only the largest subclass of mononuclear non-heme oxygenases but also the largest subclass of the previously described cupin superfamily. They possess a modified cupin sequence motif of HXD/EXnH, with the His and Asp or Glu residues coordinating the Fe2+. As members of the cupin superfamily they maintain the DSBH fold characteristic of the cupins, with metal and 2OG 13 binding occurring within the DSBH. The 2OG oxygenases catalyze an extensive array of oxidation reactions for a diverse group of substrates. 2OG oxygenases couple the oxidative decarboxylation of 2OG to the oxidation of the primary substrate. The most common reaction catalyzed is hydroxylation, and in fact this is the only reaction known to by catalyzed by 2OG oxygenases in animals (39). The catalytic versatility of the 2OG oxygenases is better represented in plants and bacteria where a much wider array of reactions are catalyzed by 2OG oxygenases including epimerization, ring closure, desaturation, epoxidation, ring opening and chlorination (Figure 1-5) (39,40). Primary substrates of these enzymes include a vast group of small molecules, as well as proteins, lipids and methylated nucleotides. Representatives of the 2OG oxygenases are present in most species and their catalytic versatility results in their involvement in many biologically important processes. 1.3.1 Biological functions of 2OG oxygenases The first 2OG oxygenases identified were prolyl and lysyl hydroxylases, which are involved in collagen biosynthesis (41,42). The family has now grown to include many biological functions including post-translational modifications, biosynthesis of plant products and antibiotics, lipid metabolism, repair of methylated DNA, regulation of gene expression and oxygen sensing. In both microorganisms and plants the oxidation of a number of small molecules for many pathways including antibiotic synthesis and fatty acid metabolism are catalyzed by 2OG oxygenases (39,40,43). For example, deacetoxycephalosporin C and deacetlycephalosporin C synthases (DAOCS and DACS, respectively) are 2OG oxygenases that catalyze sequential reactions in the synthesis of cephalosporin from isopenicillin N (43). The 2OG oxygenases SyrB2 and CmaD catalyze important chlorination reactions involved in peptide antibiotic synthesis (44) and cyclopropane ring formation (45), respectively. The 2OG oxygenase AlkB and its related 14 Figure 1-5 Examples of reactions catalyzed by 2OG oxygenases. The common ferryl intermediate in 2OG oxygenase catalysis is shown in the centre. The most common reaction, hydroxylation is highlighted in red, and specific examples of enzymes that catalyze specific reaction types are shown in green. Adapted from Clifton et al. (39). 15 proteins represent an important class of reactions. These enzymes catalyze the repair of methylated DNA through a 2OG/Fe2+ dependent hydroxylation – elimination reaction (46,47). Another important functional subclass of 2OG oxygenases is the JmjC-domain containing proteins. These proteins are identified based on sequence homology to the C-terminal domain of the jumonji transcription factor (48-51). The first JmjC-domain-containing proteins were shown to be histone lysine demethylases, which are epigenetic regulators of gene expression (52), and based on this important discovery, all proteins in the subclass were predicted to be lysine demethylases. Subsequently, a JmjC protein which regulates hypoxia response through asparaginyl hydroxylation of the hypoxia inducible factor-1α (HIF) was identified, called factor inhibiting HIF (FIH) (53), expanding the reactions for this subclass to include protein hydroxylation in general. Even more recently, hydroxylation of the hypermodified ribonucleotide hydroxywybutosine in phenylalanine tRNA was determined to be catalyzed by the JmjC protein TWY5 (54). Wybutosine represents the first non-peptide substrate for this subclass, and opens the door to greater diversity in the substrates for these enzymes. 1.3.2 Structure As with all cupin proteins, structural studies of the 2OG oxygenase family have provided a wealth of information about the mechanism and diversity of the family. The first 2OG oxygenase structure to be determined was for isopenicillin N synthase (IPNS) in complex with Mn2+ (55). This, and subsequent structures revealed the conserved DSBH structure (Figure 1-6). Typically, the DSBH consists of 2 antiparallel β-sheets, each comprised of 4 β-strands which come together to form a right-handed β-barrel, with the N-terminal strand forming the first edge of the barrel (39). This structure is referred to by a variety of names; for example jelly roll, double Greek key motif or β-barrel, which all describe a class I right-handed superhelical twist (56). The 16 Figure 1-6 Crystal structure of DAOCS (PDB ID 1UOB), a representative 2OG oxygenase. (A) Ribbon diagram of the DAOCS structure, and (B) topological representation. The DSBH strands are coloured purple, additional β-strands are grey and α-helices are in blue. In the ribbon diagram the bound Fe2+ is shown as an orange sphere, with 2OG shown in teal. Adapted from (39). 17 strands alternate sequentially between the two β-sheets, with the exception of strands 4 and 5 that are in the same sheet connected by a ‘hairpin’ loop (Figure 1-6A). This hairpin loop can be a simple β-turn or a more complex extended loop that can act in substrate binding as is observed in TauD, a 2OG oxygenase involved in metabolism of taurine (57). Different structural and functional subfamilies of the 2OG oxygenases are defined by the secondary structural elements that surround the DSBH. In all structures elucidated to date, the DSBH is extended by at least one β-strand (39). Further diversity is found at the N- and Cterminal sides of the DSBH where other domains involved in protein function or oligomerization are found. As with the cupins, 2OG oxygenases are observed in different oligomeric states, with the most common being monomeric and dimeric. 1.3.3 2OG and Fe2+ binding The DSBH forms a rigid scaffold for the binding of both the Fe2+ ion and the 2OG cosubstrate. The conserved HXD/E(X)nH motif is responsible for Fe2+ binding, with the conserved H and D/E residues located on strand 2 of the DSBH, and the distal His found on strand 7 (Figure 1-7A). This 2-His-1-carboxy metal binding triad is found not only in the 2OG oxygenase, but in all non-heme iron dependent oxygenases that have been characterized (58). In the absence of substrate binding, there are three Fe2+ coordination sites unoccupied. The Fe2+ is coordinated in an octahedral or distorted octahedral geometry in the apo and substrate bound enzymes. Crystal structures of apo DAOCS (1RXF) (59) and TauD (1OTJ) (60) revealed that these sites are occupied by three water molecules. Binding of 2OG to Fe2+ in a bidentate manner displaces two of the water molecules, leaving the third coordination site open for coordination with molecular oxygen or substrate (39). The 2-oxo group of the 2OG moiety consistently binds trans to the carboxyl ligand, while the 1-oxo group is found to alternate between being trans to the distal His 18 Figure 1-7 Binding of Fe2+ and 2OG by 2OG oxygenases. (A) The binding site of both Fe2+ and 2OG in the active site of DAOCS (PDB ID 1UOB) coloured as in Figure 1-6. The Fe2+ion is coordinated by the HXD/EXnH motif, the 1- and 2-oxo groups of 2OG and a water molecule. The 5-carboxylate is coordinated by hydrogen bonding with Arg and Ser residues. (B) Comparison of TauD and DAOCS mode of 2OG ligation to iron. In TauD, the 1-oxo group is trans to the proximal His (green) and in DAOCS the 1-oxo group is trans to distal His (blue). Open binding sites are shown by an L (red). In both cases the 2-oxo group is trans to the Asp residue. Adapted from (39). 19 residue and trans to the first His (Figure 1-7B). This differential binding dictates the stereospecificity of the enzymes. The coordination of the 5-carboxylate of 2OG is mediated through interaction with a basic residue (Arg/Lys) and an alcohol (Ser/Thr) or phenol group (Tyr). There is far greater variety in the residues that bind to 2OG but their location within the DSBH is conserved. 1.3.4 General reaction mechanism All 2OG oxygenases are believed to catalyze the decarboxylation of 2OG by facilitating a dioxygen reaction to form succinate, carbon dioxide and a reactive oxidizing species, which is responsible for the oxidation of the substrate (Figure 1-8). There is significant variation observed in the reactions that follow the formation of the reactive oxidizing species, as is expected given the large number of reactions catalyzed by 2OG oxygenases (Figure 1-5). However, the formation of the reactive oxidizing species is believed to follow the same sequential method, which was proposed based on kinetic studies of thymidine 7-hydroxylase (61). In this reaction mechanism 2OG binds to the Fe2+ in one of the two positions discussed previously (Figure 1-8A and B). This is followed by dioxygen binding, which creates an Fe3+-superoxo or Fe3+-peroxo species that creates a persuccinate or cyclic peroxide species by nucleophilic attack on the 2-oxo group of 2OG. The resulting species collapses, resulting in the creation of a ferryl intermediate (FeIV=O) as the reactive oxidizing species. Direct evidence for the formation of the FeIV=O intermediate was observed in spectroscopic studies of TauD with a trapped FeIV species (62-64). From the formation of this species the mechanisms diverge depending on the type of oxidation reaction that is occurring, accounting for the catalytic diversity of this enzyme class. 20 A B Figure 1-8 General mechanism for 2OG oxygenase formation of a reactive ferryl intermediate. Two mechanisms are shown based on the differential binding of the 1-oxo moiety of 2OG (A) trans to the proximal histidine (Hisn) or (B) trans to the distal histidine (Hisn+x). Adapted from (39). Iron is coordinated by the conserved HXD/EXnH motif. Binding of 2OG to iron displaces water, which is followed by substrate binding and binding of O2. Decarboxylation of 2OG results in the formation of a persuccinate or cyclic peroxide intermediate, which collapses to form a reactive ferryl intermediate. From here the reaction differs based on the function of the particular enzyme. 21 1.4 Di-iron oxygenases The di-iron oxygenase subclass of the non-heme iron oxygenases possesses a carboxylate-bridged di-iron center which activates O2 for cleavage of stable bonds. The conventional di-iron oxygenases are an ever-expanding family that presently includes the R2 subunit of the class I ribonucleotide reductases (RNR-R2), p-amino benzoate N-oxygenase (AurF), the stearoyl acyl carrier protein Δ9-desaturase, hemerythrin, and also the bacterial multicomponent monooxygenases, such as soluble methane monooxygenase (sMMO), toluene/oxylene monooxygenase (ToMO) and phenol monooxygenase (36,65-68). A non-conventional diiron oxygenase, myo-inositol oxygenase (MIOX), has been recently identified which appears to have arisen by convergent evolution (69,70). These enzymes all use the potent di-iron cluster to cleave highly stable C-H, O-H or C-C bonds. 1.4.1 Mechanistic insights In the ground state, conventional di-iron oxygenases possess a fully reduced, di-iron (II/II) form of the cofactor, which reduces O2 to the peroxide oxidation state. A peroxide-bridged di-iron (III/III) intermediate has been observed for several reactions (71-76), and in several cases the O-O bond is thought to be cleaved to generate high-valent iron clusters for catalysis (36,6567,70). For example, a di-iron (IV/IV) complex is formed in sMMO to cleave the C-H bond of methane (65,66,72,77-79). Similarly, RNR-R2 utilizes a di-iron (III/IV) complex to form a tyrosyl radical, which activates the protein for nucleotide reduction with the RNR-R1 subunit (8089). In all conventional cases, catalysis results in a di-iron (III/III) cluster, requiring an external reducing agent such as NAD(P)H to return to the active state (65,66,66). The non-conventional MIOX catalyzes the conversion of myo-inositol to D-glucuronic acid utilizing a mixed valent diiron (II/III) cluster to initiate the reaction (90-92). Binding of O2 putatively results in the 22 formation of a superoxo-di-iron (III/III) species that cleaves a C-H bond which ultimately leads to the cleavage of a C-C bond and regeneration of the active di-iron (II/III) state without external reducing equivalents (69). The only other di-iron containing enzyme known to possess a mixedvalent di-iron site is purple acid phosphatase, however this enzyme is not an oxygenase (93). 1.4.2 Structure of di-iron oxygenases and the di-iron site Di-iron oxygenases are globular proteins that are almost entirely α-helical in nature. The di-iron site is located between two antiparallel helix pairs, which provide the ligands for iron coordination (Figure 1-9) (69,94). This site is located deep within the core of the protein, and is thought to protect the cell from the radical and oxidizing intermediates that are formed during catalysis. This is exemplified by the hydrophobic and oxidation-resistant nature of the RNR-R2 and sMMO active sites, which prevent non-specific oxidation of the active site of the protein, providing an ideal environment for the reaction to occur (94). Though there is very little overall structure homology between MIOX and the conventional di-iron oxygenases, the core α-helical bundle surrounding the di-iron site is very similar (69). The only exception to the α-helical core structure is purple acid phosphatase, which is an α/β sandwich protein with its active site on the surface of the protein; however, as previously stated, this enzyme is not an oxygenase and therefore does not require such a buried active site (93). Conventional di-iron oxygenase proteins possess tandem EXXH motifs which provide carboxylate and imidazole groups for iron ligation (94-98). The di-iron site is highly symmetrical, with 2 bridging carboxylates, 2 additional carboxylates and 2 His residues ligating the iron (Figure 1-10A). This electron rich active site would be beneficial in the stabilization of the high- 23 Figure 1-9 Representative di-iron oxygenase structures. (A) The conventional sMMO structure (PDB ID 1MMO) and (B) MIOX (PDB ID 2HUO). The core α-helices that make up the di-iron binding site are coloured in purple, iron ions are shown as orange spheres. 24 Figure 1-10 Representative di-iron binding sites. (A) The di-iron site of sMMO (PDB ID 1MMO) is comprised of 2-His and 4-carboxyl groups, and in the structure is bridged by an Asp residue, a water molecule and an acetate (beige). (B) The hemerythrin (PDB ID 1HMD) di-iron site is comprised of 5-His and 2-carboxyl groups, the irons are bridged by 2-Glu residues and a water. (C) The di-iron site of MIOX (PDB ID 2HUO) is comprised of 4-His and 2-Asp residues. The irons are bridged by an Asp residue and a water molecule. Remaining coordination sites are occupied by the substrate, myo-inositol (beige). Bridging residues are coloured teal in all cases. 25 valent intermediates formed in conventional mechanisms such as those in RNR-R2 and sMMO (36,94). The di-iron site can be tailored to the function of the particular enzyme as is seen with the oxygen carrier protein hemerythrin, which has a slightly modified di-iron site with 5 His and 2 carboxylates coordinating the Fe2+ ions (99), and with the recently identified MIOX protein, which has an asymmetric di-iron site comprised of 4 His and 2 carboxylates and a bridging hydroxyl (69). 1.5 Significance and Implications The identification of a metal cofactor opens new realms for understanding a protein, both at the structural and functional levels. A common theme found in the characterization of the metalloproteins discussed above is the structural conservation of metal-binding sites and the power of structural analysis in functional assignment. In this thesis, we present three examples where identification of metal ion co-factor(s), combined with structural analysis, provided important functional and mechanistic insights for novel microbial metalloenzymes. The three metalloenzymes examined here are EcSI, a novel Mn2+-dependent cupin sugar isomerase from Escherichia coli pathogenic strain O157:H7; YcfD, a novel 2OG oxygenase; and PhnZ a di-iron oxygenase that utilizes a novel mechanism for cleaving stable carbon-phosphorus (C-P) bonds. For each enzyme the X-ray crystal structure led to the identification of metal ion cofactor(s), and subsequently led to the elucidation of a function and/or catalytic mechanism. Biochemical evidence from site-directed mutagenesis of active site residues, activity assays and cell growth experiments was obtained to confirm the predicted function. Several important features of metalloenzymes are highlighted by these three proteins as well as the importance of structure in elucidating their function and mechanism. 26 1.5.1 Specific Objectives 1. Through determination of the X-ray crystal structure of Z5688 from E. coli O157:H7, elucidate its function and evaluate its potential role in pathogenicity. 2. Utilizing the structure of the conserved bacterial 2OG oxygenase YcfD, determine its function and its role in E. coli cell growth. 3. Determine the X-ray crystal structure of PhnZ, and provide a structure-based mechanism for the novel C-P bond cleavage reaction. 27 Chapter 2 Structure-Based Annotation of a Novel Sugar Isomerase from the Pathogenic E. coli O157:H7 2.1 Abstract Prokaryotes can use a variety of sugars as carbon sources in order to provide a selective survival advantage. The gene z5688 found in the pathogenic E. coli O157:H7 encodes a ‘hypothetical’ protein of unknown function. Sequence analysis identified the gene product as a putative member of the cupin superfamily of proteins, but no other functional information was known. We have determined the crystal structure of the Z5688 protein at 1.6 Å resolution and identified the protein as a novel E. coli sugar isomerase (EcSI) through overall fold analysis and secondary structure matching. Extensive substrate screening revealed that EcSI is capable of acting on D-lyxose and D-mannose. The complex structure of EcSI with fructose allowed the identification of key active site residues, and mutagenesis confirmed their importance. The structure of EcSI also suggested a novel mechanism for substrate binding and product release in a cupin sugar isomerase. Supplementation of a non-pathogenic E. coli strain with EcSI enabled cell growth on the rare pentose D-lyxose. 28 2.2 Introduction The ability of prokaryotes to use a variety of different sugars as carbon sources provides a selective advantage for survival. Several pathways for the metabolism of sugars such as Dlyxose and D-mannose to form intermediates of both glycolysis and the pentose phosphate pathway have been proposed based on genetic and physiological observations (100-104). The pentose phosphate pathway, an important pathway for the production of energy, comprises glucose-6-phosphate dehydrogenase, lactonase and 6-phosphogluconic acid dehydrogenase. Together, these enzymes constitute a central pathway for producing metabolic energy in the form of NADPH. The pentose phosphate pathway first oxidizes glucose-6-phosphate to ribulose-5phosphate which in turn is epimerized to xylulose-5-phosphate and ribose-5-phosphate (101,105107). These products are then utilized as precursors for glycolytic intermediates. The enzymes responsible for the initial entry of sugars into conserved metabolic pathways are generally highly similar to those utilized in the catabolism of more common carbon sources and typically differ only by minor mutations when compared with their respective paralogues, with the exception of D-lyxose isomerases which represent a novel family of sugar metabolizing enzymes with little homology to other known enzymes (108). Typically D-mannose enters the metabolic process through a phosphotransfer system (PTS) as mannose-6-phosphate which is then isomerized to fructose-6-phosphate by phosphomannose isomerase. However, mannose isomerases (MI, E.C. 5.3.1.7), which catalyze the reversible isomerization of mannose to fructose, have been identified from a number of organisms including Agrobactermium radiobacter M-1 (109), Myobacterium smegmatis (110), Pseudomonas saccharophilia (111), and E. coli (103,104). Despite the fact that the DNA and amino acid sequences for these MIs have yet to be fully elucidated, these enzymes are used for 29 the production of mannose for commercial applications. Several MIs have been found in the search for the rarer D-lyxose isomerase. D-Lyxose isomerases (LI, E.C. 5.3.1.15) catalyze the reversible isomerization of D-lyxose to D-xylulose, an intermediate of the pentose phosphate pathway (112). D-Lyxose is a rare pentose found in bacterial glycolipids that is not commonly utilized amongst microorganisms (113,114). Though it is known that a number of microorganisms can survive on D-lyxose as a carbon source, its metabolism has been poorly understood until recently. Despite the induction of expression of these MIs when D-lyxose is provided as the sole carbon source, their low levels of activity towards this sugar suggested that they are not solely responsible for D-lyxose isomerization. The presence of an LI was also suggested from partially purified extracts of Aerobacter aerogenes PRL-R3, but the protein responsible was never determined (112). More recently, a D-lyxose isomerase was identified in a newly isolated, Gram-positive bacterium, Cohnella laevoribosii RL-39 (108). This enzyme was active towards D-lyxose, D-mannose and L-ribose in a manganese-dependent reaction, with an apparent preference for D-lyxose. A number of sugar isomerases have been classified as members of the cupin superfamily, one of the most functionally diverse classes of proteins. It comprises both enzymatic and nonenzymatic proteins, including isomerases and epimerases involved in bacterial cell wall synthesis, transcription factors, and non-enzymatic seed storage proteins in plants (10,14,115-117). Two consensus sequences have been defined for the cupin family, a GX5HXHX3,4EX6G metal binding motif and a GX5PXGX2HX3N motif, which are both found in the β-barrel fold that is characteristic of the cupin family. In this work, we have structurally and functionally characterized the gene product of z5688 from pathogenic E. coli O157:H7. Z5688 is a protein of unknown function that 30 possesses both cupin signature motifs, though further functional classification was not possible based on the primary sequence. We present the three-dimensional crystal structure of Z5688 and show it to retain the highly conserved cupin domain structure. Extensive analysis of conserved structural features and in vitro substrate analysis permitted the identification of Z5688 as a novel E. coli sugar isomerase (EcSI), with a preference for D-lyxose and D-mannose. The EcSI structure in complex with fructose revealed key residues in the active site. Additionally, we present the kinetic analysis of both wild type and mutant enzymes confirming the classification of this protein as a sugar isomerase, the importance of active site residues and propose a mechanism for substrate selectivity. Finally, we show that overexpression of EcSI enables cell growth on D-lyxose as the sole carbon source. 