Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Epigenetics of diabetes Type 2 wikipedia , lookup
Cell-free fetal DNA wikipedia , lookup
No-SCAR (Scarless Cas9 Assisted Recombineering) Genome Editing wikipedia , lookup
Genomic imprinting wikipedia , lookup
Gene therapy wikipedia , lookup
Gene expression profiling wikipedia , lookup
Public health genomics wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Skewed X-inactivation wikipedia , lookup
Gene expression programming wikipedia , lookup
Y chromosome wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Epigenetics of neurodegenerative diseases wikipedia , lookup
Down syndrome wikipedia , lookup
Neocentromere wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Oncogenomics wikipedia , lookup
Frameshift mutation wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Saethre–Chotzen syndrome wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Designer baby wikipedia , lookup
Neuronal ceroid lipofuscinosis wikipedia , lookup
Microevolution wikipedia , lookup
DiGeorge syndrome wikipedia , lookup
X-inactivation wikipedia , lookup
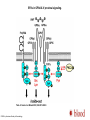
Thrombocytopenia Platelet Counts • Vary with – Age (slight decrease in mean with increased age) – Sex (Slightly lower mean in men) Consequences of Misdiagnosis of Inherited Thrombocytopenia • Wrong therapy • Wrong prognosis • Missed opportunity for genetic counseling Inherited Thrombocytopenias with Adulthood Presentation • Italian Experience • Mean age of diagnosis – 35 yrs MYH9-related disease – 33 yrs monoallelic Bernard-Soulier syndrome – 30 yrs ANKRD26-related thrombocytopenia Baluini CL et al. JTH. 2013;11:1006-1019 Inherited Thrombocytopenias with Adulthood Presentation – – – – – – – – MYH9-related disease monoallelic Bernard-Soulier syndrome ANKRD26-related thrombocytopenia Familial platelet disorder with predisposition to AML (RUNX1) ITGA2B/ITGB3-associated thrombocytopenia TUBB1-related thrombocytopenia CYCS-related thrombocytopenia Thrombocytopenia associated with sitosterolemia* Red=syndromic vs. Blue=non-syndromic *Autosomal recessive (all others dominant) MYH9-RELATED DISEASE MHY9-Related Disease • MHY9 gene at chromosome 22q12-13 • Autosomal dominant • Non-syndromic syndromic over time – Cataracts, sensorineural deafness, altered liver enzymes, and/or progressive glomerulonephropathy (ESRD) • Associated features – Giant platelets • No spontaneous bleeding MYH9 Gene • Encodes heavy chain of non-muscle myosin IIA – aka. Myosin 9 • Non-muscle myosin IIA – Motor protein involved in cellular force contraction • N-terminal motor domain – Mechanical translocation along actin filaments – Mutations here severe thrombocytopenia and full syndrome • Alpha-helical rod domain – Intermediate phenotype • C-terminal non-helical domain – Mildest phenotype with only mild thrombocytopenia MHY9-Related Disease: Pathophysiology • Disease phenotype – Caused by dominant-negative effect of the mutated myosin-9 – NOT caused by the loss of myosin-9 function MHY9-Related Disease: Pathophysiology • Non-hematologic manifestions uncertain • Thrombocytopenia – Reduced number and branching of proplatelets • Leads to fewer number of release platelets from megakaryocytes – Impaired migration of megakaryocytes from the osteoblastic niche to the vascular niche – Premature release of platelets away from the vascular sinusoids MYH9-Related Disease Mutations in which domain(s) is/are most likely associated with this phenotypic morphology? Journal of Thrombosis and Haemostasis Volume 11, Issue 6, pages 1006-1019, 3 JUL 2013 DOI: 10.1111/jth.12196 http://onlinelibrary.wiley.com/doi/10.1111/jth.12196/full#jth12196-fig-0003 MHY9-Related Disease: Diagnosis • Anti-myosin-9 antibodies – Used to identify the inclusion bodies • Present in all neutrophils of all affected patients MONOALLELIC BERNARD-SOULIER SYNDROME Benard-Soulier Syndrome • Genes Associated: – GP1BA gene at chromosome 17p13 – GB1BB gene at chromosome 22q11 – GP9 at chromosome 3q21 Glycoprotein Ib-IXA-V Receptor SFKs in GPIb-IX-V proximal signaling. Yotis A. Senis et al. Blood 2014;124:2013-2024 ©2014 by American Society of Hematology Biallelic Benard-Soulier Syndrome • Autosomal recessive (>50 mutations described) • Genes mutations in BOTH alleles of either – GP1BA gene at chromosome 17p13 – GB1BB gene at chromosome 22q11 – GP9 at chromosome 3q21 • Non-syndromic • Associated features – Giant platelets • Spontaneous bleeding Biallelic Benard-Soulier Syndrome • BOTH alleles must be mutated – Heterozygotes • Slightly enlarged platelets OR • No phenotypic abnormality Biallelic Benard-Soulier Syndrome: Pathophysiology • Mutations prevent receptor assembly in the Golgi apparatus and migration to surface – Absence of GP 1B-IXA-V receptor – Decreased vWF binding – Decreased platelets adhesion at site of vascular injury • Thrombocytopenia with giant platelets BUT – Bleeding is worse than the platelet numbers would suggest Biallelic Benard-Soulier Syndrome: Pathophysiology • Mutations prevent receptor assembly in the Golgi apparatus and migration to surface – Absence of GP 1B-IXA-V receptor – Decreased vWF binding – Decreased platelets adhesion at site of vascular injury • Thrombocytopenia with giant platelets BUT – Bleeding