2.3 Materials and Methods 2.3.1 Protein expression, purification, crystallization, and X-ray diffraction data collection Recombinant EcSI having an N-terminal hexa-histidine tag was expressed in DL41 (DE3) E. coli in Terrific Broth medium under the control of a T7 promoter. A selenomethionine derivative was also produced in DL41 (DE3) E. coli grown in Le Master medium (118). Proteins were purified by affinity chromatography using batch elution from nickel-nitrilotriacetic acid agarose resin (Qiagen, Hilden, Germany) resulting in yields of approximately 60 mg and 3 mg of pure native and seleno-substituted protein per liter of E. coli culture, respectively. Each protein was concentrated using an Amicon Centricon (Millipore, Billerica, MA) with a molecular weight cutoff of 10 kDa to 10.5 mg/mL following dialysis against 20 mM HEPES (pH 7.5) and 150 mM NaCl and stored at -80 ˚C. Protein for enzymatic analysis was purified in a similar manner, using a His-Trap HP chromatography column (Amersham Biosciences, Uppsala, Sweden). Crystals for 31 EcSI were grown using the hanging drop vapor diffusion method. The final crystallization condition for the selenomethionine derivative contained 0.1 M sodium acetate, pH 4.65, 10% (w/v) PEG 400, and 10 mM phenol. To obtain diffraction quality EcSI co-crystals, D-mannose was mixed with EcSI in a 10:1 molar ratio. The final crystallization condition for the co-crystals contained 0.1 M sodium acetate, pH 4.65, and 10.5% (w/v) PEG 550 monomethylether. All diffraction data were collected under cryogenic conditions (100 K), with 25% glycerol added to the mother liquor as a cryoprotectant. Diffraction data for the selenomethionine derivative were collected on the X3A beamline at Brookhaven National Laboratory Synchrotron Light Source using a MAR 165mm CCD Detector. Diffraction data for the EcSI co-crystal were collected on beamline 14-1 at the Stanford Synchrotron Radiation Laboratory using a MAR 325 mm CCD Detector. DENZO and SCALEPACK (119) were used to integrate, scale and merge the reflections. Both crystals belonged to the monoclinic space group P21, with unit cell dimensions of a = 53.4, b = 75.3, and c = 61.8 Å, β = 106.3° for the apo structure, and a = 53.4, b = 75.7, and c = 62.0 Å, β = 106.5° for the complex structure. Both structures comprised 2 molecules in the asymmetric unit with Matthews Coefficients of 2.1 Å3Da-1 (39.6% solvent) and 2.0 Å3Da-1 (40.6% solvent) for the apo and the complex crystals, respectively (120). 2.3.2 Structure solution and refinement The crystal structure of EcSI was solved by the single-wavelength anomalous dispersion method using a selenomethionine derivative. Heavy atom site identification and density modification were completed using HKL2MAP (121) and 419 of 454 residues were built using Arp/wArp (122). The remaining residues were built manually, and the completed model was refined using CNS (123) and REFMAC5 (124) at 1.6 Å. A total of 500 water molecules and 2 manganese molecules were included in the final structure. The structure factors and coordinates 32 have been deposited in the Protein Data Bank under the accession code 3KMH. The Secondary Structure Matching (SSM) server at the European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm) was used to compare the 3D structure to the Protein Data Bank, and search for similar structures (125). Since the apo crystal and the fructose complex crystals were isomorphous, the data was first directly refined against the apo coordinates using REFMAC5(124) and fructose was subsequently added based on the unambiguous difference density, which was subjected to further refinement. 2.3.3 Inductively coupled plasma-mass spectrometry EcSI was concentrated to 3.5 mg/mL in 20 mM HEPES, pH 7.5, 150 mM NaCl, using an Amicon Ultra centrifugal filter with 10 kDa molecular weight cut off, and the flow-through was collected. The content of Fe2+, Mn2+, Co2+, Ni2+, Cu2+, Zn2+, Ca2+ and Mg2+ for both the protein sample and the buffer flow-through were measured. An average of two measurements was used after the buffer background was subtracted for each measurement. Inductively coupled plasma (ICP) mass spectrometry analysis was carried out at the Royal Military College of Canada in conjunction with the Analytical Services Unit at Queen's University, Kingston, ON, Canada. 2.3.4 Generation of EcSI mutants The QuikChange site-directed mutagenesis kit (Stratagene, Cedar Creek, TX) was used to generate the mutants: K90A, K108A, E110A, F178A, E186A, D193A, and R205A following the standard protocol. 2.3.5 Enzymatic assays Phosphoglucose isomerase activity was measured as previously described (126). Briefly, the formation of glucose-6-phosphate (G-6-P) from fructose-6-phosphate (F-6-P) was measured 33 by coupling the reaction with the reduction of NADP+ by G-6-P dehydrogenase (Sigma, St. Louis, MO). The standard assay contained 20 mM Tris (pH 7.5), 0.5 mM NADP+, 3 mM MgCl2, 5 - 3000 µM F-6-P, and 0.35 units of glucose-6-phosphate dehydrogenase. The activity was observed by measuring the increase in NADPH absorption at 340 nm at 37 °C. For the analysis of all other sugar substrates, unless otherwise stated, the reaction was performed in 50 mM HEPES buffer (pH 7.5) containing 10 mM D-lyxose and 0.094 U ml−1 of enzyme in the presence of 2 mM Mn2+ at 50 °C for 10 min. After incubation, the reaction was stopped by the addition of HCl to a final concentration of 200 mM. The enzyme solution was centrifuged, and filtered for further analysis. One unit of enzyme activity was defined as the amount of enzyme required to produce 1 µmol of D-xylulose from D-lyxose per min at 50 °C and pH 7.5. 2.3.6 Determination of specific activity and kinetic parameters To measure the specific activities for the aldose substrates, enzyme reactions were performed for the native and mutant enzyme at 50 °C in 50 mM HEPES buffer (pH 7.5) containing 10 mM monosaccharide for 10 min with an enzyme concentration of 0.094 - 3 U ml−1. The specific activity was defined as the produced amount of ketose as a product per enzyme amount per reaction time. Various concentrations of D-lyxose, D-mannose, and L-gulose (from 12.5 mM to 150 mM) were used to determine kinetic parameters of the enzyme. The reaction was carried out in 50 mM HEPES buffer (pH 7.5) at 50 °C for 10 min. The enzyme kinetic parameters, Km (mM) and kcat (s−1), for substrates were determined by fitting the data to the Michaelis-Menten equation. 34 2.3.7 Metal ions, pH, and temperature effects To investigate the effect of metal ions on D-lyxose isomerase, the enzyme assay was carried out in the presence of 10 mM ethylenediaminetetraacetic acid (EDTA) or in the presence of 1 mM of each metal ion including Co2+, Mn2+, Mg2+, Ca2+, Zn2+, Cu2+, Fe2+, or Ba2+. A parallel measurement which received no treatment with EDTA or metal ions was utilized as the control. The reactions were performed at 50 °C for 10 min. The effect of the concentration of Mn2+ was further investigated at the range of 0 to 4 mM. To find the optimal pH of the enzyme, pH was varied from 6.5 to 8.5 using 50 mM piperazine-N,N’-bis(2-ethanesulfonic acid) (PIPES) buffer (pH 6.5-7.5) and 50 mM HEPES buffer (pH 7.5-8.0). The effect of temperature on the enzyme activity was evaluated in 50 mM HEPES buffer (pH 7.5) at temperatures ranging from 40 to 60 °C. 2.3.8 Isomerization product identification The concentration of the monosaccharide products was determined using a Bio-LC system (Dionex ICS-3000, Sunnyvale, CA) with an electrochemical detector and a CarboPac PAI column. The column was eluted at 30°C with 200 mM sodium hydroxide at a flow rate of 1 mL min−1. The concentrations of sugar phosphates were determined by the same system. The column was eluted at 30°C with a Na-acetate gradient of 75 mM NaOH and 75 mM NaOH/500 mM Naacetate. The gradient was increased to 100 mM between 0 to 35 min, to 150 mM between 35 and 38 min, to 350 mM between 38 and 65 min and then to 500 mM for 75 min. The flow rate was 1 ml min−1. 2.3.9 Growth complementation experiments Growth complementation experiments were conducted as previously described (108). Briefly, BL21 (DE3) E. coli were transformed with either empty pET15b vector, or the vector 35 containing the z5688 gene. Cells were grown at 37°C on M9 minimal media with agar supplemented with 0.4% of each monosaccharide of interest, and induced with 1 mM IPTG. 2.3.10 PDB accession numbers The coordinates and structure factors determined for EcSI in this study have been deposited in the Protein Data Bank under the accession number 3KMH, and those for the EcSIfructose complex have been deposited under the accession number 3MPD. 2.4 Results 2.4.1 Crystal structure of EcSI The crystal structure of EcSI was solved by single-wavelength anomalous dispersion method using a selenomethionine derivative crystal (Figure 2-1 A). The space group was determined to be primitive monoclinic (P21), with a dimer in the asymmetric unit. The model was refined to 1.6 Å with Rcryst and Rfree values of 19.1% and 21.3%, respectively. In the final model, 96.8% of residues were in the most favoured regions of the Ramachandran plot, and 3.2% were within allowed regions (Table 2-1). No density corresponding to the N-terminal hexa-histidine tag or the C-terminal residues 224 - 227 was observed for either of the two molecules. Furthermore, residues 203 – 207 in molecule A could not be modeled due to insufficient density though these were well-ordered in molecule B. There was insufficient density for the side chain of R60 in molecule B, thus the side chain was truncated at the C beta. The structure contained 500 water molecules, a phenol molecule, an acetate ion and 2 glycerol molecules, which were from the crystallization and cryoprotectant conditions. There was strong density located at the conserved cupin metal site, which was identified as manganese by ICP-mass spectrometry (data not shown). 36 Figure 2-1 The crystal structure of EcSI from E. coli O157:H7. (A) The cartoon representation of the EcSI dimer. Molecule A (top left) is shown with α-helices in dark teal and β-strands in purple, molecule B (bottom right) is shown with α-helices in cyan and with β-strands in magenta. The R-205 loop (residues 203-207) is highlighted in green. Manganese atoms bound in each metal binding site are shown as orange spheres. The secondary structure is labeled for molecule A. Inset: The conserved metal binding residues are shown, along with the manganese atom (yellow), and the two waters coordinating the manganese (blue spheres). The 2Fo-Fc electron density at 1σ contour level is shown in blue, and 5σ contour level is shown in magenta. (B) The phenol molecule bound in the active site of molecule B. Inset: The phenol molecule makes contact with D193 and R203, with bond lengths of 2.59 Å and 2.75 Å, respectively. The Fo-Fc omit map density for the phenol molecule is shown at 3σ contour level in blue. 37 Table 2-1 EcSI data collection and refinement statisticsa Apo EcSI Fructose Complex Wavelength (Å) 0.97561 0.97946 Space group P21 P21 Unit Cell: a ,b, c (Å) 53.4, 75.3, 61.8 53.4, 75.7, 62.0 β (°) Molecules in the asymmetric unit Solvent content (%) 106.3 106.5 2 2 46.7 40.6 Resolution range (Å) 30 – 1.54 (1.6 - 1.54) 50 – 1.9 (1.98 – 1.91) Unique reflections 62650 (6177) 35627 (2947) Completeness (%) 98.4 (97.6) 97.1 (80.4) I/σ 32.1 (5.7) 29.4 (4.0) b 0.052 (0.304) 0.081 (0.356) 7.3 (7.4) 6.8 (4.7) Rmerge Average redundancy Refinement statistics Sigma cutoff for refinement None None c Rcrys 0.191 (0.237) 0.189 (0.245) c Rfree 0.213 (0.267) 0.235 (0.289) Number of reflections used Number of reflections in test set (%) Number of non-hydrogen atoms used in refinement Number of Amino Acids (Molecule A/B) Number of Ligand Molecules 59447 33821 5.1 5.0 4036 4018 219/223 226/227 1 2 Number of water molecules 500 344 RMSD bond lengths (Å) 0.007 0.009 RMSD bond angles (°) 1.073 1.226 96.8 96.2 Ramachandran Plot Preferred (%) Allowed (%) 3.2 3.8 a Values in parentheses are for the outermost shell b Rmerge = Σ|Iobs - <I>| / ΣIobs, where Iobs is the intensity measurement and <I> is the mean intensity for multiply recorded reflections. c Rcryst and Rfree = Σ|Fobs - Fcalc| / Σ|Fobs| for reflections in the working and test sets, respectively. 38 The overall fold of EcSI is a cupin-type β-barrel (Figure 2-1). The N-terminus forms a 20-residue α-helix followed by three shorter α-helices that lead into the cupin domain. The cupin domain comprises 2 antiparallel β-sheets, one containing 8 strands (β1-3, 12, 5, 10, 7, 8) and the other consisting of 5 strands (β13, 4, 11, 6, 9). The β-barrel forms a deep pocket that is predominantly hydrophobic. The conserved manganese-binding site is located within this pocket formed by the β-barrel and the metal is coordinated by H103, H105, E110, H171 and two water molecules (Figure 2-1 A inset). In the case of molecule B, the phenol molecule is also bound in the pocket. The hydroxyl group of phenol makes contact with D193 and R205 with respective bond lengths of 2.59 Å and 2.75 Å (Figure 2-1B). The contact between the phenol and R205 stabilizes the flexible loop between residues 203 and 207 that is not visible in molecule A. EcSI forms a dimer in the asymmetric unit (Figure 2-1 A); and dynamic light scattering data confirm the existence of an EcSI dimer in solution (data not shown). In the crystal, the two molecules are intimately associated and are related by 2-fold non-crystallographic symmetry. The dimerization interface has a buried surface area of 1550.3 Å2, which accounts for 14.3% of the overall solvent-accessible area as calculated using the PISA server (http://www.ebi.ac.uk/msdsrv/prot_int/pistart.html)(127). The dimerization is facilitated through a disulfide bond between C86 of molecule A and C86 of molecule B. The protein interface is further stabilized through an extensive network of 20 hydrogen bonds involving both main chain and side chain atoms. Additional stability is provided by the formation of 15 intermolecular salt-bridges at the interface. 2.4.2 EcSI is structurally similar to cupin phosphoglucose isomerases Structural alignments using the SSM server (125) revealed high structural homology shared by EcSI and numerous cupin proteins, including oxylate decarboxylase (PDB ID: 1O4T, RMSD 1.93 Å) (128), Jack Bean canavalin (PDB ID: 1CAV, RMSD 1.69 Å) (129), as well as 39 two members of a novel class of cupin phosphoglucose isomerases (cPGIs) from Thermococcus litoralis (PDB ID: 1J3P, RMSD 2.08 Å) and Pyrococcus furiousus (PDB ID: 1PLZ, RMSD 2.9 Å) (130). The cupin β-barrel of EcSI aligned well with the cPGIs, while the α-helices differed significantly (Figure 2-2 A). Despite the lack of sequence similarity between EcSI and the cPGI proteins (sequence identity = 10%, Figure 2-2 B), key conserved structural features were apparent; these included retention of highly conserved metal coordination residues and location of the metal-site in relation to the active site entrance. There was also strong retention of the chemical characteristics of the putative active site such as the overall shape and depth of the site, as well as the largely hydrophobic nature of the active site in both enzymes. Following this lead, we tested and indeed detected the isomerization of fructose-6-phosphate to glucose-6-phosphate. However, the preliminary results indicated that the catalytic efficiency was very low (~0.01 mM1 -1 s ) using a glucose-6-phosphate dehydrogenase coupled reaction (126). We therefore postulated that EcSI may be a sugar isomerase with alternative substrate specificity, which is also supported by the phenol bound in molecule B as sugar-binding enzymes are often found to bind phenol as a substrate analogue, for example, UDP-galactose 4-epimerase which binds UDP-phenol (131). 2.4.3 Identification of substrate preference for EcSI A panel of available sugar substrates was screened for EcSI, including D- and L-forms of pentoses and hexoses. Among the aldose substrates, the specific activity was highest for D-lyxose, followed by D-mannose, L-gulose, D-talose, L-ribose, L-allose, and a similar trend was observed for the reverse ketose-aldose reactions (Table 2-2). No activity was found for the other aldoses and ketoses, D-glucose-6-phosphate, D-mannose-6-phosphate, or D-ribose-5-phosphate as a substrate. Similar to D-lyxose isomerases from C. laevoribosii (108) and A. aerogenes (112), EcSI showed activity for aldose substrates with the C2 and C3 hydroxyl groups in the left-hand 40 Figure 2-2 Structural comparison of EcSI to cupin phosphoglucose isomerases. (A) Alignment of EcSI (magenta) with the cPGI from T. litoralis (PDB ID: 1J3P, cyan), r.m.s. deviation 2.08 Å. Metal binding residues are shown as sticks, iron and manganese ions are shown as orange and grey spheres, respectively. (B) Structure-based sequence alignment of EcSI with T. litoralis cPGI. Conserved residues are marked with asterisks, while sequence segments that aligned well structurally are capitalized. The secondary structure elements are shown above and below the sequence, coloured to match Figure 2-2A. Metal binding residues are highlighted in red. Both alignments were generated using DaliLite (132). 41 Table 2-2 Specific activity of EcSI Substrate Aldose Ketose Aldosephosphate Product Specific activity (U/mg) D-Lyxose D-Xylulose 5.80 ± 0.11 D-Mannose D-Fructose 4.73 ± 0.05 L-Gulose L-Sorbose 1.55 ± 0.015 D-Talose D-Tagatose 0.20 ± 0.001 L-Ribose L-Ribulose 0.095 ± 0.003 L-Allose L-Psicose 0.01 ± 0.000 D-Idose D-Sorbose ND D-Altrose D-Psicose ND D-Arabinose D-Ribulose ND L-Xylose L-Xylulose ND D-Lyxose 35.2 ± 0.38 D-Fructose D-Mannose 1.19 ± 0.001 L-Ribulose L-Ribose 0.18 ± 0.001 L-Sorbose L-Gulose 0.024 ± 0.003 D-Tagatose D-Talose 0.006 ± 0.000 L-Psicose L-Allose 0.0004 ± 0.000 D-Sorbose D-Idose ND D-Psicose D-Altrose ND D-Ribulose D-Arabinose ND L-Xylulose L-Xylose ND D -Xylulose D-Ribose-5- D-Ribulose-5- D-Glucose-6- D-Fructose-6- D-Mannose-6phosphate D-Fructose-6- phosphate phosphate phosphate phosphate phosphate ND ND ND Data represent the means of three separate experiments with standard deviation. ND means specific activity was not detected by the analytical methods used in this study. 42 configuration (Fischer projections), such as D-lyxose, D-mannose, L-gulose, D-talose, L-ribose, Lallose (Figure 2-3), indicating that the enzyme was potentially a D-lyxose isomerase. Further specificity was noted towards substrates with the C4 hydroxyl oriented in the right-hand configuration (D-lyxose, D-mannose, and L-gulose). The isomerization reactions for all three substrates followed Michaelis-Menten kinetics (Figure 2-4). The Michaelis–Menten parameters (Km and Vmax), turnover rates (kcat), and catalytic efficiencies (kcat/Km) of the enzyme for D-lyxose, D-mannose, and L-gulose are presented in Table 2-3. The Km for D-lyxose (16.1 mM) was about 1.2-fold lower than that for D-mannose (19.8 mM), and 3.4-fold lower than that for L-gulose (55.2 mM). The kcat (13.7 s−1) using D-lyxose was about 1.1-fold higher than that using D-mannose (12.7 s−1) and 1.5-fold higher than L-gulose (8.78 s-1). As a result, the kcat/Km (850 M–1 s–1) for D-lyxose was about 1.3-fold higher for D-mannose (640 M–1 s–1) and 5.3-fold higher than for L-gulose (159 M-1s-1). These results indicate that there is no significant preference between D-lyxose and D-mannose as substrates for this enzyme. 2.4.4 Effects of metal ions, pH, and temperature on the activity of EcSI Many sugar isomerases including the D-lyxose isomerases from A. aerogenes and C. laevoribosii (108,112) and the cPGI enzymes (26), have been reported to be metal-dependent enzymes. In addition, the majority of cupin enzymes are known to require divalent cations for catalytic activity (reviewed in (116)). The activity of E. coli sugar isomerase was assayed in the absence or presence of each metal ion at a concentration of 1 mM or after treatment with 10 mM EDTA. Treatment with EDTA failed to chelate the metal, as there was no inhibition of activity observed likely due to the depth of the metal-binding site and hence tight metal binding in the protein. Since EDTA was unable to remove the metal it is unlikely that it is free to exchange with other metal ions and thus we were unable to accurately examine the effect of metal on the activity 43 Figure 2-3 Schematic representation of aldose–ketose isomerization reactions catalyzed by E. coli D-lyxose isomerase. The boxed structures indicate the preferred hydroxyl configurations, the activity order of enzyme is D-lyxose (the aldose substrate with hydroxyl groups oriented in the same direction at C2, C3) > D-mannose (C2, C3) > L-gulose (C2, C3, C5) > D-talose (C2, C3, C4) > L-ribose (C2, C3, C4) > L-allose (C2, C3, C4, C5). 44 Figure 2-4 Michaelis-Menten Plot. The initial velocity (v) was measured for varying substrate concentrations ([S]) of D-lyxose ( ), D-mannose ( ) and L-gulose ( ). Data are the mean of two experiments; error bars represent the standard deviation. 45 Table 2-3 Kinetic parameters of EcSI Substrate Km (mM) Vmax (U/mg) kcat (s–1) D-Lyxose 16.1 ± 0.10 14.1 ± 0.06 13.7 ± 0.05 850 ± 8.73 D-Mannose 19.8 ± 0.24 13.1 ± 0.02 12.7 ± 0.02 640 ± 8.73 L-Gulose 55.2 ± 2.65 9.09 ± 0.20 8.78 ± 0.19 159 ± 4.15 Data represent the means of three experiments with standard deviation. 46 kcat/Km (M-1 s-1) of EcSI. However, a slight increase in activity was observed in the presence of Mn2+, likely due to the effect of metal saturation. Thus the isomerization of D-lyxose by EcSI was performed in the presence of 2 mM Mn2+ in all subsequent experiments. The activity of the enzyme was examined over a pH range of 6.5 to 8.0 at 50°C. The maximal enzyme activity was recorded at pH 7.5. The effect of temperature on enzyme activity was investigated, and the maximum activity was observed at 50°C (Data not shown). 2.4.5 Complex structure of EcSI with D–fructose bound Native EcSI protein was crystallized in the presence of a ten times molar excess of of Dmannose. Complex crystals were grown which belonged to the same space group as the apo crystals. Density was observed in the active site of both molecules in the asymmetric unit that corresponded to a single D-fructose molecule, the product of D-mannose isomerization (Figure 2-5A). The fructose molecule is bound in both active sites through interactions with K90, K108, E110, E186, D193, N197, as well as the Mn2+ ion (Figure 2-5B). In the better-ordered molecule B, R205 is also making a hydrogen bond to the O4 of the fructose, while in molecule A only partial density for the R205 loop was observed due to disorder, with no density observed for the D204 residue. The complex structure of EcSI with product was very similar to the apo structure overall, with an RMSD of 0.9 Å. Several local differences between the two structures were observed, including the observed density for the C-terminal tail of both molecules in the fructosebound structure. The final model was refined to 1.9 Å with Rcryst and Rfree values of 18.9% and 23.5%, respectively (Table 2-1). 47 Figure 2-5 Complex structure of EcSI with fructose. (A) The fructose molecule bound in molecule B is shown with the Fo-Fc omit map; the density is shown at 3σ contour level in blue, metal binding residues are shown in light pink. The C atoms are numbered in the fructose molecule. (B) A stereo view of fructose bound in the active site of EcSI. The fructose moiety makes contact with a number of surrounding residues. Fructose molecule is shown in yellow; residues binding to fructose are shown as magenta sticks, and R205 is highlighted in green. The Mn2+ ion is shown as an orange sphere. Hydrogen bonds are shown in grey; for clarity only those contacts between the fructose and the protein are shown. 48 2.4.6 Active site mutations Based on the complex structure of EcSI with fructose, seven residues within the active site were selected for mutation and subsequent D-lyxose isomerase activity analysis (Table 2-4). Consistent with prior studies of cupin enzymes, mutation of the metal binding residue E110 to alanine resulted in a significant decrease in activity (10,14,26,116,133-136). The mutation of three residues (K108, E186, and D193) resulted in complete loss of isomerase activity. The K108 residue was found to be in contact with O2 of the fructose molecule, indicating that it may play a role in the catalytic mechanism. Mutation of E186 to alanine could impact the enzyme activity in several ways: firstly, by general disruption of the chemical characteristics of the pocket; and secondly, by removing the stabilizing interaction with K108. The significant reduction of turnover caused by mutation of D193, as well as K90, combined with the fact that these residues are in contact with hydroxyl groups not involved in the isomerization, suggests that these residues are important for the optimal orientation of the substrate in the active site. To ensure that the loss of activity of the mutants is not due to misfolding, we carried out circular dichroism experiments for selected mutants and showed they have virtually identical spectra to the wild type protein (data not shown). Interestingly, the R205A mutation reduced the specific activity to 9% of the wild type enzyme. Based on the structures of EcSI with phenol and fructose, it was apparent that R205 plays a role in substrate selectivity. As a result, the activity of the R205A mutant enzyme for hexoaldoses and pentoaldoses, such as the D- and L-forms of lyxose, xylose, arabinose, ribose, galactose, idose, gulose, talose, mannose, glucose, altrose and allose was measured. It was observed that the enzyme was active only for D-lyxose. 49 Table 2-4 Specific activities of the wild-type and mutant EcSI Enzyme Specific activity (U mg–1) Relative activity (%) Wild 5.80 ± 0.09 100 ± 1.8 K108A ND* ND E110A 0.08 ± 0.00 1.40 ± 0.01 F178A 5.81 ± 0.01 100 ± 0.2 E186A ND ND D193A ND ND R205A 0.52 ± 0.00 9.00 ± 0.02 K90A 0.04 ± 0.00 0.69 ± 0.00 *ND means specific activity was not detected by the analytical methods used in this study 50 2.4.7 Substrate binding and product release Sequence alignment of EcSI with homologues, including the C. laevoribosii D-lyxose isomerase, revealed the conservation of a number of residues (Figure 2-6A). Despite the fact that EcSI and C. laevoribosii D-lyxose isomerase possesses only 19.8% sequence identity, many of the conserved residues are in the cupin domain of the protein, including those residues involved in metal binding, and in the binding of fructose. Also found to be strictly conserved is the R205 residue located on a loop observed to be highly flexible in both the apo and the fructose bound structure (Figure 2-6A). Comparison of the apo and the fructose-bound structures demonstrated the importance of this loop for substrate binding and product release. In molecule A of the EcSI structure, no density was apparent for the R205 loop, in molecule B, however, density was observed for the loop region (L203 to L208) and permitted the unambiguous modeling of the loop including the R205 side chain. At this site, the terminal NH2 group of the R205 side chain interacts with the phenol molecule that is well-ordered in the active site. In the case of the fructose-bound structure, partial density was observed for the R205 loop in molecule A in an open conformation (Figure 2-6B). The R205 loop in molecule B is once again found to be in the closed conformation with the terminal NH2 group interacting with the O4 of the fructose molecule where the enzyme has shown steric selectivity (Figure 2-6C). Comparison of these two molecules suggested a potential model for substrate binding and stabilization in the active site. The hydroxyl of the phenol molecule and the C4 hydroxyl of the fructose molecule contact with R205, stabilizing the R205 loop (L203 to L208) in the B molecule of the two structures respectively, representing the closed substrate-bound form. In both structures, molecule A is primed for product release and binding of a new substrate molecule and, thus, in an open conformation (Figure 2-6B). Though density is visible for the loop residues of both closed forms 51 Figure 2-6 Substrate stabilization and product release mechanism. (A) Amino acid sequence alignment of EcSI (accession no. Q8X5Q7) with various homologous proteins. The proteins are identified by their Swiss-Prot accession number. Jan_3303, a hypothetical protein from Jannaschia sp. CCS1 (accession no. Q28M42), D-lyxose isomerase from C. laevoribosii (accession no. A3E7Z6), and a putative D-lyxose isomerase from Providencia stuartii (accession no. B2Q2PO). Conserved residues are marked with an asterisk, and the metal binding residues are highlighted in red. Conserved residues lining the active site are coloured in blue, while R205 is coloured in green. Secondary structure elements are shown above the sequence coloured and numbered as in Figure 2-1A. Alignment was performed using ClustalW (137). (B) The surface representation of the EcSI active site. The open form of the R205 loop is shown as orange sticks, the closed form is shown as green sticks. (C) The omit map density for the R205 in the open form and (D) the closed form is shown at 2σ. Residues 203 to 208 were omitted from both molecules of the asymmetric unit, followed by refinement and map calculation to generate the omit map. Due to the inherent flexibility of the open form, the density is much weaker. 52 and the open form of the fructose bound structure, they are still highly flexible as indicated by their elevated B-factors ranging from 40-50 Å2 for the closed forms, and 50-60 Å2 for the open form (as compared to the mean B-factor for entire molecule of 22.6 Å2 for the apo structure, and 25.7 Å2 for the fructose-bound structure). 2.4.8 Growth complementation assay In order to investigate whether EcSI could confer non-pathogenic E. coli BL21 (DE3) the ability to grow on D-lyxose as the sole carbon source, we carried out a cell growth complementation assay using a previously established method (108). Cells were streaked on M9 minimal media supplemented with either of the preferred substrates, D-mannose or D-lyxose, or with D-glucose as a positive control and grown at 37 °C. Both the vector control and the cells expressing EcSI grew on D-glucose and D-mannose in 24 hours, demonstrating that E. coli already possess a mechanism for D-mannose utilization. Only the cells expressing EcSI were able to grow on D-lyxose after 48 hours, while no growth of the vector control was observed (Figure 2-7). 2.5 Discussion Primary sequence analysis identified Z5688 as a member of the cupin superfamily of proteins, which is comprised of structurally related, functionally divergent proteins, but provided no further insight into the function of this hypothetical protein. The crystal structure confirmed that Z5688 possessed the β-barrel structure that is characteristic of the cupin superfamily. As with many cupin proteins, Z5688 was found to form a dimer with the two monomers related by 2-fold symmetry. In many cupin dimers, such as those formed by cPGIs (130), auxin-binding protein (138) and oxalate oxidase (22), it is observed that an N-terminal segment of one subunit is inserted to the ß-sheet of the other subunit resulting in an intimately connected symmetrical 53 Figure 2-7 The z5688 gene from E. coli O157:H7 confers the ability for E. coli BL21 (DE3) to grow on D-lyxose. (A) Cells streaked on M9 minimal media supplemented with D-glucose and (B) D-mannose after 24 hours growth at 37 °C (C) Cells streaked on M9 minimal media supplemented with D-lyxose after 48 hours growth at 37 °C. 54 dimer. However, this phenomenon is not observed in Z5688. The dimer forms through side-chain and backbone interactions between residues belonging to the large β-sheet of each subunit, resulting in a symmetrical dimer with both active sites accessible to solvent at opposite sides of the protein. Structural similarity searches revealed that the protein is similar in fold to a number of cupin proteins. Among these, two members of a novel class of cupin PGIs stood out due to structural conservation of the metal-binding residues and the chemical similarity of the hydrophobic pocket defining the active site. This homology led to the initial suggestion of Z5688 as a cupin sugar isomerase. Although PGI activity was observed, the activity was too low to establish that glucose-6-phosphate was the ideal substrate. Subsequent substrate screening and enzymatic analysis allowed the identification of the preferred substrates as D-lyxose and Dmannose. The EcSI showed highest activity towards aldose and ketose substrates with the C2 and C3 hydroxyl groups oriented in the left-handed configuration (Fischer projection), and the C4 hydroxyl in the right-handed orientation such as D-lyxose, D-mannose and D-gulose (Figure 2-3). The enzyme exhibited little or no activity towards other aldoses, ketoses, and sugarphosphates. This discrimination against phosphorylated sugars is explained by examination of the active site in comparison to the cPGI from P. furiosus. The cPGI has two positively charged residues, as well as two tyrosine residues that have been shown to interact with the phosphate moiety in inhibitor complex structures (130,134). The equivalent positions in EcSI are occupied by hydrophobic residues and smaller polar groups; therefore, the EcSI active site would not accommodate the negative charge of the phosphate group. The order of substrate preference for EcSI is: D-lyxose > D-mannose > L-gulose > Lribose > L-allose. Similar substrate specificity has been observed in D-lyxose isomerases from C. 55 laevoribossii and A. aerogenes (108,112). Other sugar isomerases have demonstrated a preference for substrates with the C2 and C3 positions in the left-hand orientation including L-ribose isomerase from Acinetobacter sp. (139), mannose-6-phosphate isomerase from B. subtilis (140) and mannose-6-phosphate isomerase from G. thermodenitrificans (141); however, the order of substrate preference differs amongst the enzymes. The EcSI enzyme showed similar activity towards D-lyxose and D-mannose with a turnover and catalytic efficiency for D-lyxose as a substrate (13.7 s-1 and 850 M-1s-1) only 1.1- and 1.3-fold higher than those for D-mannose (12.7 s-1 and 640 M-1s-1), respectively. This dual specificity of EcSI is not often observed amongst various sugar isomerases; however there are examples such as the bifunctional phosphoglucose/phosphomannose isomerases found in archaea (126,142). EcSI exhibits relatively high Km values for both D-lyxose and D-mannose (16.1 and 19.8 mM); however, these are comparable to other sugar isomerases such as the D-lyxose isomerase from C. laevoribosii (22.4 and 34.0 mM), and may account for the promiscuity of the enzyme (108). The catalytic rates determined for EcSI with both D-lyxose and D-mannose were low when compared to the D-lyxose isomerase from C. laevoribosii (108), and the cPGIs from hyperthermophilic archaea; however, they were similar to those cPGIs found in mesophilic homologues (26). The low activity could indicate that there may be a better substrate for this enzyme, such as a disaccharide. E. coli BL21 (DE3) overexpressing the EcSI protein were shown to grow on both D-mannose and D-lyxose (Figure 2-7). It is known that E. coli possess a mechanism for growth on D-mannose; however, they are unable to metabolize D-lyxose. The fact that BL21 (DE3) E. coli transformed with the EcSI gene could survive on D-lyxose as a sole carbon source indicates that, potentially, this enzyme could be involved in the metabolism of rare sugars by the pathogenic E. coli O157:H7, which would provide a selective advantage in the competition for resources between the different 56 bacterial strains. However more research is required to determine the ideal substrate, and the physiological role of the enzyme. Enzyme catalyzed aldose-ketose isomerization involves the transfer of a hydrogen between the C1 and C2 carbon atoms, which can occur by one of two mechanisms: through a direct hydride shift or via a cis-enediol intermediate, where the hydrogen is transferred in the form of a proton by a catalytic base (see additional figure, Figure 2-8). There has been much debate as to the catalytic mechanism of the structurally related cPGI family. Evidence has been presented for both a cis-enediol-based mechanism, as well as for a hydride transfer mechanism of catalysis (134,135,143,144). In the case of EcSI, analysis of the active site residues and mutant analysis indicate that the isomerization likely proceeds via a cis-enediol intermediate. In order for a hydride shift mechanism to be employed, a very specific hydrophobic environment is required to prevent exchange of the transferred hydrogen with the solvent, as is exemplified by xylose isomerase in which a tryptophan residue prevents solvent exchange by forming a hydrophobic barrier at the transfer site (145). The active site of EcSI is highly polar at the substrate binding site, and no large hydrophobic residue is close to the fructose in the complex structure (Figure 2-5B). Both triose phosphate isomerase and the conventional PGIs proceed via a cis-enediol based mechanism utilizing a glutamic acid residue (146-148). In the cPGI from P. furiosus the proton is proposed to be abstracted by the metal binding E97, which is equivalent to E110 in the EcSI structure (Figure 2-2B) (130,143,144). The predominate form of monosaccharide’s in solution is the closed ring form (at 25 °C), indicating the potential need for a ring opening step (149). In the case of the cPGIs no residue is present to facilitate ring opening; however, these enzymes are found in hyperthermophilic archaea which grow at such extreme temperatures that most sugars are likely to exist in their linear form. Conventional PGIs, however, catalyze the ring 57 opening step. In the crystal structure of the rabbit PGI in complex with fructose-6-phosphate it was found that a histidine residue (H388) contacts the ring oxygen (O5), and it was proposed to act as an acid catalyst for the ring opening (150). Examination of the fructose-bound structure of EcSI revealed that one of the metal binding histidine residues (H103) is also positioned near the O5 of the fructose ring, and could potentially act in a similar manner to H388 in the rabbit PGI. Once the ring is opened, E110 can abstract a proton, and the cis-enediol intermediate is formed. This highly unstable intermediate requires stabilization while isomerization occurs. In the conventional rabbit PGI, stabilization is facilitated by a positively charged residue (R272) while, in the cPGIs as well as EcSI, it is likely that the metal ion provides the positive charge necessary for stabilization (130). Further stabilization in EcSI is likely provided by K108. Mutation of these residues, as well as other key residues involved in orienting the substrate was found to compromise the activity of the enzyme, confirming their importance in catalysis (see Table 2-4). The D-lyxose isomerases from both C. laevoribosii and A. aerogenes have been reported to require a metal ion for activity (108,112). Specifically, it was reported that Mn2+ activated the A. aerogenes LI most efficiently, and that treatment with EDTA inhibited the reaction completely. In this work, we show that the metal bound to EcSI was Mn2+ by ICP-MS, and EDTA was a poor inhibitor of the D-lyxose isomerization reaction. Many cupin enzymes require a divalent cation for activity, and some superfamily members, including the phosphoglucose isomerase family possess a very high affinity for their metal cofactors such that they are not readily chelated by the addition of EDTA (26). Structural similarity between EcSI and cPGIs from P. furiosus and T. litoralis was most apparent at the metal binding site, and the location of the metal ion within the cupin domain was similar, suggesting the lack of inhibition by EDTA observed for EcSI is due to a similar high affinity binding of the metal. This is further supported 58 by the ICP-mass spectrometry data which did not detect other divalent cations bound in the analyzed samples. The crystal structure of EcSI revealed that, similar to other cupin sugar isomerases, the protein is a symmetrical dimer (Figure 2-1A). The two molecules of the dimer are virtually identical, with only one key difference that cannot be attributed to the crystal packing: the loop region between residues 203 and 207 was observed in two different conformations, open and a closed (Figure 2-6). In the case of the closed conformation, R205 makes contact with the hydroxyl at the C4 position of the substrate, closing the active site, and blocking solvent entry. After isomerization occurs, the loop moves to the open conformation allowing for product release and binding of a new substrate molecule. Sequence comparison showed strict conservation of R205 between homologous proteins, including D-lyxose isomerase from C. laevoribosii, indicating that this residue is important for the substrate binding and product release mechanism of these enzymes (Figure 2-5A). Interestingly, mutation of this residue to an alanine results in an enzyme that is specific only for D-lyxose, confirming the importance of this loop for substrate binding. In summary, we have determined that Z5688, or EcSI, is a novel sugar isomerase specific for D-lyxose and D-mannose through analysis of the crystal structure and substrate screening. Our work has raised several important questions regarding this novel enzyme with regard to the role for its apparent dual specificity, its lack of similarity to known mannose isomerases and the need to isomerize D-lyxose in the natural environment of E. coli O157:H7. Certainly, additional study is necessary to fully delineate the in vivo role for EcSI. However, this work supports the notion that divergence in metabolic pathways may be critical in defining niches for pathogenic bacteria that are distinct from their non-pathogenic counterparts. 59 Figure 2-8 Schematic representation of the two potential mechanisms for enzyme catalyzed aldose-ketose isomerization. A) The direct hydride shift mechanism involves a direct transfer of a hydride (red) from the C2 to the C1 position. B) The cis-enediol mechanism involves the abstraction of the C2 proton (red) by a base (B) to form the cis-enediol intermediate. The proton on the base is free to exchange with solvent (indicated by a red to blue H) before being donate to the C1 of the intermediate. Adapted from (143). 60 Chapter 3 Crystal Structure and Preliminary Functional Analysis of YcfD, a Novel 2-oxogluterate/ Fe2+-Dependent Oxygenase Involved in Translational Regulation in E. coli 3.1 Abstract The 2-oxoglutarate/Fe2+-dependent dioxygenases (2OG oxygenases) are a large family of proteins that share a similar overall three-dimensional structure and catalyze a diverse array of oxidation reactions. The Jumonji C (JmjC)-domain containing proteins represent an important subclass of the 2OG oxygenase family that typically catalyze protein hydroxylation. However, recently other reactions have been recently identified, including tRNA modification. The E. coli gene, ycfD, was predicted to be a JmjC-domain containing protein of unknown function based on primary sequence. We have determined the crystal structure of YcfD at 2.7 Å resolution, confirming that YcfD is structurally similar to known JmjC proteins, and showed 2OG binds to YcfD using isothermal titration calorimetry. Structural homology to ribosomal assembly proteins combined with GST-YcfD pull-down of ribosomal protein L-16 indicate that YcfD is involved in regulation of bacterial ribosome assembly. Furthermore, overexpression of YcfD is shown to inhibit cell growth signifying a toxic effect on ribosome assembly. 61 3.2 Introduction The 2-oxoglutarate/Fe2+ dependent oxygenases (2OG oxygenases, EC 1.14.11.-) constitute the largest group of mononuclear, non-heme, ferrous iron-dependent oxidizing enzymes. The founding members of the 2OG oxygenase family were prolyl and lysyl hydroxylases which are essential to collagen formation (41,42). Since then, 2OG oxygenases have been shown to be widely-distributed, evolutionarily-conserved enzymes catalyzing a diverse array of reactions. The most common reaction catalyzed by this class of enzyme couples the oxidative decarboxylation of 2OG to the hydroxylation of the primary substrate, which may include protein, methylated nucleotides, lipids, or a wide variety of small molecules (40). Other reactions catalyzed by 2OG oxygenases include such oxidation reactions as ring formation or expansion, desaturation, carbon-carbon bond cleavage, and epimerization (13),(40). Due to their catalytic versatility, 2OG oxygenases are involved in a wide range of biologically important processes such as protein modification, nucleic acid repair, lipid metabolism, as well as, secondary metabolite production in plants and microbes. The 2OG oxygenase family of enzymes is characterized by the presence of a conserved HX(D/E)XnH sequence motif that coordinates the catalytically essential Fe2+ ion. This sequence motif, along with the 2OG-binding site, are found within the signature double-stranded β-helix (DSBH), or cupin fold, that is common to all 2OG oxygenases and related enzymes (reviewed in (39)). The DSBH is made up of two β-sheets consisting of 4 strands each. Within the DSBH, the order of the β-strands is maintained and the ligands coordinating the Fe2+ are in a similar conformation. It is the secondary structure elements surrounding the DSBH that differentiate the 2OG oxygenases further into structural subfamilies, and define their individual functions. To 62 date, the structures of 17 2OG oxygenases have been solved, providing a wealth of functional and mechanistic information. An extensive subclass of 2OG oxygenases has been identified based on sequence similarity to the C-terminal domain of the jumonji transcription factor, termed JmjC-domain containing proteins (48-51). Initially, the JmjC proteins were classified as epigenetic regulators of gene expression through histone-lysine demethylase activity (52). Since then JmjC proteins have been associated with a variety of cellular functions. For example, factor inhibiting hypoxia inducible factor 1 (FIH) is a JmjC-domain containing protein that regulates the hypoxic response through hydroxylation of the β carbon of an Asn residue in the hypoxia inducible factor (HIF) 1α transcription factor (53). Recently, TYW5, an enzyme that catalyzes the hydroxylation of the hypermodified nucleoside, hydroxywybutosine, in tRNAPhe was identified as a JmjC-domain containing protein (54). TYW5 was the first JmjC protein to catalyze the hydroxylation of a substrate other than protein; its discovery has expanded the catalytic potential for the remaining JmjC domain containing proteins that have not yet been functionally annotated. The ycfD gene product of E. coli is a highly conserved microbial protein of unknown function. Based on sequence analysis, it is predicted to be a member of the JmjC-domain containing family; however the sequence alone cannot define a more specific function. Through determination of the 3-dimensional (3-D) crystal structure of YcfD and analysis of its binding of 2OG, we confirm that YcfD is a member of the 2OG family of enzymes. We show that YcfD interacts with the ribosomal protein L-16 (RL-16) from E. coli which, when combined with structure analysis, provides initial evidence that YcfD is involved in translational regulation. Further, we demonstrate that overexpression of YcfD leads to a decrease in cell growth consistent with a role for YcfD in the essential process of translation. 63 3.3 Materials and Methods 3.3.1 Cloning and generation of YcfD variants The ycfD gene (GenBank NC000913) was amplified by PCR using E. coli K12 genomic DNA as a template. Taq DNA polymerase was used with the oligonucleotides 5’-GC GGA TCC ATG GAA TAC CAA CTC ACT CTT-3’ and 5’-GCG GCC GCT CCC TCC GAA GAA CCA ATA CCC-3’ as the forward and reverse primers, respectively, to engineer BamHI and NotI restriction sites (underlined). The PCR product was then subcloned using the TOPO® TA Cloning Kit (Invitrogen, Carlsbad, CA, USA), followed by ligation into the BamHI-NotI site of pET21b and pGEX 4T3 vectors. These constructs produce YcfD with a C-terminal hexahistadine tag or an N-terminal GST tag, respectively, under the control of a T7 promoter. The correct gene sequence was confirmed by sequencing of both strands. The QuikChange site-directed mutagenesis kit (Stratagene, Cedar Creek, TX, USA) was used to generate S116A and R140A variants, as well as, an E146A, K147A double variant (YcfDekaa) using the standard protocol. The double varient was predicted by the Surface Entropy Reduction prediction (SERp) server (http://services.mbi.ucla.edu/SER/) to confer enhanced crystallizability (151). 