is worse than the platelet numbers would suggest Biallelic Benard-Soulier Syndrome: Diagnosis • PFA-100 – Both collagen and ADP prolonged • Platelet aggregation assays – Decreased aggregation with Ristocetin • Flow cytometry – Absence of GP IB-IXA-V Monoalleleic Bernard-Soulier Syndrome • Autosomal dominant • Genes Associated: – GP1BA gene at chromosome 17p13 – GB1BB gene at chromosome 22q11 – GP9 at chromosome 3q21 • Non-syndromic • Associated features – Giant platelets with GP1BA mutations – Large platelets with GP1BB and GP9 mutations • No spontaneous bleeding Monoalleleic Bernard-Soulier Syndrome • Autosomal dominant – Far fewer mutations described • Genes mutations in ONE allele of either – GP1BA gene at chromosome 17p13 – GB1BB gene at chromosome 22q11 • Non-syndromic • Associated features – Large to Giant platelets • No spontaneous bleeding Monoallelic Benard-Soulier Syndrome: Clinical Features • Thrombocytopenia with giant platelets – Mean platelet count 81 x 109/L • No or trivial bleeding tendency usually – Less commonly more severe thrombocytopenia and bleeding seen • 5% of cases in one series had been treated for ITP Monoallelic Benard-Soulier Syndrome: Pathophysiology • WHY monoallelic forms uncertain – DiGeorge’s syndrome (22q11 microdeletion) has 50% expression but no giant plaletets or thrombocytopenia – Hypothesis • Mutations must exert some dominant effect ? Bernard-Soulier Syndrome: Why Macrothrombocytopenia? • GP IB-IXA-V needed for platelet production – Inhibition blocks proplatelet formation • Proplatelet tips are larger • a-tubulin distribution altered – NO effect on differentiation or maturation ANKRD26-RELATED THROMBOCYTOPENIA ANKRD26-Related Thrombocytopenia • Gene associated – ANKRD26 gene at chromosome 10p2 • Autosomal dominant • Non-syndromic • Associated features – Normal size platelets – Increase risk of acute leukemia (?) • No spontaneous bleeding ANKRD26-related thrombocytopenia: Pathophysiology • Mutations in the 5’-UTR of ANKRD26 on 10p2 • How leads to disease physiology – uncertain – Possibly enhance expression of ANKRD26 • Deletions and partial inactivations do not lead to thrombocytopenia – Bone marrow examinations • Suggest possible dysmegakaryopoiesis • Many smaller or dystrophic megakaryocytes – Hypolobulated nuclei ANKRD26-related thrombocytopenia: Clinical Features • Thrombocytopenia with normal size platelets – Mean platelet count 47 x 109/L • No or mild bleeding tendency usually • Less commonly severe thrombocytopenia (<10 x 109/L) ANKRD26-Related Thrombocytopenia Journal of Thrombosis and Haemostasis Volume 11, Issue 6, pages 1006-1019, 3 JUL 2013 DOI: 10.1111/jth.12196 http://onlinelibrary.wiley.com/doi/10.1111/jth.12196/full#jth12196-fig-0004 ANKRD26-related thrombocytopenia: Clinical Features • 30-fold increase in acute leukemia (?) – Compared with the normal population FAMILIAL PLATELET DISORDER AND PREDISPOSITION TO ACUTE MYELOGENOUS LEUKEMIA Familial Platelet Disorder and Predisposition to AML • • • • RUNX1 gene at chromosome 21q22 Autosomal dominant Non-syndromic Associated features – Normal-size platelets – HIGH risk of AML and MDS • No spontaneous bleeding Familial Platelet Disorder and Predisposition to AML: Pathophysiology • RUNX1 gene at chromosome 21q22 – Encodes for RUNX1 (aka CBFa2) • Core Binding Factor – Heterodimeric transcription complex • CBFa2 important for dimerization and DNA binding • CBFb helps stabilize CBFa2 – Protecting it from degradation – Enhancing its DNA affinity • Mutations usually lead to haploinsufficiency – Some dominant negative mutations also reported • Even higher risk of hematologic malignancy Familial Platelet Disorder and Predisposition to AML: Pathophysiology • RUNX1 gene at chromosome 21q22 – Encodes for RUNX1 (aka CBFa2) • Core Binding Factor – Heterodimeric transcription complex • CBFa2 important for dimerization and DNA binding • CBFb helps stabilize CBFa2 – Protecting it from degradation – Enhancing its DNA affinity – Regulates expression of many hematopoietic-specific genes • Inversely regulates MYL9 and MYH10 • Directly regulates MYH9 • Thus suggests megakaryopoiesis may be effected in part by deregulation of myosin IIA and IIb defective proplatelet formation • Mutations usually lead to haploinsufficiency – Some dominant negative mutations also reported • Even higher risk of hematologic malignancy Familial Platelet Disorder and Predisposition to AML: Clinical Features • Thrombocytopenia with normal size platelets – Platelets may look a little paler – Mild thrombocytopenia • Reduced platelet aggregation with collagen and epi – Platelet counts NOT ALWAYS reduced • Uncertain if also have qualitative platelet defects • No or mild bleeding tendency usually • Easy bruising since childhood in some but not all – Hemoglobin and WBC may be elevated Familial Platelet Disorder and Predisposition to AML: Clinical Features • Increased risk of hematologic malignancies – ESPECIALLY MDS and AML • 40% of affected individual • Mean age of onset 33 years Inherited thrombocytopenias frequently diagnosed in adults Journal of Thrombosis and Haemostasis Volume 11, Issue 6, pages 1006-1019, 3 JUL 2013 DOI: 10.1111/jth.12196 http://onlinelibrary.wiley.com/doi/10.1111/jth.12196/full#jth12196-fig-0005