3.3.2 Protein expression and purification Recombinant YcfD with a C-terminal hexahistidine tag was expressed in BL21 DE3 cells in LB broth (Bioshop Canada Inc, Burlington, ON, Canada). A selenomethionine derivative was also produced in DL41 DE3 cells grown in LeMaster media (118). Cells were grown at 37°C to an optical density of 0.8 at 600 nm (OD600 = 0.8). Protein expression was induced by the addition of 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG, Bioshop Canada Inc, Burlington, ON, Canada) and cells grown for an additional 3 hours. Proteins extracted from the cell lysates were 64 purified by affinity chromatography using nickel-nitrilotriacetic acid agarose resin (Qiagen, Hilden, Germany) using batch elution. Protein-containing fractions were pooled and dialyzed against 50 mM Tris pH 8.0, 300 mM NaCl. The dialyzed protein was then subjected to sizeexclusion chromatography using a Superdex 200 column and FPLC (GE Healthcare Lifesciences, Uppsala, Sweden). Protein fractions were pooled and concentrated to ~10 mg/mL in an Amicon Centricon (Millipore, Billerica, MA, USA) with a molecular weight cut-off of 30 kDa. Approximately 150 mg of native and 30 mg selenomethionine derivative proteins were obtained per litre culture. All variant proteins were expressed and purified in a similar manner. For pull-down assays a glutathione S-transferase (GST) tagged YcfD was expressed using BL21 DE3 cells in LB broth. The protein was purified over glutathione sepharose resin (GE Healthcare Lifesciences, Uppsala, Sweden). Nonspecifically bound protein was removed by extensive washes with phosphate buffered saline (PBS) before elution of the GST-tagged protein with 10 mM reduced glutathione in PBS. The eluted protein was concentrated to ~10 mg/mL using an Amicon Centricon and stored at -80°C, following flash-freezing in liquid nitrogen. 3.3.3 Crystallization and X-ray data collection YcfDekaa crystals were grown at room temperature by vapour diffusion. Initial crystals were obtained by sitting drop vapour diffusion from the JCSG core II suite condition 78 (Qiagen, Hilden, Germany) with 0.07 M sodium acetate pH 4.6, 5.6% (w/v) PEG 4000 and 30% (v/v) glycerol. In order to obtain diffraction-quality selenomethionine derivative crystals, multiple rounds of microseeding were performed as previously described (152). Briefly, a crystal was washed 3 times in well solution, then transferred to 100 μL cold stable solution (0.07 M sodium acetate pH 4.6, 8.6% (w/v) PEG 4000 and 30% (v/v) glycerol). The crystal was crushed and then further diluted with 100 μL stable solution to make a seed stock. Final crystals were grown by 65 sitting drop vapour diffusion against 0.07 M sodium acetate pH 4.6, 3.6% (w/v) PEG 4000 and 30% glycerol. Drops were formed with 2 μL protein mixed with 2 μL well solution and 0.5 μL of a 1/10 dilution of seed stock in a microbridge with 5 μL of Fluorinert (Hampton Research, Aliso Viejo, CA, USA). Multiple anomalous dispersion (MAD) diffraction data were collected under cryogenic conditions (100 K) at the Stanford Synchrotron Radiation Lightsource (Stanford, California, U.S.A.) beamline 9-2 using a Marmosaic 325 CCD detector. Data were indexed, integrated and scaled using XDS (153). Crystals belonged to the tetragonal space group P43212 with unit cell dimensions a = b = 75.71, c = 210.90 Å. There was one YcfD molecule in the asymmetric unit with a Matthews coefficient of 3.48 Å3Da-1 (64.7% solvent) (120). 3.3.4 Structure solution and refinement The structure of YcfD was solved by the MAD method. The positions of nine out of ten selenium atoms were determined, followed by density modification and automatic building using autoSHARP (154). In total, 278 of 381 residues were built automatically. The remaining residues were built manually in iterative cycles using Coot (155) followed by refinement using PHENIX (156). The structure was initially refined using standard refinement strategy and followed by subsequent Translation/Liberation/Screw (TLS) refinement in PHENIX. In addition to the protein, the final model contained 59 water molecules, an iron ion and a single glycerol from the crystallization condition. The PDBeFOLD Structural Similarity server at the European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm/) was used to compare the 3-D structure of YcfD to structures in the Protein Data Bank (PDB) (125). 66 3.3.5 Metal identification Inductively coupled plasma optical emission spectrometry (ICP-OES) was used to identify bound metal in YcfD. A total of 1 mL of YcfD, concentrated to 10.7 mg/mL in buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl), as well as 1 mL of buffer were digested with 2 mL nitric acid and heated for 15 min. The digested samples were made up to 12.5 mL with water and filtered. The amounts of Fe2+, Mn2+, Co2+, Ni2+, Cu2+, Zn2+, Ca2+ and Mg2+, as well as S as an internal control, were measured in both the protein and buffer by ICP-OES at the Analytical Services Unit at Queen's University and compared against known standards (Kingston, ON, Canada). 3.3.6 Isothermal titration calorimetry Isothermal titration calorimetry (ITC) was performed using a VP-ITC isothermal titration calorimeter (GE Healthcare, Piscataway, NJ, USA) in the Protein Function Discovery (PFD) Facility, Queen’s University (Kingston, ON, Canada). Concentrated samples of YcfD, R140A and S116A variants were dialyzed overnight against 50 mM Tris, pH 7.4, 150 mM NaCl, and 10 μM FeSO4. The proteins were then diluted with the same buffer to 105, 84, or 82 μM, respectively and loaded into the reaction cell. A solution of 1 mM 2OG dissolved in the same buffer was injected into the reaction cell in 10 μL aliquots at 300 s intervals using a rotating stirrer-syringe for a total of 29 injections. The data was analyzed using ORIGIN 7.0 (OriginLab Corporation , Microcal, Northampton, MA, USA). 3.3.7 Pull-down assay and mass spectrometry E. coli BL21 DE3 cells were grown until an OD600 of approximately 0.6 in LB broth and harvested. Cell pellets were resuspended in 20 mM Tris-HCl, pH 8.0, 200 mM NaCl (wash buffer), containing 0.1% Triton-X 100 and protease inhibitors, and lysed by sonication. The 67 resulting lysate was clarified by centrifugation for 10 min at 13000 rpm. The pull-down experiment was set up as follows: 900μL of lysate was combined with 100 μL of equilibrated glutathione sepharose resin and 25 μg of GST-YcfD. A control containing only resin and lysate was set up in parallel. The pull-down mixture was allowed to equilibrate for 3 hours at 4°C. The equilibrated resin was loaded onto a micro spin column (Bio-Rad Laboratories (Canada), Mississagua, ON, Canada) and washed extensively with wash buffer. Bound proteins were eluted with 100 μL of 10 mM reduced glutathione in wash buffer, resolved using 12% SDS-PAGE and visualized by silver staining. Subsequently, the protein band was subjected to “in-gel” trypsin digest for MALDI-TOF mass spectrometry on a Voyager DePro mass spectrometer (Applied Biosystems Corporation, Foster City, CA, USA) followed by tandem sequencing (MS/MS) mass spectrometry using a Waters qTOF Global Ultima mass spectrometer coupled to a Waters CSPLC XE chromatography system (Waters Corporation, Milford, MA, USA). Resulting peptide sequences were used to identify the protein using Mascot (157). Mass spectrometry was performed in the PFD Faclity, Queen’s University (Kingston ON, Canada). 3.3.8 Colony forming assay BL21 (DE3) cells were transformed with a pET21b vector containing either the ycfD or ygiN, a gene encoding an unrelated protein used as a control and their growth was compared to that of the ΔycfD JW114.2 (158) cell line (a ycfD gene knockout cell line). A single colony for each gene was inoculated into 5 mL of LB media containing appropriate antibiotic. Cells were grown at 37°C to an OD600 of approximately 1.2 and diluted 10000 times in fresh LB media. An aliquot of 100 μL of the diluted culture was spread on LB-agar plates containing 0.2 mM IPTG and appropriate antibiotic, and the cells were then allowed to grow at 37°C for 18 hours. The 68 ability of the resultant E. coli to form colonies was then assessed qualitatively by visual inspection. 3.4 Results and Discussion 3.4.1 Crystal structure of YcfD The crystal structure of recombinant YcfD was solved by MAD using a single selenomethionine labeled crystal (Figure 3-1). The final structure was refined to 2.7 Å with an R and Rfree value of 18.5% and 22.9%, respectively (Table 3-1). The space group was P43212 with a single YcfD molecule in the asymmetric unit. The majority of residues were found to be in the preferred or allowed regions of the Ramachandran plot (95.5% and 4.2%, respectively) with one outlier, D109. Upon examination of the environment of D109, it is found to be in a region of random coil and the main chain carbonyl groups are involved in hydrogen bonds with the side chain of R310 on one side, and an ordered solvent molecule on the other, thus constraining it in a slightly distorted conformation. Electron density was not observed for a flexible loop comprised of residues 149 – 159 or for residues 373 to the end of the histidine tag. Side chain density was missing for a number of residues and, as such, these side chains were truncated. The structure contained 59 water molecules and a single glycerol molecule. There was strong density observed in the predicted metal-binding site which was modeled as iron, based on similarity to known 2OG oxygenases, and confirmed by ICP-OES. The N-terminal domain of YcfD contains the signature double stranded β-helix (DSBH) or β-barrel that is characteristic of 2OG-dependent oxygenases. The core DSBH is made up of 2 sheets, one comprised of 5 strands and the second comprised of 3. The second sheet is extended by an ordered loop that could form an additional strand upon substrate and/or co-factor binding. As is observed in a number of 2OG oxygenases the first sheet is also extended by 3 additional 69 Figure 3-1 Crystal structure of YcfD. (A) A cartoon representation of YcfD. The α-helices are coloured blue, and β-strands are green. Inset: the conserved metal ion-binding residues are shown, along with the bound iron ion (orange sphere) and two ligating waters (red spheres). (B) The dimer of YcfD that is formed with a symmetry molecule. The symmetry related molecule in light colours. The orientation of the active site opening is indicated by orange arrows. 70 Table 3-1 Data collection and refinement statistics for MAD data Data Collection Space Group P43212 Unit Cell: a, b, c (Å) 75.73, 75.73 , 210.90 Peak Inflection Remote Wavelength (Å) 0.97913 0.97927 0.91837 Resolution (Å) 20.0-2.7 (2.79-2.70) 7.0 (50.1) 20.0-2.7 (2.79-2.70) 6.9 (47.3) 20.0-2.7 (2.79-2.70) 7.0 (47.9) 11.5 (2.1) 11.4 (2.1) 11.7 (2.0) Unique Reflections 31993 31997 31988 Completeness (%) 99.4 (99.5) 99.5 (99.8) 99.4 (99.6) 3.6 (3.6) 3.6 (3.6) 3.6 (3.7) b Rmerge I/σI Redundancy Refinement Statistics Sigma cutoff for refinement None c Rcryst 0.185 (0.324) c Rfree 0.229 (0.391) Number of reflections used 17602 Number of reflections test set (%) 5.0 Number of non-hydrogen atoms 3003 Number of amino acids 361 Number of iron molecules 1 Number of water molecules 117 2 Mean B factor (Å ) 39.6 RMSD bond lengths (Å) 0.010 RMSD bond angles (°) 1.247 Ramachandran Plot Preferred (%) 95.5 Allowed (%) 4.2 Outliers (%) 0.3 a Values in parentheses are for the outermost shell Rmerge = Σ|Iobs - <I>| / ΣIobs, where Iobs is the intensity measurement and <I> is the mean intensity for multiply recorded reflections. c Rcryst and Rfree = Σ|Fobs - Fcalc| / Σ|Fobs| for reflections in the working and test sets, respectively. b 71 strands (39). Iron ion coordination in 2OG oxygenases has been observed to occur in an octahedral or distorted octahedral manner with the iron ion ligated by the conserved facial triad from HX(D/E)XnH at the centre of the DSBH. This facial triad consisting of 2-His and 1carboxylate residues ligating the iron ion is not only conserved among the 2OG oxygenase but also in most non-heme mononuclear Fe2+ enzymes (159). As expected, the conserved metal binding triad of YcfD — H125, D127 and H187 — are found to ligate an iron ion along with two ordered water molecules. The sixth coordination site of the YcfD iron ion is vacant in the crystal structure, which is likely due to resolution limitations as, in similar crystal structures, this site is typically occupied by a water molecule (59),(60). The 2OG cofactor is known to chelate Fe2+ in a bidentate manner and the water molecules likely correspond to the positions of the 2-oxo and 1carboxylate groups of 2OG molecule when it is bound (59),(160). Immediately following the cupin domain are three α-helices that form an α-helical arm, which upon analysis of crystal packing was revealed to mediate dimerization of YcfD with a symmetry related molecule from neighbouring asymmetric unit (Figure 3-1B). The YcfD dimer is formed by intimate hydrophobic interactions between the α-helical arms of each subunit with the two monomers related by rotation around an axis resulting in the active site of each monomer being oriented in opposite directions (Figure 3-1B). The resulting buried surface area of 6150 Å2, which represents 19.5% of the total solvent accessible surface of the protein as calculated by the PDBePISA server (http://www.ebi.ac.uk/msd-srv/prot_int/pistart.html) (127), is large in comparison to proteins with a similar molecular weight (161). A similar dimer has been observed in other 2OG oxygenases including hypoxia-inducible factor asparagine hydroxylase (FIH) (162) and human TYW5 (163), with the YcfD dimer more closely resembling that of FIH as, in both 72 dimers, the active sites point in opposite directions, while in TYW5, the active sites are oriented perpendicularly. The C-terminus of YcfD forms a small domain comprised of four α-helices and four short β-strands. This domain is not commonly found in all 2OG oxygenases, and may be important in determining the function of YcfD. As is discussed later, this domain is conserved among structural homologues and may be of importance for functional determination. 3.4.2 YcfD binds 2OG To examine whether YcfD binds 2OG, we used ITC to assess the interaction. The binding of 2OG to YcfD was observed to occur in an exothermic manner (Figure 3-2A). The dissociation constant (Kd) was determined to be 55.6 ± 2.6 μM, which is slightly weaker than those previously reported for other 2OG oxygenases (164). Though the 2OG binding affinity is lower than other similar enzymes, it can still be considered physiologically relevant as the concentration of 2OG in E. coli has been reported to range from 0.2 mM to 2.3 mM. However, we cannot rule out the possibility that YcfD may co-purify with small amount of 2OG bound as has been observed for two recombinant 2OG oxygenases (165),(166). This would result in an artificially weaker affinity due to partial occupation of the 2OG binding site. As shown in other 2OG oxygenases, the 2OG cofactor binds within the DSBH with its 1carboxylate and 2-oxo groups coordinating the iron ion. Hydrogen bonds between the 5carboxylate and a basic residue (Arg or Lys) as well as a Ser or Thr residue provide further stability for 2OG binding. In YcfD, based on sequence conservation and structural analysis, R140 and S116 were identified as the likely 2OG-binding residues (Figure 3-2D). Active site variants possessing alanine substitutions for either S116 or R140 were prepared and assessed for their ability to bind 2OG using ITC under the same conditions as the native protein. In both cases, the 73 Figure 3-2 Isothermal titration calorimetry of YcfD binding to 2-oxoglutarate (A) ITC data for native YcfD titrated with 2OG. Both the baseline-corrected raw data (top) and a graph of the integrated heat change per injection (bottom) are shown. The fitted curve (red), obtained using non-linear regression, was used to calculate the dissociation constant (Kd = 55.6 ± 2.6 μM). (B) and (C) ITC data for S116A and R140A variants. No heat change was observed upon titration with 2OG, indicating lack of interaction. D) Location of S116 and R140 residues within the active site of YcfD is shown in relation to the metal binding site. 74 alanine substitution resulted in a loss of binding under the same conditions and confirmed the critical role of these residues in 2OG binding (Figure 3-2B,C). 3.4.3 YcfD is structurally similar to human ribosome assembly proteins A structure homology search of the PDB using PDBeFOLD (125) revealed that YcfD is similar to a number of 2OG oxygenases. The top match was a putative asparaginyl hydroxylase from Bacillus subtilis (PDB ID: 1VRB, RMSD = 2.72 Å); unfortunately this provided little functional insight into YcfD due to a lack of functional data associated with the 1VRB structure. YcfD is also homologous to a human tRNA modification protein, TYW5 (PDB ID: 3AL6, RMSD = 2.60 Å). The two enzymes share the DSBH catalytic domain and have similar helical arms that promote dimerization, but TYW5 does not possess a domain similar to the C-terminal domain of YcfD. The TYW5 enzyme hydroxylates wybutosine, a nucleoside found in phenylalanine tRNA, and must bind to the tRNA molecule; as such it is largely positively charged at the active site (49),(163) (Figure 3-3D). Since YcfD possesses a very different charge distribution at the active site and has an extra domain, tRNA modification is an unlikely function for YcfD. Of more functional interest was the structural homology of YcfD to the catalytic domains of Mina53 (PDB ID: 2XDV, RMSD = 2.19 Å) and human NO66 (PDB ID: 4DIQ, RMSD = 2.96 Å). Mina53, or Myc-induced nuclear antigen, also known as NO52, is a 53 kDa nuclear protein with observed concentration in the nucleolus that is upregulated in a number of cancers in response to transcriptional activation by c-Myc, and is known to be involved in cellular proliferation (167-175). Mina53 interacts with precursors to both the large and small ribosomal subunit leading to the conclusion the it likely plays an essential role in ribosome biogenesis with evidence that it plays a role in the later stages of preribosomal particle processing (176). Human 75 NO66 is a conserved nucleolar protein that immunoprecipitates with preribosomal particles but is absent from cytoplasmic ribosomes and, thus, has been proposed to be involved in ribosome biogenesis (177). The active site of YcfD is observed to be a deep pocket localized at the metalbinding site within the cupin domain. It has two patches of charge, but is otherwise largely hydrophobic (Figure 3-3A). Comparison of the active site pockets in human NO66 and Mina53 revealed a very similar pocket in both shape and charge (Figure 3-3B,C). The overall sequence homology between YcfD and both Mina53 and NO66 is only 15%; however, structure-based sequence alignments reveals conservation of the metal and 2OG-binding residues as well as a number of active site residues, providing further evidence that YcfD may be involved in bacterial ribosomal assembly (Figure 3-4). 3.4.4 YcfD Pulls-Down Ribosomal Protein RL-16 of the 50S subunit Both structural homologues NO52 and NO66 have been demonstrated to immunoprecipitate ribosomal proteins (176,177). To investigate whether YcfD is also involved in ribosomal biogenesis, we carried out pull-down experiments. Lysates from BL21 (DE3) E. coli were mixed with recombinant GST-YcfD and subjected to “pull-down” using glutathione sepharose resin. SDS-PAGE analysis showed a single band which co-eluted from the resin with the GST-YcfD, implying a putative interaction (Figure 3-5A). Tandem sequencing mass spectrometry resulted in the sequences of two peptides (Figure 3-5B), which led to the identification of the band as ribosomal protein L16 (RL-16) (Mascot score 174). The peptide sequences represented approximately 25% of the overall sequence of RL-16, indicating a significant match (Figure 3-5B). 76 Figure 3-3 Structural comparisons of (A) YcfD, (B) Mina53 (PDB ID 2XDV), (C) NO66 (PDB ID 4DIQ) and (D) TYW5 (PDB ID 3AL6). The cartoon representation is shown on the left, coloured as in Figure 3-1, and the surface representation is shown on the right, with areas of negative charge coloured red and positive charge coloured blue. All four proteins have a similar fold. Examination of the active site pocket reveals that, despite low sequence homology, the overall shape, size, localization and charge distribution are maintained in the active sites of YcfD, Mina53 and NO66, but is vastly different in TYW5. 77 Figure 3-4 Structure based multiple sequence alignment of YcfD, Mina53 and NO66. Secondary structure elements of YcfD are shown above the alignment and are coloured as in Figure 3-1; arrows and cylinders represent β-strands and α-helices, respectively. Conserved residues are marked with an asterisk, while semi-conserved residues are marked with a colon, periods indicate similar residues. The metal and 2OG binding residues are highlighted in red. Sequences were aligned using Expresso (178). 78 Figure 3-5 YcfD interacts with a ribosomal protein. (A) BL21 DE3 cell lysates were pulleddown with GST-YcfD and glutathione sepharose resin (lane 3-5) or a control of glutathione sepharose resin alone (lane 2). Pull-down with GST-YcFD consistently resulted in a 16 kDa (arrow) band that was not seen in the control or in the GST-YcfD alone fraction (lane 1). (B) Sequences of two peptides resulting from an in-gel digest of the identified band followed by mass spectrometry with their respective Mascot scores (157). (C) The amino acid sequence of RL-16 is shown with the two peptides highlighted in red. 79 The role of RL-16 is to organize the architecture of the aminoacyl-tRNA binding site within the 50s subunit of the bacterial ribosome (179). It is an essential component of the bacterial ribosome since a lack of RL-16 resulted in defects in many aspects of ribosome function, including association with the 30S subunit (180), binding of aminoacyl-tRNA (181), peptidyl transferase activity (182),(183), peptidyl-tRNA hydrolysis activity(184) and interactions with antibiotics (185-187). Structural studies show that RL-16 is inserted between two helices of the 23S rRNA that, together with RL-16, form part of the aminoacyl-tRNA binding site (188),(189). Interestingly, RL-16 is known to be a late-assembly component of the 50S subunit (190), which is consistent with evidence that NO52 is involved in the late stages of ribosomal biogenesis. The pull-down of RL-16 combined with the structural homology to ribosomal assembly proteins provides strong evidence that YcfD is likely a bacterial regulator of ribosome biogenesis. Further experiments are required to validate and examine the interaction of YcfD with RL-16, and to determine what specific role YcfD is playing in the regulation of ribosome assembly. 3.4.5 Growth effects of YcfD overexpression Previous studies have demonstrated that induction of GFP-tagged YcfD expression inhibits cell growth in E. coli (191). To examine the effects of YcfD on E. coli growth and viability further, a colony-forming assay was used to compare the growth of a knock-out strain, ΔycfD (158), to cells overexpressing YcfD to an overexpression control in the same vector. Cells from overnight cultures were diluted 10000 times and streaked onto LB-agar plates with appropriate antibiotic and 0.2 mM IPTG. Induction of YcfD expression resulted in a dramatic loss of colony formation (Figure 3-6). The overexpression control, E. coli quinol monooxygenase, showed significantly more colonies than YcfD, indicating that the loss of cell growth is not due to 80 Figure 3-6 Overexpression of YcfD inhibits cell growth. BL21 DE3 cells transformed with (A) pET21b-ycfD, (B) pET21b-YgiN expression vector or (C) ΔycfD E. coli cells were plated on LB agar plates containing 0.2 mM IPTG and incubated at 37°C overnight. The plates were qualitatively assessed to compare colony formation ability. 81 protein overexpression, as both expression vectors produce similar levels of protein. The ΔycfD strain produced a large number of colonies indicating the ycfD gene is not required for cell survival. A role for YcfD in regulation of ribosome assembly through modification of RL-16 is consistent with the observed growth defects upon induction of YcfD overexpression. As previously noted, RL-16 is involved in late ribosome assembly and organization of the aminoacyl-tRNA binding site in the 50S subunit. Though in vitro reconstitution of ribosomes deficient in RL-16 have been shown to be capable of peptide synthesis (192), no E. coli mutant strain deficient in RL-16 has been identified, despite a systematic search, indicating that, in vivo, RL-16 is likely essential to bacterial ribosome function (190). Furthermore, the yeast mitochondrial homologue of RL-16 has been shown to be an essential ribosomal protein through gene disruption experiments (193). As such the toxic effects of YcfD could be an indication that it inhibits ribosome assembly, and thus ribosome function, through disruption of RL-16 addition to the assembling 50S subunit. Our current speculation is that if YcfD were to act to modify RL16 (e.g. hydroxylation), such a modification would impede RL-16’s insertion into 23S rRNA through, for example, steric hindrance or chemical incompatibility. 3.4.6 Summary Here we present the X-ray crystal structure of a novel 2OG/Fe2+ dependent dioxygenase, YcfD. We confirmed that YcfD does in fact bind to 2OG, and identified the residues responsible for binding. Based on structure homology to human ribosomal assembly proteins and the fact that YcfD pulls-down a component of the 50S ribosomal subunit, we propose that YcfD is involved in regulating bacterial ribosome assembly. Further investigation into the exact mechanism of regulation is required. 82 Chapter 4 Identification of a Novel Di-iron Oxygenase, PhnZ, with a Unique Mechanism for C-P bond Cleavage 4.1 Abstract Phosphorus, in the form of inorganic phosphate is essential for many biological functions and can be a limiting nutrient in a number of microbial environments. In these environments organophosphonates, which possess a highly stable carbon-phosphorus (C-P) bond in the place of the more labile ester bond of organophosphates, are found to make up a significant portion of the total dissolved phosphorus. In order to utilize organophosphonates as a phosphate source, microorganisms require mechanisms to liberate phosphate from organophosphonates. Recently, PhnY and PhnZ have been identified as a novel enzyme pair for C-P bond cleavage. PhnZ, a member of the HD superfamily of metal dependent hydrolases, was identified as the enzyme responsible for C-P bond cleavage in an iron dependent reaction. Here, we present the X-ray crystal structure of PhnZ in both its apo and substrate-bound forms. The crystal structures revealed a previously unknown di-iron binding site and structural homology to myo-inositol oxygenase, which led to the annotation of PhnZ as a novel di-iron oxygenase. Based on the substrate-bound structure a number of key residues were identified, and their contribution to activity was assessed. The structures also revealed a potential role for an active site tyrosine residue (Y24) in the protection of the di-iron cofactor from oxidation, and a mechanism for active site closure upon substrate binding. Our work has enabled us to propose a structure-based enzymatic mechanism for C-P bond cleavage. 83 4.2 Introduction Phosphorous, as an element, is essential for many biological processes including regulation of cellular signaling pathways, generation of metabolic energy, and is a critical component of nucleic acids and membrane phospholipids. Typically, in biological systems phosphorous is found fully oxidized as inorganic phosphate (Pi) and its organic esters, amides, and anhydrides. Organophosphonates represent an important alternative source of Pi, which occur as both natural products and synthetic components of agricultural and medicinal agents (194,195). They possess a highly stable carbon-phosphorus (C-P) bond and constitute the primary source of Pi in numerous phosphate-poor environments, particularly in aquatic ecosystems where Pi concentrations may drop to picomolar levels. In such environments organophosphonates are found to comprise a large portion of the total soluble phosphorous (196,197). Microorganisms living in these Pi-limiting environments have evolved mechanisms to utilize organophosphonates in the place of Pi containing compounds. The most abundant naturally occurring organphosphonate, 2-aminoethylphosphonic acid (AEP), is found to replace phosphocholine to form phosphonolipids in the membrane of numerous organisms, including phytoplankton (194,198,199). In order to use organophosphonates as a source of Pi microorganisms require a mechanism to cleave the C-P bond. To date three mechanistic classes of C-P bond cleavage enzymes have been identified (194,196,200). In the first mechanism alkylphosphonates with a β-carbonyl group undergo C-P bond cleavage through a direct nucleophilic attack on the phosphorus center, resulting in the formation of a carbanion intermediate that is stabilized by a Schiff base or a metal ion. Included in this class of enzymes are such examples as phosphonatase, phosphonopyruvate hydrolase, phosphoenolpyruvate phosphomutase, and phosphonoacetate hydrolase (196,201). The second 84 mechanism for C-P bond cleavage in microbes is catalyzed by C-P lyase, which is encoded by the 14-gene phn operon (phnCDEFGHIJKLMNOP) (202). C-P lyase catalyzes the cleavage of C-P bonds in a variety of organophosphonates through reaction with ribose to form 5-phospho-α-Dribosyl-1-alkylphosphonate, which yields 5-phospho-α-D-ribosyl-1,2-cyclic phosphate and an alkane after C-P bond cleavage (203). C-P bond cleavage has recently been shown to be catalyzed by the [4Fe-4S] cluster containing PhnJ enzyme through a radical reaction using Sadenosylmethionine as a cofactor (204). Recently, a third class of phosphonate utilization enzymes was discovered from a functional screen of marine metagenomic DNA. In this screen a fosmid library of the metagenomic DNA was screened for genes capable of complementing the growth of a Δphn E. coli strain on AEP as a sole phosphate source (205). Along with identifying the presence of a gene encoding for a phosphonatase enzyme, the screen identified genes phnY and phnZ encoding a novel pair of C-P bond cleaving enzymes. The PhnY and PhnZ pair is specific for AEP, and together cleave the C-P bond to release Pi (Figure 4-1). PhnY is homologous to the 2oxoglutarate/FeII-dependent oxygenase family of enzymes that include phytanoyl-CoA dioxygenases and halogenases (205). Recent work has shown that PhnY catalyzes the hydroxylation of the α-carbon of AEP to form 2-amino-1-hydroxyethylphosphonic acid (1-OHAEP) (200). PhnZ is homologous to an uncharacterized subclass of the HD hydrolase superfamily of proteins, which are characterized by the presence of a conserved HD motif that is known to coordinate a metal-ion required for activity (206). Interestingly, the phnZ gene is found to be associated with the phn operon in several bacterial species (205). PhnZ has been shown to catalyze the oxidation of 1-OH-AEP to Pi and glycine in an Fe(II) dependent manner (200). 85 Figure 4-1 A general reaction scheme for PhnY/PhnZ catalyzed C-P bond cleavage 86 The oxidative PhnZ reaction represents a new strategy for C-P bond cleavage for metabolic release of Pi. In order to further understand the mechanism and function of this enzyme, the X-ray crystallographic structure of both the apo and substrate-bound PhnZ were determined. The structures revealed that PhnZ is a di-iron oxygenase. Based on the substrate bound structure key residues for enzyme function were identified. Comparison of the apo and substrate-bound structures further revealed a large conformational change upon substrate binding which results in active site closure. This work provides novel insights into this C-P bond cleavage mechanism and expands upon the known reactions catalyzed by the HD family of hydrolases. 4.3 Materials and Methods 4.3.1 Expression and purification Recombinant PhnZ with a C-terminal hexahistidine tag was expressed and purified as previously described (200). Briefly, protein was expressed using BL21 (DE3) E. coli in LuriaBertani broth. A selenomethionine derivative was also produced using DL41 (DE3) E. coli grown in LeMaster media (118). PhnZ and its variants were purified by affinity chromatography using batch elution from nickel–nitrilotriacetic acid agarose resin (Qiagen, Hilden,Germany). Protein containing fractions were pooled and further purified by size-exclusion chromatography using a Superdex 200 column (GE Healthcare Life Sciences, Uppsala, Sweden). Protein fractions were pooled and concentrated to 16 mg mL-1 by centrifugation in an Amicon Centricon (Millipore, Billerica, MA, USA). The expression and purification of the native and selenomethione proteins resulted in 15 and 13 mg of pure protein samples per litre of culture, respectively. All mutant PhnZ proteins were produced as described for the native PhnZ. 87 4.3.2 Crystallization and X-ray data collection Crystals for the selenomethionine derivative of PhnZ were grown using the hanging drop vapour diffusion method, and typically grew after 5-7 days at room temperature. The final condition contained 2.4 M ammonium sulfate and 0.15 M potassium sodium L-tartrate in the well. The reservoir solution was mixed in a 1:1 ratio (5 μL + 5 μL) with 740 μM PhnZ (16 mg ml-1) and 1 μL of 5% n-octyl-β,D-glucoside (BOG) was added to the drop. For co-crystallization with substrate, PhnZ was mixed with racemic mixture of 2-amino-1-hydroxyethylphosponic acid in a 1:20 molar ratio and grown using sitting drop vapour diffusion with a 1:1 ratio of protein : substrate complex to well (1 μL + 1 μL). The final crystallization condition contained 0.1 M BisTris, pH 6.5 and 20% (w/v) polyethylene glycol (PEG) 5000 monomethylether. All diffraction data were collected at 100 K, using the well solution plus 20% glycerol as a cryoprotectant. Diffraction data for both apo and substrate-bound crystals were collected at the 23 ID-B beamline at the Advance Photon Source (Argonne National Laboratory, Chicago, IL, USA) equipped with a MARMOSAIC 300 CCD detector. Data were indexed and scaled using XDS (153). The selenomethionine derivative crystals belonged to the primitive orthorhombic space group P21212 with a unit cell of a = 104.7, b = 76.0 and c = 60.7 Å. The substrate-bound crystals belonged to the primitive orthorhombic space group P212121 with a unit cell of a = 66.6, b = 74.9 and c = 75.8 Å. Both structures had two molecules in the asymmetric unit with Mathews coefficients of 2.8 Å3Da-1 (55.5% solvent) and 2.2 Å3Da-1 (42.8% solvent) for the apo and substrate-bound structures, respectively (120). 4.3.3 Structure solution and refinement The structure of PhnZ was solved by the multiple anomalous dispersion (MAD) method using a single selenomethionine crystal that diffracted to 1.7 Å resolution. AutoSHARP (154) 88 was used to determined the position of 14 of the 15 selenium atoms, carry out phase extension, and automatically build 343 of the 392 residues of PhnZ. The remaining residues were manually built in Coot (155) and iterative refinement was performed using PHENIX (156). The Dali server (http://ekhidna.biocenter.helsinki.fi/dali_server/) was used to search for structural homologues of PhnZ in the protein data bank (PDB) (207,208). The 2.1 Å structure of substrate-bound PhnZ was solved by molecular replacement using MOLREP (209) using a single PhnZ monomer as a search model then refined using PHENIX. Missing loops were manually built in Coot and 1-OH-2AEP was subsequently added to the unambiguous difference density in the active site. Coordinates for R-2-amino-1-hydroxyethylphosphonic acid were generated using Prodrg2 server (210). 4.3.4 Generation of PhnZ variants The standard protocol for the QuikChange site-directed mutagenesis kit (Stratagene, Cedar Creek, TX, USA) was used to change PhnZ active-site residues to yield the following variants: Y24F, Y24E, E27A, H34A, H58A, D59A, H62A, H80A, H104A, and D161A. Expression and purification of all PhnZ variants were the same as the native PhnZ. 4.3.5 Detection of product formation Formation of glycine by PhnZ and active site variants was detected as previously described (200). Briefly, 5 μM enzyme was combined with 1 mM (±)-2-amino-1hydroxyethylphosphonic acid and allowed to react for 16 hours at 30°C. A sample was removed and mixed with 10 mM sodium dithionite, 5 mM EDTA and 20% (v/v) D2O, and subjected to 31P NMR to assess product formation. The relative amount of product was qualitatively assessed. 89 4.4 Results 4.4.1 Structure determination The novel marine phosphonate utilization enzyme, PhnZ, was recombinantly expressed and purified from E. coli. Interestingly, upon concentration the protein developed a pink colour which was visible above 2 mg mL-1 and increased in intensity with increasing protein concentration, indicating that the colour was associated with the protein. Crystals of apo PhnZ were found to maintain this pink colour (Figure 4-2). Addition of a 20 times molar excess of the racemic substrate caused a colour change from pink to yellow, resulting in the formation of yellow crystals. The crystal structure of PhnZ was solved to 1.7 Å resolution by MAD using selenomethionine labeled enzyme. The structure was refined to an Rcryst and Rfree of 17.2 and 19.8%, respectively (Table 4-1). Crystals belonged to the P21212 space group with two molecules of PhnZ per asymmetric unit. The final model contained 378 of 392 residues, with no electron density apparent for residues of the N-terminal methionine or the C-terminal histidine tag in both molecules. Additionally, electron density was missing for a number of side chains and as such they were truncated at the β-carbons (i.e. Ala). In the structure, 97.6% of residues were in the preferred and 2.4% in the allowed regions of the Ramachandran plot with no outliers. The final model contained 376 water molecules, two L-tartrate molecules, an n-octyl-β-D-glucoside (BOG) molecule, a glycerol molecule, 2 sulphates and a sodium ion from the crystallization buffer. There were also 2 peaks of strong electron density at the HD motif in each active site indicating the presence of bound metal ions. These metal ions were modeled as iron based on the detection of 1.2 mol of Fe per mol PhnZ, and by ICP-MS (200). ICP-MS also showed 0.5 mol of calcium per 90 Figure 4-2 Crystals for (A) apo PhnZ and (B) PhnZ in the presence of (±)-2-amino-1hydroxyethylphosphonic acid were grown aerobically by vapour diffusion. The apo protein is pink in colour, which changes to yellow in the presence of substrate. Black bar represents 0.1 mm. 91 Table 4-1 PhnZ data collection and refinement statistics. Data Collection Space Group Unit Cell: a, b, c (Å) P21212 P212121 104.67, 76.04, 60.71 66.63, 74.91, 55.81 Peak Inflection Remote Wavelength (Å) 0.9794 0.9796 0.9778 1.0332 Resolution (Å) 20.0-1.70 (1.74-1.70) 5.7 (53.8) 20.0-1.70 (1.74-1.70) 5.7 (66.6) 20.0-1.90 (1.95-1.90) 5.3 (44.8) 20.0 – 2.10 (2.20-2.10) 11.1 (62.9) 14.2 (2.4) 14.4 (2.0) 17.1 (3.1) 12.0 (3.6) Unique Reflections 102392 103096 73994 22671 Completeness (%) 99.4 (99.7) 99.4 (99.7) 99.3 (98.4) 99.6 (99.7) 3.5 (3.4) 3.5 (3.4) 3.5 (3.4) 7.3 (7.4) b Rmerge I/σI Redundancy Refinement Statistics Sigma cutoff for refinement None None Rcryst 0.172 (0.247) 0.196 (0.219) Rfree 0.198 (0.274) 0.253 (0.304) 53980 22669 5.1 5.0 3461 2962 189/189 184/183 4 4 376 138 Mean B factor (Å ) 21.1 31.4 RMSD bond lengths (Å) 0.009 0.012 RMSD bond angles (°) 0.941 0.993 Preferred (%) 97.6 96.3 Allowed (%) 2.4 3.7 Outliers (%) 0 0 c c Number of reflections used Number of reflections test set (%) Number of non-hydrogen atoms Number of amino acids Number of iron molecules Number of water molecules 2 Ramachandran Plot a Values in parentheses are for the outermost shell Rmerge = Σ|Iobs - <I>| / ΣIobs, where Iobs is the intensity measurement and <I> is the mean intensity for multiply recorded reflections. c Rcryst and Rfree = Σ|Fobs - Fcalc| / Σ|Fobs| for reflections in the working and test sets, respectively. b 92 mol PhnZ, however refinement with Ca2+ was not successful. It is likely that PhnZ is not fully saturated upon purification, and only the fully saturated enzyme is crystallized. The crystal structure of PhnZ bound to the substrate 1-OH-AEP was solved by molecular replacement using the apo PhnZ structure. A monomer of PhnZ was used to search for 2 copies in the asymmetric unit. The final model was refined to an Rcryst and Rfree of 19.6 and 25.3% and contained 367 of 392 residues. Electron density was not observed for the N-terminal methionine and the C-terminal tag in both molecules. Additionally no electron density was observed for residues116-117 and E139 of molecule A as well as residues 25-30 and D66 of molecule B. In the final model 96.3% and 3.7% of residues were in the preferred and allowed regions, respectively, of the Ramachandran plot. 4.4.2 Overall structure of PhnZ The overall structure of PhnZ is comprised of 11 α-helices folded into a single domain (Figure 4-3A). In addition to the metal ion density within each active site of the apo protein, there was unmistakable density for a bound L-tartrate molecule from the crystallization buffer (Figure 4-3B). In the active site of each molecule of the substrate bound PhnZ there was unambiguous density for the R-enantiomer of the 1-OH-AEP (Figure 4-3C). The selection of the R-enantiomer is consistent with unpublished results that PhnZ is competent with the R- but not the S-enantiomer (Fern McSorley, personal communication). In both the apo and the substrate-bound structures, PhnZ was a dimer in the asymmetric unit, which is inconsistent with size-exclusion results indicating that it is a monomer in solution. In the case of the apo protein the crystallographic dimer is formed by interaction of a molecule of BOG between α10 of molecule A and α9 of molecule B. The BOG is bound through hydrogen bonding and hydrophobic interactions to both molecules (Figure 4-3D). Further stabilization is provided by side chain interactions from α2 of 93 Figure 4-3 Crystal structure of PhnZ. (A) The cartoon representation of the overall monomeric structure of apo PhnZ. The helices are labeled α1-α11, with the helices that provide Fe ion ligands highlighted in pink. Fe ions are shown as orange spheres. (B) Electron density for the bound L-tartrate (green sticks) and (C) R-2-amino-1hydroxyethylphosphonic acid (yellow sticks). The electron density represents an omit map contoured at 2σ (D) Dimer interface of Apo PhnZ where ligands are shown as sticks, with the BOG and sulphate (SO4) involved in dimerization labelled. Molecule A is coloured as in part A while Molecule B is solid teal. (E) Dimer of substrate bound PhnZ, Helices that contribute to metal binding are colour purple in molecule A, while molecule B is solid blue. Residues involved in dimerization are shown as sticks. Hydrogen bonds are shown as yellow dashed lines. 94 molecule A and α5 /α6 of molecule B. The dimer is additionally supported through hydrogen bonding with solvent molecules and sulphate ions from the crystallization buffer. The crystallographic dimerization of the substrat-bound PhnZ occurs in a different manner than that of the apo PhnZ. The interaction between the two monomers is much less intimate with 3 hydrogen bonds occurring directly between side chains of α2/α10 of molecule A and α1 of molecule B, and 3 hydrogen bonds coordinated by an ordered water molecule (Figure 4-3E). 4.4.3 PhnZ is a di-iron oxygenase Structural homology searches using Dali (207) revealed that PhnZ is similar to a number of HD domain containing proteins of unknown function. Interestingly PhnZ is most similar to mouse myo-inositol oxygenase (MIOX, PDB ID: 2HUO, RMSD = 2.7Å) (Figure 4-4). MIOX is an HD domain containing protein that catalyzes the oxidative cleavage of a carbon-carbon bond in myo-inositol to form D-glucuronic acid, and has recently been shown to be a di-iron, Fe(II) / Fe(III) dependent oxygenase (91). The α-helical core of the two proteins align very well, with the most significant difference being that PhnZ lacks the extensive series of loops that are predicted to be essential for MIOX interaction with the next enzyme in the myo-inositol catabolic pathway (69). Also, in the MIOX structure, which is bound to its substrate, there is a loop closing the active site to solvent, while the active site of the apo PhnZ is open to solvent. Despite overall low sequence conservation between MIOX and PhnZ (16% identity) there is remarkable conservation of the metal-binding residues within the active site in both the residue type and geometry (Figure 4-4B). This homology, combined with the fact that electron density for two iron ions is observed in the active site of PhnZ identifies PhnZ as a novel di-iron oxygenase. 95 Figure 4-4 Structural homology to MIOX reveals PhnZ is a di-iron oxygenase. (A) Overall structural alignment. The apo PhnZ structure is coloured as in Figure 4-3A, MIOX (PDB ID 2HUO ) is coloured in light blue. Fe ions are modeled as orange (PhnZ) or Yellow (MIOX) spheres (B) Alignment of Fe ion binding residues coloured as in part A. Water molecules are shown as red (PhnZ) or pink (MIOX). Alignment prepared using DaliLite (132). 96 4.4.4 The di-iron site The two Fe ions are bound within a pocket near the core of the apo PhnZ structure, which is enclosed by 2 sets of antiparallel helices (α2/α3 and α5/α6) as well as a fifth helix (α10) that contribute the ligands for coordination of the Fe ions (Figure 4-3A). As is observed in MIOX (69), the Fe ions are doubly bridged by the carboxylate group of the aspartate residue of the HD motif (D159), which symmetrically binds through Oδ1 and Oδ2 and a bridging water or hydroxide molecule for which clear difference density is observed. The distance between the two Fe ions is similar in both PhnZ molecules of the apo structure (3.76 and 3.72 Å) and PhnZ molecule A of the substrate bound structure (3.74 Å). These distances are consistent with previously reported di-iron structures (36,69). In molecule B of the substrate bound structure the inter-iron distance is slightly longer at 3.83 Å. The first Fe ion (Fe1) is ligated in a distorted octahedral geometry by Y24, H34, H58, D59, D161 and the bridging water molecule. In the case of molecule A of the substrate bound structure the Y24 ligation site is occupied by a water molecule. PhnZ ligates the second Fe ion (Fe2) in an octahedral geometry through interactions with D59, H80 and H104 side chains as well as the bridging water. In the apo structure Fe2 is further coordinated in a bidentate fashion by adjacent carboxylate and α-hydroxyl oxygens of tartrate (O1 and O2). These sites are occupied in a similar fashion by the α-hydroxyl and phosphoryl oxygen of 1-OH-AEP in the substrate bound structure. The iron-ligand distances range from 1.87 – 2.38 Å (Table 4-2). In molecule B of the substrate bound structure the di-iron site is found to be more asymmetric than for the other molecules, with a 0.2 Å shift of the bridging water molecule closer to the Fe2 ion, and a slight 0.1 Å shift of the bridging carboxylate toward Fe1. 97 Figure 4-5 The di-iron site of tartrate bound (A) and 1-OH-AEP bound (B) PhnZ. Side chains of Fe ion ligands are shown as sticks, and the Fe ions are shown as orange spheres labeled Fe1 and Fe2. The water molecule that displaces Y24 in the substrate bound structure (OW104) is labeled. For clarity, the bridging water is unlabelled. 98 Table 4-2 Fe ion-ligand inter-atomic distances Distance (Å) Iron Ligating Apo_A Apo_B 1-OH-AEP_A 1-OH-AEP_B Fe2 3.76 3.72 3.74 3.83 Y24 2.17 2.24 - 2.19 H34 2.11 2.15 2.22 2.13 H58 2.17 2.18 2.06 2.10 D59 2.08 2.06 2.10 2.02 D161 2.15 2.11 2.08 2.26 Bridging-OW 2.09 2.14 2.15 2.38 OW 104 - - 2.17 - D59 2.08 2.10 2.19 2.17 H80 2.22 2.23 2.25 2.19 H104 2.21 2.20 2.15 2.16 TLA_O1 2.14 2.14 - - TLA_O2 2.04 2.06 - - C1O - - 2.22 2.11 PO4 - - 2.29 2.23 Bridging-OW 2.20 2.10 2.03 1.87 Residue Fe1 Fe2 99 4.4.5 Co-Crystal Structure of PhnZ with substrate reveals conformational change upon substrate binding The two molecules of the crystallographic dimer for both the apo and the substrate-bound structure align well with an RMSD of 0.3 and 0.8 Å, respectively. The substrate-bound and apo PhnZ also align well with an RMSD of 1.7 Å between the two A molecules and 1.5 Å between substrate-bound molecule B and apo molecule A. There are two sites of significant conformational change that occur upon substrate binding that act to close off the active site of PhnZ (Figure 4-6). The first involves a loop connecting α1 and α2 (residues 21-30, the Y24 loop). In the apo structure this loop is in an open conformation with Y24 contacting Fe1 and E27 exposed to the solvent. In molecule A of the substrate-bound structure the loop is now in a closed conformation with Y24 flipped out to the solvent and E27 contacting the 2-amino group of the substrate. E27 likely forms a strong electrostatic interaction to the substrate 2-amino group, which is likely positively charged (pKa ~ 9) in the active site. The loop represents the main site of change between molecules A and B of the substrate-bound structure. In molecule B there is no electron density for residues 25- 30 indicating that the loop is highly mobile. Y24 contacts Fe1 as it does in the apo structure. The second conformational change involves the region between α3 and α5 (residues 5990). In the apo structure D59 is found to be at the end of α3. Upon substrate binding a slight shift results the addition of a single turn to the C-terminus of the helix. The entire region shifts down and closes over the opposite side of the active site to the Y24 loop. In molecule B of the substrate-bound structure there is once again missing density for a single residue (D66) in this region, but the loop is in the closed conformation like the A molecule. The two loops in molecule A come together in the substrate-bound structure to close over the active site and are held there 100 Figure 4-6 Conformational changes observed upon substrate binding. (A) Substrate-bound molecule A (purple) aligned with the apo PhnZ (teal), areas of conformational change are highlight in pink and green, respectively. (B) Surface representation of the apo structure with the cartoon diagram of the substrate bound structure. The conformational change upon substrate binding closes the active site pocket. (C) Alignment of substrate bound molecule A (purple) with MIOX (2HUO, light blue). The conformational change upon substrate binding (pink) results in similar configuration to the loops predicted to play a similar role in MIOX (dark blue). 101 through the interaction of E27 with the substrate and hydrophobic interactions with the Y24 loop. Both of the conformational changes observed upon substrate binding result in a conformation that is more similar to the myo-inositol bound MIOX (Figure 4-6C). 4.4.6 Investigation of key active site residues Both L-tartrate and the substrate R-1-OH-AEP are bound in a similar location in the active site and coordinated by the same set of residues (Figure 4-7). The C1 of the tartrate occupies the position of the C1 of the substrate, with the rest of the tartrate binding where the phosphate of the substrate is found to bind. Interestingly, in molecule A of the substrate bound structure, E27, which is solvent exposed in the apo structure, flips into the active site to interact with the 2-amino group of the substrate, thereby closing the active site. This contact is made possible by the fact that Y24 simultaneously flips out into the solvent. A water molecule is observed to occupy the position of the phenolic oxygen in a similar manner to the water that is predicted to represent the binding site of molecular oxygen in the MIOX structure (69). H62 is found to hydrogen bond with the C1-OH of both the L-tartrate and R-1-OH-AEP in an similar position to the catalytically important K127 of MIOX (69). Based on the co-crystal structure of PhnZ with substrate a number of active site residues were targeted for mutational analysis (Table 4-3). The ability of the active site variants to convert (±)-1-OH-AEP to glycine was assessed by 31P-NMR and qualitatively compared to wild-type PhnZ as previously described (200). Alanine substitutions for metal binding residues resulted in a loss of activity, confirming the importance of both iron ions for catalysis. In some cases substitution of metal ion binding residues (H58, D59 and H80) with alanine resulted in unstable protein that precipitated upon concentration so activity was not assessed. A Y24F substitution 102 Figure 4-7 Comparison of tartrate and substrate binding. (A) A stereo diagram of the apo PhnZ active site. (B) A stereo diagram of the substrate-bound PhnZ active site. Residues involved in metal and ligand binding are shown as sticks. The Fe ions are shown as orange spheres, number 1 and 2. 103 Table 4-3 Activity of active site mutants of PhnZ as measured by 31P-NMR Mutation Activity Colour Proposed Fe1/Fe2 oxidation state Wild Type +++ Pink (II/III) H34A - Yellow (III/III) H58A N/D N/D N/D D59A N/D N/D N/D H80A N/D N/D N/D H104A - Yellow (III/III) D161A + Clear No iron Y24F +++ Yellow (III/III) Y24E + Yellow (III/III) E27A - Pink (II/III) - Pink (II/III) Metal Binding Residues Y24 Loop Residues Ligand Binding H62A a N/D – not determined due to instability or insolubility of protein +++ Wild type activity + low activity - no activity 104 resulted in fully active protein while the Y24E variant showed a significant decrease in activity. Interestingly, substitution of E27 and H62 with an alanine resulted in no product formation. 4.5 Discussion PhnZ, along with PhnY, was identified in a screen for marine bacterial enzymes capable of cleaving the highly stable C-P bond of organophosphonates. The two enzymes comprise a novel pathway for the metabolic cleavage of the C-P bond in AEP, one of the most abundant organophosphonates in nature. Sequence analysis identified PhnZ as a member of the HD hydrolase class of enzymes, which require a divalent metal-ion for catalytic activity. Previous studies indicate that PhnZ requires Fe2+ as a cofactor for conversion of 1-OH-AEP to glycine and Pi (200). The crystal structure of PhnZ, both in its apo and substrate bound forms, were determined, revealing a di-iron active site and providing insight into the mechanism of C-P bond cleavage by this enzyme. Interestingly, in the apo structure, PhnZ is bound to L-tartrate in a similar position to that found for the R-enantomer of the substrate. Unpublished work has shown that PhnZ can cleave the C-P bond of the R-enantiomer but not the S-enantiomer. Strong electron density was observed in the active site associated with the HD motif for not one but two bound metals. Combined with the fact that the closest structural homologue to PhnZ is MIOX, a di-iron oxygenase, the above indicates that PhnZ is a novel di-iron oxygenase. 4.5.1 Di-iron oxygenases Di-iron oxygenases, such as plant fatty acyl desaturases, bacterial multi-component monooxygenases (MMO) and the R2 subunit of conventional class I ribonucleotide reductases (RNR) are a class of non-heme iron oxygenases that activate molecular oxygen using a carboxylate-bridged di-iron cluster to break stable C-H or O-H bonds in their respective substrates (36,65-67,70). These conventional di-iron oxygenases all utilize a fully reduced di-iron 105 (II/II) form of the cofactor. Though MIOX is known to be a di-iron oxygenase, it shows little sequence or structural homology to known members of the di-iron family. Instead, it is homologous to proteins of unknown function belonging to the HD-domain superfamily of metal ion dependent hydrolases (69). Extensive spectroscopic and kinetic analysis of MIOX have led to the discovery that it employs a mixed-valence antiferromagnetic di-iron (II/III) cofactor in its catalytically active state (90). 4.5.2 Di-iron binding site in PhnZ The di-iron binding site and the active site as a whole are highly conserved between MIOX and PhnZ despite low overall sequence homology. The ligands which coordinate the Fe ions in di-iron dependent enzymes define their catalytic activity. For example the RNR, MMO and fatty acyl desaturases ligate the two Fe ions via two His residues and four carboxylates resulting in a higher proportion of negatively charged ligands that are likely necessary to stabilize the predicted high-valence Fe intermediates of the reaction mechanism (36,94,97). The di-iron oxygen carrier protein hemerythrin on the other hand has a more positively charged ligand composition with four His residues and two carboxylates (99). MIOX and PhnZ share four His residues and two Asp residues, which is more similar to hemerythrin than the to RNR oxygenases, indicating that both enzymes potentially do not utilize high-valence Fe intermediates (69). The conservation of the di-iron site between MIOX and PhnZ suggests the PhnZ likely also employs a mixed-valence di-iron (II/III) site as its catalytically active state, with Fe1 as the likely Fe(II) site as predicted for MIOX (69). The only other structurally characterized di-iron enzyme known to employ a mixed-valence cofactor is purple acid phosphatase (93). This is a structurally distinct α/β enzyme that is not an oxygenase and employs a different set of ligand residues. Thus purple acid phosphatase is not functionally analogous to PhnZ. 106 4.5.3 Active site and catalytic mechanism The structure of PhnZ demonstrates, for the first time, that PhnZ possesses a di-iron cofactor and that the substrate (and a substrate analogue, L-tartrate) binds to the Fe2 site in a bidentate manner with a bridging water or hydroxyl molecule. This supports an oxidative C-P bond cleavage mechanism that is analogous to the C-C bond cleavage reaction catalyzed by MIOX (70,200). Although there is much remaining to be elucidated regarding the complete mechanism of PhnZ, the structure provides several important insights, most importantly the conformational change observed upon substrate binding (Figure 4-8). In the L-tartrate bound or apo structure the active site is in an open conformation. The Y24 loop in the open conformation is oriented such that E27 is exposed to the solvent and Y24 is ligating Fe1 of the di-iron site (Figure 4-8A). The position of the phenolic oxygen of Y24 would prevent binding by molecular oxygen as observed at this position in MIOX (69,70). For this reason we initially hypothesized a potential role for a tyrosyl radical in C-P bond cleavage by analogy to the tyrosyl radical formed in the diiron site of RNR (211). However, site-directed mutagenesis indicated that this was unlikely (see below). A surprising result was observed upon binding the R-enantiomer of the substrate in the active site. A large conformational change is observed in two loop regions that close off the active site similar to the active site closure observed in MIOX (69,212). The conformational change in the loop containing Y24 and E27 results in E27 closing over the active site and forming an electrostatic interaction with the 2-amino group of the substrate. This coincides with Y24 flipping out into the solvent and the phenolic oxygen being replaced by a water molecule in a position that is in an analogous position to the predicted binding site of O2 in MIOX (Figure 4-8B)(69). This change was only observed in the A molecule of the substrate bound structure. In 107 the B molecule Y24 remains in coordination with Fe1, and the rest of the loop is disordered. This molecule also shows higher asymmetry in the coordination by the two bridging species, with the bridging carboxylate shifted slightly closer to Fe1 and the bridging water molecule shifted significantly closer to Fe2. There is also a slight increase in the inter-iron distance (~0.1 Å), while the coordination in both the apo and the substrate bound molecule A are more symmetrical, closely resembling the mouse MIOX bound to substrate. A similar asymmetric ligation was observed in the crystal structure of human MIOX bound to an inhibitor, and was attributed to the fact that this structure was inhibitor bound, which could alter the environment of the active site (212). In the case of the substrate bound PhnZ structure, a minor contact of two hydrogen bonds with an ordered solvent molecule and a single side chain interaction with a neighboring molecule stabilize the closed conformation of molecule A, while the molecule B active site is solvent exposed. Though the closed conformation of one PhnZ molecule is stabilized through a crystal contact it most likely represents a biologically relevant form as it more closely resembles the active site of substrate bound MIOX. Moreover, E27 provides a productive binding interaction with the substrate, while triggering expulsion of Y24 from the active site. This removes the steric obstacle to O2 binding at this Fe ion which was observed in the L-tartrate bound structure of PhnZ. The second conformational change occurs in the loop between α3 and α5. This loop is anchored on each end by metal binding residues (D59 and H80) (Figure 4-6). The loop shifts over the active site forming hydrophobic interactions with the closed conformation of the loop containing Y24 and E27. The new conformation is further stabilized by hydrogen bonds between the main chain of two sections of the loop that fold back on each other. Within this region is the critical substrate binding residue, H62. Binding of substrate to this residue as well as the metal 108 Figure 4-8 Key steps in the PhnZ catalyzed C-P bond cleavage of 1-OH-AEP, modified from McSorley et al. (200). (A) The hydroxyl of Y24 protects the reduced state Fe1. (B) Substrate binding to Fe2 results in E27 moving into the active site to coordinate the 2-amino group of the substrate, causing Y24 to move out of the active site. (C) With Y24 out of the way an Fe(III)-superoxo species is formed at the Fe1 site by reduction of molecular oxygen. (D) The Fe(III)-superoxo intermediate removes the hydrogen from the C1 carbon initiating C-P bond cleavage, resulting in the production of glycine and Pi. Figure generously provided by David L. Zechel. 109 ligating residues likely initiates this conformational change. As previously stated, the two conformational changes combine to block the active site from the solvent, which would protect the highly reactive free radical intermediates that are formed in the predicted mechanism. It would also expedite the reaction by holding the reactants in the active site. 4.5.4 Active site substitutions Based on the substrate bound structure a number of active site residues were targeted for site-directed mutagenesis to examine their importance for activity. The PhnZ variants H34A, H104A and D161A were found to be inactive while other metal ligand substitutions (D59A, H80A and H58A) resulted in unstable variants that precipitated upon purification. This highlights the importance of both metal binding sites for not only enzyme activity but also stability. Substitution of the substrate binding residue H62 for alanine was also found to abolish activity of PhnZ. This residue hydrogen bonds to the C1-OH of the substrate in an analogous manner to the binding of K127 of MIOX to the O1 of myo-inositol. The Lewis-acid interaction between the C1OH and the Fe(III) ion can be expected to reduce the pKa of the hydroxyl (pKa ~16). This would allow H62 to act as a general base and ionize the hydroxyl group, which would serve to activate the C1-H bond for cleavage. Such a mechanism would explain the essential role of H62 for catalytic activity. Interestingly, an alanine substitution for E27 resulted in a complete loss of activity. Upon substrate binding E27 was found to close the Y24 loop over the active site through an interaction with the 2-amino group of the substrate. This change coincided with the displacement of Y24 from the active site and its ligand interaction with Fe1 replaced with a solvent molecule. PhnZ Y24F was observed to maintain activity while a Y24E substitution showed a significant drop in activity. Together these results indicate that Y24 is not directly involved in the reaction 110 mechanism, such as by forming a tyrosyl radical. This is because Y24F cannot form a radical, and the Y27E variant is expected to bind to the Fe1 with higher affinity, which would inhibit the conformational change required to allow O2 binding and subsequent closure of the active site. This combined with the fact that Y24 is observed to coordinate the predicted Fe(II) ion of the mixed-valence site suggests a role for Y24 in preventing the premature oxidation of the Fe1 ion. This could be very important for maintaining the active form of PhnZ, which does not use external reducing equivalents to reform Fe(II) in the active site (200). 4.5.5 Structural insights for substrate specificity Both crystal structures presented here provide insight into the substrate specificity of PhnZ. It is clear from the binding mode of both L-tartrate and 1-OH-AEP that a bidentate mode is required for iron coordination and orientation of the substrate in the active site. Moreover, although a racemic substrate mixture was used for the formation of PhnZ-substrate co-crystals, only the R-enantiomer bound, which is consistent with earlier findings that PhnZ turns over only 50% of racemic substrate (200) and that PhnZ can only cleave the C-P bond of the R-enantiomer (Fern McSorley, personal communication). Active site analysis shows that the R-enantiomer binds with the C1-hydrogen in close proximity (~2.2 Å) to where O2 is thought to bind, aligning it properly for the initiation of the reaction. Finally, the requirement of the interaction between E27 and the substrate 2-amino group to close the active site was observed structurally and confirmed by site-directed mutagenesis, emphasizing the requirement of this electrostatic interaction for activity. The fortuitous binding of an L-tartrate molecule during the crystallization of the apo protein provided many initial insights to the mechanism of PhnZ prior to the determination of the substrate bound structure, including the mode of substrate binding to the di-iron centre. Though the PhnY/PhnZ enzyme pair have been shown to be specific for AEP (205), the binding of tartrate 111 presents an interesting insight into potential inhibitors for PhnZ or for the possibility of engineering PhnZ to cleave the C-P bond of other organophosphonates. This would be beneficial on multiple levels but particularly in regards to environmental concerns given that commercially utilized toxic organophosphonates are building up in a different ecosystems (213). 112 Chapter 5 General Discussion and Summary 5.1 Importance of metals in metalloproteins Metalloproteins represent a major portion of the total proteome. Unfortunately, despite a large number of metalloprotein structures, and extensive sequence and biochemical analyses, it remains difficult to predict the presence of a metal ion-binding site within a protein and its implication in the protein’s function. It is not uncommon that extensive experiments are required before a metal ion cofactor is identified. This additional effort, however, often pays off considerably: once a protein is identified as a metalloprotein, new dimensions to both the protein’s structure and its function can be explored. Within the context of a metalloenzyme, for example, metal ions offer virtually unlimited catalytic potential and identification of a metal ion cofactor provides important insights into the catalytic mechanism of the enzyme. Here, we have described the structural and functional characterization of three novel microbial metalloenzymes. These results highlight the significance of identifying a protein as a metalloenzyme, as well as the importance of the metal ion cofactor for both activity and stability. For all three enzymes, the presence of bound metal ions was identified from strong difference densities in the electron density maps for the crystal structures. In the cases of EcSI and YcfD, it was known from sequence analysis that the proteins were of the cupin superfamily, and thus potentially bound a metal ion. YcfD in particular was identified as a JmjC-domaincontaining protein which is known to be of the 2OG/Fe2+-dependent dioxygenase family. For PhnZ, although it was previously observed to co-purify with a bound metal ion (200), the crystal structure revealed a second metal ion binding site that was critical for its annotation as a di-iron 113 oxygenase. None of the protein expression systems were supplemented with excess metal ion, and no metal-ions where added to the purification buffers, indicating that all three proteins copurified from the cell with metal ion co-factors. Since crystallography can be considered a form of purification it is possible that the proteins were not fully saturated with metal ion, and the metal ion-bound proteins were selected for by the process of crystal formation. The fact that the proteins co-purify with their metal cofactors illustrates the importance of the metal ion for protein stability and the affinity for the metal ion, which are not mutually exclusive. In the case of PhnZ, substitution of several iron-binding residues with alanine resulted in a loss of protein stability, while for both EcSI and PhnZ substitution of metal ion-binding residues with alanine resulted in a loss of activity. With regards to activity, the structures of EcSI and PhnZ revealed that in both cases the substrate was bound directly to the metal ion, indicating a vital role for the metal ion in catalysis. The sugar isomerization catalyzed by EcSI is proposed to proceed via a cis-endiol intermediate. The coordination of the substrate by the manganese ion would provide a positive charge to stabilize the highly negative charge of the enolate intermediate, allowing isomerization to occur. In the PhnZ-catalyzed C-P bond cleavage of 1-OH-AEP, the two iron ions play distinct roles. The substrate of PhnZ binds directly to the Fe2 atom (predicted to be in the Fe3+ ion state), with the αhydroxyl and phosphoryl oxygen groups binding in a bidentate manner. This binding orients the substrate for removal of the hydrogen from C1 by O2 to initiate C-P bond cleavage. The positive charge of the Fe3+ would help activate the substrate for attack by ionizing the α-hydroxyl with the help of the catalytically essential H62 residue, which in turn would activate the C1-H for removal. The Fe2 is also predicted to play a role in stabilizing the intermediates formed in the reaction. The Fe1 atom is predicted to be in the Fe2+ oxidation state, and does not contact the 1114 OH-AEP substrate. Instead, it is responsible for O2 binding and activation. Together, these succinctly exemplify the catalytic power and versatility of metal-ions. 5.2 Structure and function The three structures presented here also highlight the importance of the 3-D structure for functional determination, particularly for proteins of unknown function, and for elucidating enzymatic mechanisms. Genomic sequencing projects have identified many genes that encode for proteins of unknown function. Primary sequence homology is often found to be insufficient for functional annotation of novel proteins, particularly for proteins belonging to sequentially and functionally diverse superfamilies such as the cupin and non-heme iron oxygenase families. Elucidation of the 3-D structure of a protein provides information regarding location and composition of active site, potential protein-ligand and protein-protein interaction sites, and information regarding the nature of potential ligands. Because the overall fold of a protein is often better conserved than the primary sequence (as is the case with the 2OG oxygenases) comparison of protein structure is a powerful tool to identify distant evolutionary relationships, which, when combined with biochemical data, aids in a positive functional assignment. The E. coli proteins, EcSI and YcfD, are both examples of genes that were annotated by sequencing projects as hypothetical proteins, with no sequence homology to proteins of known function. From sequence analysis, EcSI was identified as a cupin protein, but due to the diversity of the cupin superfamily further functional analysis based on sequence was difficult. Structural comparisons identified a structural homology to cPGIs, though EcSI only possessed nominal cPGI activity. This is consistent with phylogenetic analysis that revealed that cupin proteins of the same function typically bind the same metal for activity (15). The cPGIs have been shown to be Fe2+- or Ni2+-dependent enzymes, and the optimal activity of EcSI was observed in the 115 presence of Mn2+ (26). However, this crucial homology led to the investigation of the sugar isomerase activity of EcSI and the subsequent identification of D-lyxose and D-mannose as the preferred monosaccharide substrates. YcfD was identified as a member of the JmjC-domaincontaining subclass of the 2OG oxygenases, which are among the most functionally diverse classes of proteins. The crystal structure confirmed that the YcfD fold is homologous to the basic 2OG oxygenase fold, and that the 2OG and Fe2+ binding sites are conserved. Structural homology to MINA53 and NO66, two human proteins that have been shown to be involved in ribosomal assembly (176,177), together with evidence that YcfD interacts with the E. coli ribosome, resulted in the putative functional assignment of YcfD as a regulator of ribosome assembly. Prior to structure determination, the function of PhnZ as a novel C-P bond cleaving enzyme was already established; however, the exact mechanism of catalysis remained unclear and awaited experimental investigation. From sequence analysis PhnZ was classified as a member of the HD hydrolase superfamily of metal dependent enzymes (205,206). The crystal structure provided the first evidence that PhnZ possesses a di-iron cofactor, and structural homology to the well characterized di-iron oxygenase MIOX provided evidence that PhnZ employs a mixed-valence di-iron (II/III) cofactor to cleave the highly stable C-P bond to produce glycine and Pi as was previously predicted(200). Interestingly, in both the PhnZ and EcSI apo structures, substrate analogues from the crystallization condition were found to bind within the active sites. In both cases the bound ligand provided important clues for the binding of substrate, including the active site location and the initial identification of catalytically important residues. In the case of EcSI, binding of phenol, a known sugar analogue, offered initial clues as to the identity of the class of substrate. This phenomenon of binding a ligand from the crystallization condition is a fortuitous side effect of 116 crystallization that is not uncommon. One of the examples is PhnP, a phosphodiesterase, where (S)-malate was found to bind in the structure (214). For PhnZ, despite extensive screening, crystals were never observed in the absence of substrate or the substrate analogue, L-tartrate, which could suggest that apo protein is very flexible in the absence of a ligand. This is not surprising as it is often observed that the addition of ligands and cofactors increases the crystallizability of a protein. 5.3 Active site closure upon substrate binding Enzymes lower the activation energy of a reaction in a number of ways, including by providing an ideal site for the reaction to occur, and by preventing the diffusion of intermediates. The flexibility, shape, location, and amino acid composition of the active site all contribute to the creation of this ideal environment. In the three structures discussed here, one common feature observed is active site closure upon substrate binding (Figure 5-1). The substrate/product-bound structures of PhnZ and EcSI revealed conformational changes in the flexible loops surrounding the active site which act to close the active site upon substrate binding to limit solvent exposure. A similar conformational change resulting in active site closure has been observed in various proteins such as MIOX (69), and a number of kinases (e.g. (215-217)). Though no substratebound structure of YcfD has been obtained, it is highly likely that the YcfD active site has very limited solvent exposure upon substrate binding. This is due to the narrow shape of the active site within the DSBH of YcfD, and also due to the fact that the predicted substrate is the 16 kDa protein RL-16, which is expected to make extensive contacts with the area surrounding the active site, essentially plugging the narrow active site pocket. Crystal structures of the 2OG oxygenase FIH bound to a peptide substrate is such one example (162). The side chain of the asparagine residue that is the target of hydroxylation by FIH is bound within the active site, with the peptide 117 Figure 5-1 Active site closure upon substrate binding. (A) surface representation of EcSI with the R205 loop shown in its open (orange) and closed (green) conformation. (B) Surface representation of apo PhnZ with the closed conformation cartoon (magenta) shown closing the active site. (C) The surface representation of YcfD. Charged patches surrounding the active site likely to be involved in RL-16 binding are indicated with orange arrows. In the surface representations, red represents regions of negatively charged residues, blue represents positively charged regions. 118 backbone covering the active site, and the surrounding peptide residues making contact with the surface of FIH. The RL-16-YcfD interaction would likely mimic this, with the target residue binding in the YcfD active site, and the surrounding RL-16 residues making contact with the surface of YcfD, likely to the charged patches that surround the active site (Figure 5-1C). These active site closures would prevent diffusion of the reactants through solvent exchange, and protect any reactive intermediates which might form during catalysis. There are three models of substrate-enzyme complex formation: 1) the Fischer “lock and key” model, where the snug fit between substrate and enzyme active site involves no conformational change; 2) the Koshland induced fit model, which describes a conformational change upon ligand binding; and 3) the selected-shift model, which assumes an equilibrium of conformations for the enzyme from which the ligand selects and stabilizes a particular conformation (218-223). The conformational changes observed in EcSI and PhnZ are both examples of the selected-shift model followed by a ligand-induced conformational change to close the active site. In apo EcSI the R205 loop, which was shown to close the active site upon ligand binding, is highly flexible as demonstrated by a lack of electron density for this loop. This suggests that the loop moves through a number of different conformations between the open and closed forms of the enzyme. The ligand, either phenol or the substrate, would “select” the open conformation for binding. Subsequently, the ligand would “induce” the closed conformation by stabilizing the loop over the active site via interactions with EcSI R205. The conformational change observed in PhnZ is more complex; however the same principles apply. The apo form of PhnZ is likely to be highly flexible, alternating between open and closed conformations, due to the fact that crystallization attempts of PhnZ with no ligand bound were fruitless. The fortuitous binding of L-tartrate from the crystallization condition can be seen as mimicking the 119 “conformational selection” step, where the tartrate has selected for the open form of PhnZ. The tartrate lacks a positively charged group that is required for active site closure. In the case of substrate interaction with PhnZ, the substrate “selects” the open conformation of the enzyme in an analogous manner to tartrate. The positively charged 2-amino group of the substrate forms an ionic interaction with the side-chain of E27 on the Y24 loop. This produces a conformational change that positions the loops so that they surround the active site, allowing oxygen to subsequently bind and fully close the active site, thereby enabling the reaction to proceed. In both EcSI and PhnZ this active site closure has been shown to be essential for catalytic activity, as substitution of the residues involved in stabilizing the closed conformation results in a dramatic decrease in activity. 5.4 Future directions The three proteins discussed here have all provided insight into the structure and function of three important classes of metalloenzymes, as well as highlighting the importance of metals for catalysis. Individually, the structure-based functional analysis for each protein has provided many implications and expanded the knowledge for each respective protein. As is always the case, however, for each question answered many more are raised, and so in this section the future investigations for each protein are discussed on an individual basis. 5.4.1 EcSI E. coli O157:H7 is a potent pathogenic bacterium that has been associated with outbreaks such as the Walkerton tragedy, various manifestations of Hamburger disease, and even as part of the devastation following hurricane Katrina. Understanding the mechanisms of pathogenicity of such bacteria is vital for developing novel therapeutics to combat infection. The gene encoding for EcSI, z5688, is highly conserved amongst pathogenic species, making it a target of interest for 120 functional characterization. The identification of EcSI as a sugar isomerase with specificity for Dlyxose and D-mannose, coupled with the demonstration that over-expression of EcSI allows this bacteria to subsist on the rare pentose D-lyxose, led to the suggestion that this protein may provide a selective advantage in the competition for resources with non-pathogenic bacteria, which do not possess a mechanism for D-lyxose isomerization. More specifically, in order to successfully colonize in a given environment, E. coli O157:H7 needs to compete with other bacteria for resources, and the ability to metabolize multiple carbon sources would provide a competitive edge, as is observed in studies of E. coli O157:H7 that demonstrate this bacteria’s ability to form a biofilm in competition with the indigenous biolayer on spinach leaves (224). It would be of interest to investigate whether E. coli O157:H7 are capable of growth on D-lyxose. It is noted that there are safety challenges to work with live pathogenic strains, which needs to be carried out in specifically designed and certified laboratories. Since the isomerase activity of EcSI for D-lyxose and D-mannose was low in comparison to known D-lyxose isomerases, it is possible that a more ideal substrate exists. A more extensive screen of sugar substrates, including nucleotide sugars and disaccharides could result in the identification of a new substrate. Alternatively, examining the ability of EcSI over-expression in non-pathogenic E. coli to complement cell growth on a panel of carbon sources could provide indications as the exact substrate of EcSI. The genes immediately surrounding the EcSI gene are all hypothetical proteins, which have been assigned functionality based upon sequence homology (Figure 5-2). The three genes upstream of the EcSI gene (z5689-z5691) are predicted to encode components of a ribose ABC transporter, while the z5687 gene is predicted to be a sugar aldolase, and the z5686 gene is predicted to be a kinase of the PFK-b family of carbohydrate kinases. Annotation of the 121 Putative kinase (PFKb) Putative periplasmic ribose-binding protein Putative periplasmic Sugar ribose-binding Isomerase protein (EcSI) z5686 z5688 z5689 z5687 z5690 z5691 Putative ATP-binding protein Putative sugar aldolase ABC Transporter Figure 5-2 A schematic of the genes surrounding the gene for EcSI (z5688, pink) in the E. coli O157:H7 genome. The predicted function for each gene is indicated. 122 surrounding genes or the operon of a particular gene has been shown to aid in functional assignment (225). With that in mind, assessment of the substrate specificity of the genes surrounding EcSI could provide useful insights into the role of EcSI in E. coli O157:H7 pathogenicity and metabolism. 5.4.2 YcfD The crystal structure of YcfD was found to have structural homology to two human proteins involved in ribosomal assembly. Moreover, YcfD interacts with the ribosomal protein RL-16, an essential component of the bacterial ribosome. Together, these results provide strong preliminary evidence that YcfD is also involved in the regulation of ribosomal assembly or translation as a whole. In order to further investigate this role, the interaction between YcfD and RL-16 must first be validated using, for example, size-exclusion chromatography and/or immunoprecipitation experiments. The sequence and structural homology of YcfD to the JmjCdomain-containing subclass of the 2OG oxygenases indicates that YcfD likely hydroxylates a residue on RL-16, as hydroxylation is the most common reaction observed for this class of enzyme, though demethylation is also a viable alternative (39). To identify which residue of RL16 is the target of YcfD, as well as the type of modification that is occurring, the two proteins could be combined with 2OG and Fe2+ and subjected to mass spectrometry. The addition of a hydroxyl to an amino acid in RL-16 will result in a shift of +16 Da, while the removal of a methyl group will result in a -14 Da shift, enabling the identification of the target residue for YcfD modification. Once the target residue is identified a structural basis for the effect of the modification can be proposed based on the structure of the E. coli ribosome (189). The interaction between YcfD and RL-16 can be further studied through cocrystallization. The interaction between the two proteins is predicted to be relatively stable based 123 on the purity of the E. coli cell lysate pull-down; this lends itself well to crystal formation as a stable complex is required to obtain high-quality crystals. Alternatively, an appropriate peptide containing the target RL-16 residue can be designed for complex formation with RL-16. In both cases, measures to prevent enzymatic turnover need to be considered, which could include the use of a 2OG analog and/or substituting the modified residue of RL-16. The complex structure can provide many mechanistic and functional insights, and act as a platform for the design of inhibitors or activators of YcfD function. The potential exists that YcfD may be a therapeutic target, given that RL-16 is the target of a number of antibiotics, and resistance to antibiotics that target the ribosome has been shown to be conveyed by point mutations of RL-16 (226-229). Furthermore, it has been predicted that antibiotics bind directly to RL-16, based on the crystal structures of the bacterial ribosome as well as an RL-16 solution structure (188,230). A role for a JmjC-domain-containing proteins in translational regulation seems like a natural addition to the list of important cellular processes governed by their activity, which thus far includes RNA splicing (231), chromatin remodeling (49), tRNA modification (232) and transcriptional regulation (233). Both MINA53 and NO66 have been reported to play a role in cellular proliferation with a reduction in expression correlated to reduced proliferation (172,174,176,177,234). A similar role may exist for YcfD. However, over-expression of YcfD was observed to result in a dramatic loss of E. coli cell growth, while a YcfD knock-out (ΔycfD) maintains wild-type growth levels. This, combined with the fact that RL-16 is known to be an essential protein, indicates that YcfD is possibly inhibiting ribosome assembly (190). However, due to the fact that the human homologues are promoters of cell proliferation, and efficient translation is required for cell growth, it could be the case that YcfD is also a positive regulator of translation, and over-expression of YcfD results in higher ribosome activity, which in turn 124 produces more YcfD, and results in a dramatic redirection of cell resources to the production of YcfD. These two explanations assume a “black and white” definition of activity; in reality ribosomal assembly and translation are governed by a large number of factors and thus the function of YcfD is likely to be similarly governed. For example, the resources available to a bacterial cell are restricted to its local environment, therefore mechanisms for optimal growth in a number of environments exist, and perhaps YcfD plays a role in translation under particular conditions. Further investigation into the growth of YcfD over-expressing cells and the ΔycfD cell line under a variety of growth conditions could shed light on the precise role of YcfD in cell survival. 5.4.3 PhnZ The identification of PhnZ as a di-iron oxygenase belonging to the HD superfamily of metal-dependent hydrolases extends the known reactions catalyzed by both classes of enzymes. MIOX was the first such enzyme identified and extensive characterization of its reaction mechanism has resulted in the elucidation of a unique mixed-valence iron (II/III) cofactor as the active form for catalysis (reviewed in (235)). Conventional di-iron oxygenases are known to initiate catalysis with a reduced iron (II/II) cofactor (65,94). The valence state of the di-iron cluster is of great significance to understanding the catalytic mechanism of C-P bond cleavage by this enzyme. In the case of MIOX, the electronic properties of the metals were evaluated using electron paramagnetic resonance spectroscopy and Mössbauer spectroscopy (90-92,235). Recently, a synergistic approach using X-ray absorption spectroscopy (XAS) and X-ray crystallography was used for analysis of metalloprotein active sites (236). XAS can be used to probe local electronic and physical structures by measuring the core-level electron excitation by the absorption of X-rays. The precision and resolution of XAS is sufficient to measure fine details 125 such as the formal metal oxidation state in a metalloprotein, and when combined with the crystal structure of PhnZ, XAS could provide details as to the nature of the di-iron cofactor. The kinetic properties of native PhnZ and the active site variants will provide information about the kinetic activity of this protein, and the precise role of various active site residues in catalysis. The kinetics of this reaction can be measured through a simple enzyme coupled reaction with purine nucleoside phosphorylase (PNP), which converts 2-amino-6-mercapto-7methylpurine riboside (MESG) to 2-amino-6-mercapto-7-methylpurine and ribose-1-phosphate in the presence of Pi. The product of the reaction has an absorbance maximum of 360 nm, and an increase in A360 is directly proportional to an increase in Pi concentration (237). The kinetics of PhnZ in the presence of the PhnY enzyme, which catalyzes the formation of 1-OH-AEP, would also be of interest to determine if there is any cooperativity between the two enzymes such as substrate shuttling through transient interactions between the two proteins. The PhnY/PhnZ enzyme pair has been shown to compliment growth of Δphn E. coli with on AEP, and not with methylphosphonic acid or ethylphosponic acid (205). Consistent with this observation, PhnY was unable to hydroxylatate either substrate (200). It is clear from the structure and active site analysis of PhnZ that the 2-amino group of 1-OH-AEP is critical for function. However the identification of PhnZ genes associated with the phn operon in several bacteria that lack PhnY suggests that PhnZ may have the potential to catalyze C-P bond cleavage in the absence of PhnY, or in a variety of substrates (205). Investigation into the ability of PhnZ to catalyze C-P bond cleavage of various phosphonates with an amino group and a hydroxylated α-carbon could indicate promiscuity in the enzyme, which is of environmental interest due to concerns that the stable, commercially used oranophosphonates are building up in different ecosystems creating toxic effects (213) . Furthermore, characterization of the PhnZ active site, 126 along with protein engineering efforts, could lead to identification of variants that catalyze C-P cleavage for a variety of substrates. Analysis of PhnY activity and active site composition would be essential to this process since the α-hydroxyl is essential for substrate binding and positioning in the active site of PhnZ. 5.5 Conclusions In summary, we have presented three examples of novel microbial metalloenzymes and their structure-based functional annotation. In all three cases the crystal structure led to the identification of bound metal ion(s) that are critical for function. For EcSI and YcfD the structure was essential for the positive elucidation of previously unknown function. In the case of PhnZ the crystal structure and subsequent identification of a di-nuclear iron site played a key role in determining the catalytic mechanism and ultimately finding the product of the reaction. All three enzymes exemplify the versatility and catalytic potential of a bound metal, and clearly show how structural information can aid in functional characterization. 127 Reference List 1. Zheng, H., Chruszcz, M., Lasota, P., Lebioda, L., and Minor, W. (2008) J. Inorg. Biochem. 102, 1765-1776 2. Holm, R. H., Kennepohl, P., and Solomon, E. I. (1996) Chem. Rev. 96, 2239-2314 3. Kasampalidis, I. N., Pitas, I., and Lyroudia, K. (2007) Proteins 68, 123-130 4. Dupont, C. L., Yang, S., Palenik, B., and Bourne, P. E. (2006) Proc. Natl. Acad. Sci. U. S. A 103, 17822-17827 5. Harding, M. M., Nowicki, M. W., and Walkinshaw, M. D. (2010) Crystallography Reviews 16, 247-302 6. Shi, W., Zhan, C., Ignatov, A., Manjasetty, B. A., Marinkovic, N., Sullivan, M., Huang, R., and Chance, M. R. (2005) Structure. 13, 1473-1486 7. McCall, K. A., Huang, C., and Fierke, C. A. (2000) J. Nutr. 130, 1437S-1446S 8. Irving, H. and Williams, R. J. P. (1948) Nature 162, 746-747 9. Waldron, K. J. and Robinson, N. J. (2009) Nat. Rev. Microbiol. 7, 25-35 10. Dunwell, J. M., Purvis, A., and Khuri, S. (2004) Phytochemistry 65, 7-17 11. Dunwell, J. M. and Gane, P. J. (1998) J. Mol. Evol. 46, 147-154 12. Dunwell, J. M., Khuri, S., and Gane, P. J. (2000) Microbiol. Mol. Biol. Rev. 64, 153-179 13. Dunwell, J. M. (1998) Biotechnol. Genet. Eng Rev. 15, 1-32 14. Dunwell, J. M., Culham, A., Carter, C. E., Sosa-Aguirre, C. R., and Goodenough, P. W. (2001) Trends Biochem. Sci. 26, 740-746 15. Agarwal, G., Rajavel, M., Gopal, B., and Srinivasan, N. (2009) PLoS. One. 4, e5736 16. Cleasby, A., Wonacott, A., Skarzynski, T., Hubbard, R. E., Davies, G. J., Proudfoot, A. E., Bernard, A. R., Payton, M. A., and Wells, T. N. (1996) Nat. Struct. Biol. 3, 470-479 17. Anand, R., Dorrestein, P. C., Kinsland, C., Begley, T. P., and Ealick, S. E. (2002) Biochemistry 41, 7659-7669 18. Lane, B. G. (1994) FASEB J. 8, 294-301 128 19. Lane, B. G., Bernier, F., Dratewka-Kos, E., Shafai, R., Kennedy, T. D., Pyne, C., Munro, J. R., Vaughan, T., Walters, D., and Altomare, F. (1991) J. Biol. Chem. 266, 1046110469 20. Nunez, C., Leon, R., Guzman, J., Espin, G., and Soberon-Chavez, G. (2000) J. Bacteriol. 182, 6550-6556 21. Verhees, C. H., Huynen, M. A., Ward, D. E., Schiltz, E., de Vos, W. M., and van der, O. J. (2001) J. Biol. Chem. 276, 40926-40932 22. Woo, E. J., Dunwell, J. M., Goodenough, P. W., Marvier, A. C., and Pickersgill, R. W. (2000) Nat. Struct. Biol. 7, 1036-1040 23. Schafleitner, R. and Wilhelm, E. (2002) J. Physiol. Mol. Plant. Path 61, 339-348 24. Mahalingam, R., Gomez-Buitrago, A., Eckardt, N., Shah, N., Guevara-Garcia, A., Day, P., Raina, R., and Fedoroff, N. V. (2003) Genome Biol. 4, R20 25. Proudfoot, A. E., Turcatti, G., Wells, T. N., Payton, M. A., and Smith, D. J. (1994) Eur. J. Biochem. 219, 415-423 26. Hansen, T., Schlichting, B., Felgendreher, M., and Schonheit, P. (2005) J. Bacteriol. 187, 1621-1631 27. Sauer, M. and Kleine-Vehn, J. (2011) Plant Cell 23, 2033-2043 28. Adams, M. and Jia, Z. (2005) J. Biol. Chem. 280, 28675-28682 29. Lawrence, M. C., Izard, T., Beuchat, M., Blagrove, R. J., and Colman, P. M. (1994) J. Mol. Biol. 238, 748-776 30. Pirovani, C. P., Macedo, J. N., Contim, L. A., Matrangolo, F. S., Loureiro, M. E., and Fontes, E. P. (2002) Eur. J. Biochem. 269, 3998-4008 31. Rocha, C. S., Luz, D. F., Oliveira, M. L., Baracat-Pereira, M. C., Medrano, F. J., and Fontes, E. P. (2007) Phytochemistry 68, 802-810 32. Gopal, B., Madan, L. L., Betz, S. F., and Kossiakoff, A. A. (2005) Biochemistry 44, 193201 33. Dai, Y., Wensink, P. C., and Abeles, R. H. (1999) J. Biol. Chem. 274, 1193-1195 34. Ju, T., Goldsmith, R. B., Chai, S. C., Maroney, M. J., Pochapsky, S. S., and Pochapsky, T. C. (2006) J. Mol. Biol. 363, 823-834 35. Denisov, I. G., Makris, T. M., Sligar, S. G., and Schlichting, I. (2005) Chem. Rev. 105, 2253-2277 129 36. Solomon, E. I., Brunold, T. C., Davis, M. I., Kemsley, J. N., Lee, S. K., Lehnert, N., Neese, F., Skulan, A. J., Yang, Y. S., and Zhou, J. (2000) Chem. Rev. 100, 235-350 37. Costas, M., Mehn, M. P., Jensen, M. P., and Que, L., Jr. (2004) Chem. Rev. 104, 939-986 38. Tshuva, E. Y. and Lippard, S. J. (2004) Chem. Rev. 104, 987-1012 39. Clifton, I. J., McDonough, M. A., Ehrismann, D., Kershaw, N. J., Granatino, N., and Schofield, C. J. (2006) J. Inorg. Biochem. 100, 644-669 40. Hausinger, R. P. (2004) Crit Rev. Biochem. Mol. Biol. 39, 21-68 41. Hutton, J. J., Jr., Trappel, A. L., and Udenfriend, S. (1966) Biochem. Biophys. Res. Commun. 24, 179-184 42. Hutton, J. J., Jr., Kaplan, A., and Udenfriend, S. (1967) Arch. Biochem. Biophys. 121, 384-391 43. Prescott, A. G. and Lloyd, M. D. (2000) Nat. Prod. Rep. 17, 367-383 44. Vaillancourt, F. H., Yin, J., and Walsh, C. T. (2005) Proc. Natl. Acad. Sci. U. S. A 102, 10111-10116 45. Vaillancourt, F. H., Yeh, E., Vosburg, D. A., O'Connor, S. E., and Walsh, C. T. (2005) Nature 436, 1191-1194 46. Trewick, S. C., Henshaw, T. F., Hausinger, R. P., Lindahl, T., and Sedgwick, B. (2002) Nature 419, 174-178 47. Begley, T. J. and Samson, L. D. (2003) Trends Biochem. Sci. 28, 2-5 48. Clissold, P. M. and Ponting, C. P. (2001) Trends Biochem. Sci. 26, 7-9 49. Klose, R. J., Kallin, E. M., and Zhang, Y. (2006) Nat. Rev. Genet. 7, 715-727 50. Balciunas, D. and Ronne, H. (2000) Trends Biochem. Sci. 25, 274-276 51. Takeuchi, T., Yamazaki, Y., Katoh-Fukui, Y., Tsuchiya, R., Kondo, S., Motoyama, J., and Higashinakagawa, T. (1995) Genes Dev. 9, 1211-1222 52. Tsukada, Y., Fang, J., Erdjument-Bromage, H., Warren, M. E., Borchers, C. H., Tempst, P., and Zhang, Y. (2006) Nature 439, 811-816 53. Lando, D., Peet, D. J., Gorman, J. J., Whelan, D. A., Whitelaw, M. L., and Bruick, R. K. (2002) Genes Dev. 16, 1466-1471 130 54. Noma, A., Ishitani, R., Kato, M., Nagao, A., Nureki, O., and Suzuki, T. (2010) J. Biol. Chem. 285, 34503-34507 55. Roach, P. L., Clifton, I. J., Fulop, V., Harlos, K., Barton, G. J., Hajdu, J., Andersson, I., Schofield, C. J., and Baldwin, J. E. (1995) Nature 375, 700-704 56. Stirk, H. J., Woolfson, D. N., Hutchinson, E. G., and Thornton, J. M. (1992) FEBS Lett. 308, 1-3 57. Elkins, J. M., Ryle, M. J., Clifton, I. J., Dunning Hotopp, J. C., Lloyd, J. S., Burzlaff, N. I., Baldwin, J. E., Hausinger, R. P., and Roach, P. L. (2002) Biochemistry 41, 5185-5192 58. Bruijnincx, P. C., van, K. G., and Klein Gebbink, R. J. (2008) Chem. Soc. Rev. 37, 27162744 59. Valegard, K., van Scheltinga, A. C., Lloyd, M. D., Hara, T., Ramaswamy, S., Perrakis, A., Thompson, A., Lee, H. J., Baldwin, J. E., Schofield, C. J. et al. (1998) Nature 394, 805-809 60. O'Brien, J. R., Schuller, D. J., Yang, V. S., Dillard, B. D., and Lanzilotta, W. N. (2003) Biochemistry 42, 5547-5554 61. Holme, E. (1975) Biochemistry 14, 4999-5003 62. Price, J. C., Barr, E. W., Glass, T. E., Krebs, C., and Bollinger, J. M., Jr. (2003) J. Am. Chem. Soc. 125, 13008-13009 63. Riggs-Gelasco, P. J., Price, J. C., Guyer, R. B., Brehm, J. H., Barr, E. W., Bollinger, J. M., Jr., and Krebs, C. (2004) J. Am. Chem. Soc. 126, 8108-8109 64. Proshlyakov, D. A., Henshaw, T. F., Monterosso, G. R., Ryle, M. J., and Hausinger, R. P. (2004) J. Am. Chem. Soc. 126, 1022-1023 65. Wallar, B. J. and Lipscomb, J. D. (1996) Chem. Rev. 96, 2625-2658 66. Merkx, M., Kopp, D. A., Sazinsky, M. H., Blazyk, J. L., Muller, J., and Lippard, S. J. (2001) Angew. Chem. Int. Ed Engl. 40, 2782-2807 67. Fox, B. G., Lyle, K. S., and Rogge, C. E. (2004) Acc. Chem. Res. 37, 421-429 68. Choi, Y. S., Zhang, H., Brunzelle, J. S., Nair, S. K., and Zhao, H. (2008) Proc. Natl. Acad. Sci. U. S. A 105, 6858-6863 69. Brown, P. M., Caradoc-Davies, T. T., Dickson, J. M., Cooper, G. J., Loomes, K. M., and Baker, E. N. (2006) Proc. Natl. Acad. Sci. U. S. A 103, 15032-15037 131 70. Krebs, C., Price, J. C., Baldwin, J., Saleh, L., Green, M. T., and Bollinger, J. M., Jr. (2005) Inorg. Chem. 44, 742-757 71. Liu, K. E., Wang, D., Huynh, B. H., Edmondson, D. E., Salifoglou, A., and Lippard, S. J. (1994) J. Am. Chem. Soc. 116, 7465-7466 72. Liu, K. E., Valentine, A. M., Wang, D., Huynh, B. H., Edmondson, D. E., Salifoglou, A., and Lippard, S. J. (1995) J. Am. Chem. Soc. 117, 10174-10185 73. Bollinger, J. M., Jr., Krebs, C., Vicol, A., Chen, S., Ley, B. A., Edmondson, D. E., and Huynh, B. H. (1998) J. Am. Chem. Soc. 120, 1094-1095 74. Saleh, L., Krebs, C., Ley, B. A., Naik, S., Huynh, B. H., and Bollinger, J. M., Jr. (2004) Biochemistry 43, 5953-5964 75. Murray, L. J., Garcia-Serres, R., Naik, S., Huynh, B. H., and Lippard, S. J. (2006) J. Am. Chem. Soc. 128, 7458-7459 76. Yun, D., Garcia-Serres, R., Chicalese, B. M., An, Y. H., Huynh, B. H., and Bollinger, J. M., Jr. (2007) Biochemistry 46, 1925-1932 77. Lee, S. K., Fox, B. G., Froland, W. A., Lipscomb, J. D., and Munck, E. (1993) J. Am. Chem. Soc. 115, 6450-6451 78. Shu, L., Nesheim, J. C., Kauffmann, K., Munck, E., Lipscomb, J. D., and Que, L., Jr. (1997) Science 275, 515-518 79. Lee, S. K., Nesheim, J. C., and Lipscomb, J. D. (1993) J. Biol. Chem. 268, 21569-21577 80. Bollinger, J. M., Jr., Stubbe, J., Huynh, B. H., and Edmondson, D. E. (1991) J. Am. Chem. Soc. 113, 6289-6291 81. Bollinger, J. M., Jr., Edmondson, D. E., Huynh, B. H., Filley, J., Norton, J. R., and Stubbe, J. (1991) Science 253, 292-298 82. Sturgeon, B. E., Burdi, D., Chen, S., Huynh, B. H., Edmondson, D. E., Stubbe, J., and Hoffman, B. M. (1996) J. Am. Chem. Soc. 118, 7551-7557 83. Bollinger, J. M., Jr., Tong, W. H., Ravi, N., Huynh, B. H., Edmondson, D. E., and Stubbe, J. (1994) J. Am. Chem. Soc. 116, 8024-8032 84. Burdi, D., Sturgeon, B. E., Tong, W. H., Stubbe, J., and Hoffman, B. M. (1996) J. Am. Chem. Soc. 118, 281-282 85. Willems, J.-P., Lee, H.-I., Burdi, D., Doan, P. E., Stubbe, J., and Hoffman, B. M. (1997) J. Am. Chem. Soc. 119, 9816-9824 132 86. Riggs-Gelasco, P. J., Shu, L., Chen, S., Burdi, D., Huynh, B. H., Que, L., Jr., and Stubbe, J. (1998) J. Am. Chem. Soc. 120, 849-860 87. Burdi, D., Willems, J.-P., Riggs-Gelasco, P. J., Antholine, W. E., Stubbe, J., and Hoffman, B. M. (1998) J. Am. Chem. Soc. 120, 12910-12919 88. Stubbe, J. (2003) Curr. Opin. Chem. Biol. 7, 183-188 89. Stubbe, J., Nocera, D. G., Yee, C. S., and Chang, M. C. (2003) Chem. Rev. 103, 21672201 90. Xing, G., Barr, E. W., Diao, Y., Hoffart, L. M., Prabhu, K. S., Arner, R. J., Reddy, C. C., Krebs, C., and Bollinger, J. M., Jr. (2006) Biochemistry 45, 5402-5412 91. Xing, G., Hoffart, L. M., Diao, Y., Prabhu, K. S., Arner, R. J., Reddy, C. C., Krebs, C., and Bollinger, J. M., Jr. (2006) Biochemistry 45, 5393-5401 92. Xing, G., Diao, Y., Hoffart, L. M., Barr, E. W., Prabhu, K. S., Arner, R. J., Reddy, C. C., Krebs, C., and Bollinger, J. M., Jr. (2006) Proc. Natl. Acad. Sci. U. S. A 103, 6130-6135 93. Guddat, L. W., McAlpine, A. S., Hume, D., Hamilton, S., de, J. J., and Martin, J. L. (1999) Structure. 7, 757-767 94. Nordlund, P. and Eklund, H. (1995) Curr. Opin. Struct. Biol. 5, 758-766 95. Nordlund, P. and Eklund, H. (1993) J. Mol. Biol. 232, 123-164 96. Fox, B. G., Shanklin, J., Somerville, C., and Munck, E. (1993) Proc. Natl. Acad. Sci. U. S. A 90, 2486-2490 97. Rosenzweig, A. C., Frederick, C. A., Lippard, S. J., and Nordlund, P. (1993) Nature 366, 537-543 98. Lindqvist, Y., Huang, W., Schneider, G., and Shanklin, J. (1996) EMBO J. 15, 4081-4092 99. Holmes, M. A. and Stenkamp, R. E. (1991) J. Mol. Biol. 220, 723-737 100. Ahmed, Z. (2001) Electron. J. Biotechnol. 4, 103-111 101. Heath, E. C., Hurwitz, J., Horecker, B. L., and Ginsburg, A. (1958) J. Biol. Chem. 231, 1009-1029 102. Izumori, K., Yamanka, K., and Elbein, A. D. (1976) J. Bac 128, 587-591 103. Stevens, F. J. and Wu, T. T. (1976) J. Gen. Microbiol. 97, 257-265 133 104. Stevens, F. J., Stevens, P. W., Hovis, J. G., and Wu, T. T. (1981) J. Gen. Microbiol. 124, 219-223 105. Gunsalus, I. C., Horecker, B. L., and Wood, W. A. (1955) Bacteriol. Rev. 19, 79-128 106. Kletzien, R. F., Harris, P. K., and Foellmi, L. A. (1994) FASEB J. 8, 174-181 107. Kruger, N. J. and von Shaewen, A. (2003) Curr. Opin. Plant Biol. 6, 236-246 108. Cho, E. A., Lee, D. W., Cha, Y. H., Lee, S. J., Jung, H. C., Pan, J. G., and Pyun, Y. R. (2007) J. Bacteriol. 189, 1655-1663 109. Hirose, J., Maeda, K., Yokoi, H., and Takasaki, Y. (2001) Biosci. Biotechnol. Biochem. 65, 658-661 110. Hey-Ferguson, A. and Elbein, A. D. (1970) J. Bacteriol. 101, 777-780 111. Palleroni, N. J. and Doudoroff, M. (1956) J. Biol. Chem. 218, 535-548 112. Anderson, R. L. and Allison, D. P. (1965) J. Biol. Chem. 240, 2367-2372 113. Maclennan, A. P. (1962) Biochem. J. 82, 394-400 114. Khoo, K. H., Suzuki, R., Dell, A., Morris, H. R., McNeil, M. R., Brennan, P. J., and Besra, G. S. (1996) Biochemistry 35, 11812-11819 115. Khuri, S., Bakker, F. T., and Dunwell, J. M. (2001) Mol. Biol. Evol. 18, 593-605 116. Dunwell, J. M., Khuri, S., and Gane, P. J. (2000) Microbiol. Mol. Biol. Rev. 64, 153-179 117. Dunwell, J. M. (1998) Microb. Comp Genomics 3, 141-148 118. Hendrickson, W. A., Horton, J. R., and LeMaster, D. M. (1990) EMBO J. 9, 1665-1672 119. Otwinowski, Z. and Minor, W. (1997) Methods in Enzymology 276, 307-326 120. Matthews, B. W. (1968) J. Mol. Biol. 33, 491-497 121. Pape, T. and Schneider, T. R. (2004) J. Appl. Cryst. 37, 843-844 122. Perrakis, A., Morris, R., and Lamzin, V. S. (1999) Nat. Struct. Biol. 6, 458-463 123. Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S. et al. (1998) Acta Crystallogr. D. Biol. Crystallogr. 54, 905-921 134 124. Murshudov, G. N., Vagin, A. A., and Dodson, E. J. (1997) Acta Crystallogr. D. Biol. Crystallogr. 53, 240-255 125. Krissinel, E. and Henrick, K. (2004) Acta Crystallogr. D. Biol. Crystallogr. 60, 22562268 126. Hansen, T., Urbanke, C., and Schonheit, P. (2004) Extremophiles. 8, 507-512 127. Krissinel, E. and Henrick, K. (2007) J. Mol. Biol. 372, 774-797 128. Schwarzenbacher, R., von Delft, F., Jaroszewski, L., Abdubek, P., Ambing, E., Biorac, T., Brinen, L. S., Canaves, J. M., Cambell, J., Chiu, H. J. et al. (2004) Proteins 56, 392395 129. Ko, T. P., Ng, J. D., Greenwood, A., and McPherson, A. (1993) Acta Crystallogr. D. Biol. Crystallogr. 49, 478-489 130. Berrisford, J. M., Akerboom, J., Turnbull, A. P., de Geus, D., Sedelnikova, S. E., Staton, I., McLeod, C. W., Verhees, C. H., van der, O. J., Rice, D. W. et al. (2003) J. Biol. Chem. 278, 33290-33297 131. Bauer, A. J., Rayment, I., Frey, P. A., and Holden, H. M. (1992) Proteins 12, 372-381 132. Holm, L. and Park, J. (2000) Bioinformatics. 16, 566-567 133. Jeong, J. J., Fushinobu, S., Ito, S., Jeon, B. S., Shoun, H., and Wakagi, T. (2003) FEBS Lett. 535, 200-204 134. Berrisford, J. M., Akerboom, J., Brouns, S., Sedelnikova, S. E., Turnbull, A. P., van der, O. J., Salmon, L., Hardre, R., Murray, I. A., Blackburn, G. M. et al. (2004) J. Mol. Biol. 343, 649-657 135. Swan, M. K., Solomons, J. T., Beeson, C. C., Hansen, T., Schonheit, P., and Davies, C. (2003) J. Biol. Chem. 278, 47261-47268 136. Hansen, T., Schlichting, B., Grotzinger, J., Swan, M. K., Davies, C., and Schonheit, P. (2005) FEBS J. 272, 6266-6275 137. Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., Valentin, F., Wallace, I. M., Wilm, A., Lopez, R. et al. (2007) Bioinformatics. 23, 2947-2948 138. Woo, E. J., Marshall, J., Bauly, J., Chen, J. G., Venis, M., Napier, R. M., and Pickersgill, R. W. (2002) EMBO J. 21, 2877-2885 139. Mizanur, R. M., Takata, G., and Izumori, K. (2001) Biochim. Biophys. Acta 1521, 141145 135 140. Yeom, S. J., Ji, J. H., Kim, N. H., Park, C. S., and Oh, D. K. (2009) Appl. Environ. Microbiol. 75, 4705-4710 141. Yeom, S. J., Kim, N. H., Yoon, R. Y., Kwon, H. J., Park, C. S., and Oh, D. K. (2009) Biotechnol. Lett. 31, 1273-1278 142. Hansen, T., Wendorff, D., and Schonheit, P. (2004) J. Biol. Chem. 279, 2262-2272 143. Berrisford, J. M., Hounslow, A. M., Akerboom, J., Hagen, W. R., Brouns, S. J., van der, O. J., Murray, I. A., Michael, B. G., Waltho, J. P., Rice, D. W. et al. (2006) J. Mol. Biol. 19;358, 1353-1366 144. Wu, R., Xie, H., Cao, Z., and Mo, Y. (2008) J. Am. Chem. Soc. 130, 7022-7031 145. Whitlow, M., Howard, A. J., Finzel, B. C., Poulos, T. L., Winborne, E., and Gilliland, G. L. (1991) Proteins 9, 153-173 146. Read, J., Pearce, J., Li, X., Muirhead, H., Chirgwin, J., and Davies, C. (2001) J. Mol. Biol. 309, 447-463 147. Jeffery, C. J., Hardre, R., and Salmon, L. (2001) Biochemistry 40, 1560-1566 148. Raines, R. T., Sutton, E. L., Straus, D. R., Gilbert, W., and Knowles, J. R. (1986) Biochemistry 25, 7142-7154 149. Swenson, C. A. and Barker, R. (1971) Biochemistry 10, 3151-3154 150. Lee, J. H., Chang, K. Z., Patel, V., and Jeffery, C. J. (2001) Biochemistry 40, 7799-7805 151. Goldschmidt, L., Cooper, D. R., Derewenda, Z. S., and Eisenberg, D. (2007) Protein Sci. 16, 1569-1576 152. Luft, J. R. and DeTitta, G. T. (1999) Acta Crystallogr. D. Biol. Crystallogr. 55, 988-993 153. Kabsch, W. (2010) Acta Crystallogr. D. Biol. Crystallogr. 66, 125-132 154. Vonrhein, C., Blanc, E., Roversi, P., and Bricogne, G. (2007) Methods Mol. Biol. 364, 215-230 155. Emsley, P., Lohkamp, B., Scott, W. G., and Cowtan, K. (2010) Acta Crystallogr. D. Biol. Crystallogr. 66, 486-501 156. Adams, P. D., Afonine, P. V., Bunkoczi, G., Chen, V. B., Davis, I. W., Echols, N., Headd, J. J., Hung, L. W., Kapral, G. J., Grosse-Kunstleve, R. W. et al. (2010) Acta Crystallogr. D. Biol. Crystallogr. 66, 213-221 136 157. Perkins, D. N., Pappin, D. J., Creasy, D. M., and Cottrell, J. S. (1999) Electrophoresis 20, 3551-3567 158. Baba, T., Ara, T., Hasegawa, M., Takai, Y., Okumura, Y., Baba, M., Datsenko, K. A., Tomita, M., Wanner, B. L., and Mori, H. (2006) Mol. Syst. Biol. 2, 2006 159. Hegg, E. L. and Que, L., Jr. (1997) Eur. J. Biochem. 250, 625-629 160. Schofield, C. J. and Zhang, Z. (1999) Curr. Opin. Struct. Biol. 9, 722-731 161. Jones, S. and Thornton, J. M. (1995) Prog. Biophys. Mol. Biol. 63, 31-65 162. Elkins, J. M., Hewitson, K. S., McNeill, L. A., Seibel, J. F., Schlemminger, I., Pugh, C. W., Ratcliffe, P. J., and Schofield, C. J. (2003) J. Biol. Chem. 278, 1802-1806 163. Kato, M., Araiso, Y., Noma, A., Nagao, A., Suzuki, T., Ishitani, R., and Nureki, O. (2011) Nucleic Acids Res. 39, 1576-1585 164. Dunning Hotopp, J. C., Auchtung, T. A., Hogan, D. A., and Hausinger, R. P. (2003) J. Inorg. Biochem. 93, 66-70 165. Mishina, Y., Chen, L. X., and He, C. (2004) J. Am. Chem. Soc. 126, 16930-16936 166. McNeill, L. A., Flashman, E., Buck, M. R., Hewitson, K. S., Clifton, I. J., Jeschke, G., Claridge, T. D., Ehrismann, D., Oldham, N. J., and Schofield, C. J. (2005) Mol. Biosyst. 1, 321-324 167. Ishizaki, H., Yano, H., Tsuneoka, M., Ogasawara, S., Akiba, J., Nishida, N., Kojiro, S., Fukahori, S., Moriya, F., Matsuoka, K. et al. (2007) Pathol. Int. 57, 672-680 168. Komiya, K., Sueoka-Aragane, N., Sato, A., Hisatomi, T., Sakuragi, T., Mitsuoka, M., Sato, T., Hayashi, S., Izumi, H., Tsuneoka, M. et al. (2010) J. Cancer Res. Clin. Oncol. 136, 465-473 169. Kuratomi, K., Yano, H., Tsuneoka, M., Sakamoto, K., Kusukawa, J., and Kojiro, M. (2006) Kurume Med. J. 53, 71-78 170. Ogasawara, S., Komuta, M., Nakashima, O., Akiba, J., Tsuneoka, M., and Yano, H. (2010) Hepatol. Res. 40, 330-336 171. Tan, X. P., Zhang, Q., Dong, W. G., Lei, X. W., and Yang, Z. R. (2012) Oncol. Lett. 3, 1037-1041 172. Teye, K., Arima, N., Nakamura, Y., Sakamoto, K., Sueoka, E., Kimura, H., and Tsuneoka, M. (2007) Oncol. Rep. 18, 841-848 137 173. Tsuneoka, M., Fujita, H., Arima, N., Teye, K., Okamura, T., Inutsuka, H., Koda, Y., Shirouzu, K., and Kimura, H. (2004) Clin. Cancer Res. 10, 7347-7356 174. Tsuneoka, M., Koda, Y., Soejima, M., Teye, K., and Kimura, H. (2002) J. Biol. Chem. 277, 35450-35459 175. Zhang, Q., Hu, C. M., Yuan, Y. S., He, C. H., Zhao, Q., and Liu, N. Z. (2008) Int. J. Biol. Markers 23, 83-88 176. Eilbracht, J., Kneissel, S., Hofmann, A., and Schmidt-Zachmann, M. S. (2005) Eur. J. Cell Biol. 84, 279-294 177. Eilbracht, J., Reichenzeller, M., Hergt, M., Schnolzer, M., Heid, H., Stohr, M., Franke, W. W., and Schmidt-Zachmann, M. S. (2004) Mol. Biol. Cell 15, 1816-1832 178. Armougom, F., Moretti, S., Poirot, O., Audic, S., Dumas, P., Schaeli, B., Keduas, V., and Notredame, C. (2006) Nucleic Acids Res. 34, W604-W608 179. Teraoka, H. and Nierhaus, K. H. (1978) FEBS Lett. 88, 223-226 180. Kazemie, M. (1975) Eur. J. Biochem. 58, 501-510 181. Kazemie, M. (1976) Eur. J. Biochem. 67, 373-378 182. Moore, V. G., Atchison, R. E., Thomas, G., Moran, M., and Noller, H. F. (1975) Proc. Natl. Acad. Sci. U. S. A 72, 844-848 183. Hampl, H., Schulze, H., and Nierhaus, K. H. (1981) J. Biol. Chem. 256, 2284-2288 184. Tate, W. P., Schulze, H., and Nierhaus, K. H. (1983) J. Biol. Chem. 258, 12810-12815 185. de Bethune, M. P. and Nierhaus, K. H. (1978) Eur. J. Biochem. 86, 187-191 186. Nierhaus, D. and Nierhaus, K. H. (1973) Proc. Natl. Acad. Sci. U. S. A 70, 2224-2228 187. Teraoka, H. and Nierhaus, K. H. (1978) J. Mol. Biol. 126, 185-193 188. Yusupov, M. M., Yusupova, G. Z., Baucom, A., Lieberman, K., Earnest, T. N., Cate, J. H., and Noller, H. F. (2001) Science 292, 883-896 189. Harms, J., Schluenzen, F., Zarivach, R., Bashan, A., Gat, S., Agmon, I., Bartels, H., Franceschi, F., and Yonath, A. (2001) Cell 107, 679-688 190. Nierhaus, K. H. (1991) Biochimie 73, 739-755 191. Kitagawa, M., Ara, T., Arifuzzaman, M., Ioka-Nakamichi, T., Inamoto, E., Toyonaga, H., and Mori, H. (2005) DNA Res. 12, 291-299 138 192. Franceschi, F. J. and Nierhaus, K. H. (1990) J. Biol. Chem. 265, 16676-16682 193. Pan, C. and Mason, T. L. (1995) Nucleic Acids Res. 23, 3673-3677 194. Metcalf, W. W. and van der Donk, W. A. (2009) Annu. Rev. Biochem. 78, 65-94 195. Kafarski, P. and Lejczak, B. (2001) Curr. Med. Chem. Anticancer Agents 1, 301-312 196. Quinn, J. P., Kulakova, A. N., Cooley, N. A., and McGrath, J. W. (2007) Environ. Microbiol. 9, 2392-2400 197. Hudson, J. J., Taylor, W. D., and Schindler, D. W. (2000) Nature 406, 54-56 198. White, A. K. and Metcalf, W. W. (2007) Annu. Rev. Microbiol. 61, 379-400 199. Van Mooy, B. A., Fredricks, H. F., Pedler, B. E., Dyhrman, S. T., Karl, D. M., Koblizek, M., Lomas, M. W., Mincer, T. J., Moore, L. R., Moutin, T. et al. (2009) Nature 458, 6972 200. McSorley, F. R., Wyatt, P. B., Martinez, A., Delong, E. F., Hove-Jensen, B., and Zechel, D. L. (2012) J. Am. Chem. Soc. 201. Kononova, S. V. and Nesmeyanova, M. A. (2002) Biochemistry (Mosc. ) 67, 184-195 202. Chen, C. M., Ye, Q. Z., Zhu, Z. M., Wanner, B. L., and Walsh, C. T. (1990) J. Biol. Chem. 265, 4461-4471 203. Hove-Jensen, B., McSorley, F. R., and Zechel, D. L. (2011) J. Am. Chem. Soc. 133, 3617-3624 204. Kamat, S. S., Williams, H. J., and Raushel, F. M. (2011) Nature 480, 570-573 205. Martinez, A., Tyson, G. W., and Delong, E. F. (2010) Environ. Microbiol. 12, 222-238 206. Aravind, L. and Koonin, E. V. (1998) Trends Biochem. Sci. 23, 469-472 207. Holm, L. and Rosenstrom, P. (2010) Nucleic Acids Res. 38, W545-W549 208. Holm, L. and Sander, C. (1995) Trends Biochem. Sci. 20, 478-480 209. Vagin, A. and Teplyakov, A. (2010) Acta Crystallogr. D. Biol. Crystallogr. 66, 22-25 210. van Aalten, D. M., Bywater, R., Findlay, J. B., Hendlich, M., Hooft, R. W., and Vriend, G. (1996) J. Comput. Aided Mol. Des 10, 255-262 211. Ehrenberg, A. and Reichard, P. (1972) J. Biol. Chem. 247, 3485-3488 139 212. Thorsell, A. G., Persson, C., Voevodskaya, N., Busam, R. D., Hammarstrom, M., Graslund, S., Graslund, A., and Hallberg, B. M. (2008) J. Biol. Chem. 283, 15209-15216 213. Ternan, N. G., McGrath, J. W., McMullen, G., and Quinn, J. P. (1998) World. J. Microbiol. 14, 635-647 214. Podzelinska, K., He, S. M., Wathier, M., Yakunin, A., Proudfoot, M., Hove-Jensen, B., Zechel, D. L., and Jia, Z. (2009) J. Biol. Chem. 284, 17216-17226 215. Taylor, S. S., Radzio-Andzelm, E., Madhusudan, Cheng, X., Ten, E. L., and Narayana, N. (1999) Pharmacol. Ther. 82, 133-141 216. Hyeon, C., Jennings, P. A., Adams, J. A., and Onuchic, J. N. (2009) Proc. Natl. Acad. Sci. U. S. A 106, 3023-3028 217. Banos-Sanz, J. I., Sanz-Aparicio, J., Whitfield, H., Hamilton, C., Brearley, C. A., and Gonzalez, B. (2012) J. Biol. Chem. 287, 29237-29249 218. Sullivan, S. M. and Holyoak, T. (2008) Proc. Natl. Acad. Sci. U. S. A 105, 13829-13834 219. Weikl, T. R. and von, D. C. (2009) Proteins 75, 104-110 220. Johnson, K. A. (2008) J. Biol. Chem. 283, 26297-26301 221. Koshland, D. E. (1958) Proc. Natl. Acad. Sci. U. S. A 44, 98-104 222. Tummino, P. J. and Copeland, R. A. (2008) Biochemistry 47, 5481-5492 223. Boehr, D. D., Nussinov, R., and Wright, P. E. (2009) Nat. Chem. Biol. 5, 789-796 224. Carter, M. Q., Xue, K., Brandl, M. T., Liu, F., Wu, L., Louie, J. W., Mandrell, R. E., and Zhou, J. (2012) PLoS. One. 7, e44186 225. Adams, M. A., Suits, M. D., Zheng, J., and Jia, Z. (2007) Proteomics. 7, 2920-2932 226. Adrian, P. V., Zhao, W., Black, T. A., Shaw, K. J., Hare, R. S., and Klugman, K. P. (2000) Antimicrob. Agents Chemother. 44, 732-738 227. Aarestrup, F. M. and Jensen, L. B. (2000) Antimicrob. Agents Chemother. 44, 3425-3427 228. McNicholas, P. M., Mann, P. A., Najarian, D. J., Miesel, L., Hare, R. S., and Black, T. A. (2001) Antimicrob. Agents Chemother. 45, 79-83 229. Zarazaga, M., Tenorio, C., Del, C. R., Ruiz-Larrea, F., and Torres, C. (2002) Antimicrob. Agents Chemother. 46, 3657-3659 140 230. Nishimura, M., Yoshida, T., Shirouzu, M., Terada, T., Kuramitsu, S., Yokoyama, S., Ohkubo, T., and Kobayashi, Y. (2004) J. Mol. Biol. 344, 1369-1383 231. Webby, C. J., Wolf, A., Gromak, N., Dreger, M., Kramer, H., Kessler, B., Nielsen, M. L., Schmitz, C., Butler, D. S., Yates, J. R., III et al. (2009) Science 325, 90-93 232. Iyer, L. M., Abhiman, S., de Souza, R. F., and Aravind, L. (2010) Nucleic Acids Res. 38, 5261-5279 233. Loenarz, C. and Schofield, C. J. (2011) Trends Biochem. Sci. 36, 7-18 234. Suzuki, C., Takahashi, K., Hayama, S., Ishikawa, N., Kato, T., Ito, T., Tsuchiya, E., Nakamura, Y., and Daigo, Y. (2007) Mol. Cancer Ther. 6, 542-551 235. Bollinger, J. M., Jr., Diao, Y., Matthews, M. L., Xing, G., and Krebs, C. (2009) Dalton Trans. 905-914 236. Cotelesage, J. J., Pushie, M. J., Grochulski, P., Pickering, I. J., and George, G. N. (2012) J. Inorg. Biochem. 115, 127-137 237. Webb, M. R. (1992) Proc. Natl. Acad. Sci. U. S. A 89, 4884-4887 141