Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Long non-coding RNA wikipedia , lookup
Essential gene wikipedia , lookup
Pathogenomics wikipedia , lookup
Metagenomics wikipedia , lookup
Vectors in gene therapy wikipedia , lookup
Gene therapy wikipedia , lookup
Epigenetics of diabetes Type 2 wikipedia , lookup
Gene nomenclature wikipedia , lookup
History of genetic engineering wikipedia , lookup
Public health genomics wikipedia , lookup
Gene desert wikipedia , lookup
Minimal genome wikipedia , lookup
Polycomb Group Proteins and Cancer wikipedia , lookup
Therapeutic gene modulation wikipedia , lookup
Genomic imprinting wikipedia , lookup
Oncogenomics wikipedia , lookup
Genome evolution wikipedia , lookup
Biology and consumer behaviour wikipedia , lookup
Ridge (biology) wikipedia , lookup
The Selfish Gene wikipedia , lookup
Site-specific recombinase technology wikipedia , lookup
Nutriepigenomics wikipedia , lookup
Epigenetics of human development wikipedia , lookup
Genome (book) wikipedia , lookup
Artificial gene synthesis wikipedia , lookup
Microevolution wikipedia , lookup
Designer baby wikipedia , lookup
Classifying Lymphoma
Dataset Using Multi-class
Support Vector Machines
INFS-795 Advanced Data Mining
Prof. Domeniconi
Presented by Hong Chai
1
Agenda
(1) Lymphoma Dataset Description
(2) Data Preprocessing
- Formatting
- Dealing with Missing Values
- Gene Selections
(3) Multi-class SVM Classification
- 1-against-all
- 1-against-1
(4) Tools
(5) References
2
Lymphoma Dataset
• Alizadeh et al.(2000), Distinct Types of Diffuse Large Bcell Lymphoma Identified by Gene Expression Profiling
• Publicly available at http://llmpp.nih.gov/lymphoma/
• In microarray data,
Expression profiling of
genes are measured in
rows
Samples are columns
3
Lymphoma Dataset
• 96 samples of lymphocytes (instances)
• 4026 human genes (features)
• 9 classes of lymphoma:
DLBCL, GCB, NIL, ABB, RAT, TCL, FL, RBB, CLL
• Glimpse of data
DLCL
-0042
DLCL0031
DLCL
-0036
CLL
-60
CLL68
FL-10
FL-11
GCB
NILIgM
CLL65
GENE2406X
0.75
0.12
0.28
0.58
0.37
-1.2
0.37
-0.6
0.37
0.12
GENE3689X
0.01
-0.4
-0.6
0.37
0.09
0.78
-0.8
2.45
-0.45
0.37
GENE3133X
1.43
-0.8
0.37
-0.5
0.37
1.15
0.37
-1.29
1.26
0.67
GENE1008X
0.05
0.64
0.37
0.12
0.63
0.35
0.12
0.37
-2.29
-0.8
4
Lymphoma Dataset
5
Goal
Task: classification
• Assign each patient sample to one of 9 categories, e.g. Diffuse
Large B-cell Lymphoma (DLBCL) or Chronic Lymphocytic Leukemia
(CLL).
• Microarray data classification: an alternative to current malignancies
classification that relies on morphological or clinical variables
Medical implications
• Precise categorization of cancers; more relevant diagnosis
• More accurate assignment of cases to high risk or low risk
categories
• more targeted therapies
• Improved predictability of outcome.
6
Data Preprocessing
Missing Values Imputation
•
•
•
•
•
3% of gene expression profiles data are missing
1980 of the 4026 genes have missing values
49.1% of genes (features) involved
Some of these genes may be highly informative for
classification
Need to deal with missing values before applying to
SVM
7
Missing Value Approaches
• Instance or feature deletion
- if dataset large enough & does not distort distribution
• Replace with a randomly drawn observed value
- proved to work well (http://globain/cse/psu.edu/courses/spring2003/3-norm-val.pdf)
• EM algorithm
• Global mode or mean substitution
- will distort distribution
• Local mode or mean substitution with KNN algorithm
(Prof. Domeniconi)
8
Local Mean Imputation (KNN)
1.
Partition the data set D into two sets.
• Let the first set, Dm, contain instances with missing value(s).
• The other set, Dc, contains instances with complete values.
2.
For each instance vector x Dm
• Divide the vector into observed and missing parts as x = [xo; xm].
• Calculate the distance between xo and every instance y Dc,
using only those features that are observed in x.
• From the K closest y’s (instances in Dc), calculate the mean of
the feature for which x has missing value(s). Make substitution
with this local mean.
(Note: for nominal features use mode. n/a in microarray data)
9
Data Preprocessing
Feature Selection: Motivations
- Number of features large, instances small
- Reduce dimensionality to overcome overfitting
- A small number of discriminant “marker” genes
may characterize different cancer classes
Example: Guyon et al. identified 2 genes that yield zero leaveone-out error in the leukemia dataset, 4 genes in the
colon cancer dataset that give 98% accuracy.
(Guyon et al. Gene Selection for Cancer Classification using SVM, 2002)
10
Feature Selection
Discriminant Score Ranking
• Which gene is more informative in the 2-class
case:
+
Gene 1
+
Gene 2
11
Separation Score
• Gene 1 more discriminant. Criteria:
- Large difference of μ+ and μ- Small variance within each class
• Score function
F(gj) = | (μj+ - μj-) / (σj+ + σj-) |
12
Separation Score
• In multi-class cases, rank genes that are
discriminant among multiple classes
C1
C2
Δ
C3
• A gene may functionally relates to several
cancer classes such as C1 and C2
13
Separation Score
• Proposing an adapted score function
For each gene j
Calculate μi in each class Ci
Sort μi in descending order
Find a cutoff point with largest diff(μi, μj)
μexp-cutoff-left
σ+ σexp-cutoff-left
μ- μexp-cutoff-right
σ- σexp-cutoff-right
μ+
F(gj) = | (μj+ - μj-) / (σj+ + σj-) |
Rank genes by F(gj)
Select top genes via threshold
14
Separation Score
Disadvantage:
• Does not yield more compact gene sets;
still abundant
• Does not consider mutual information
between genes
15
Feature Selection
Recursive Feature Elimination/SVM
1. In the linear SVM model on the full feature set
Sign (w•x + b)
w is a vector of weights for each feature, x is an
input instance, and b a threshold.
If wi = 0, feature Xi does not influence classification
and can be eliminated from the set of features.
16
RFE/SVM
2. When w is computed for the full feature set,
sort features according in descending order of
weights. The lower half is eliminated.
3. A new linear SVM is built using the new set of
features. Repeat the process until the set of
predictors is non-divisible by two.
4. The best feature subset is chosen.
17
Feature Selection
• PCA comment: not common in microarray
data.
• Disadvantage: none of original inputs can
be discarded
• We want to retain a minimum subset of
informative genes to achieve best
classification performance.
18
Multi-class SVM
19
Multi-class SVM Approaches
1-against-all
• Each of the SVMs separates a single class from all
remaining classes (Cortes and Vapnik, 1995)
1-against-1
• Pair-wise. k(k-1)/2, k Y SVMs are trained. Each SVM
separates a pair of classes (Fridman, 1996)
Performance similar in some experiments (Nakajima, 2000)
Time complexity similar: k evaluation in 1-all, k-1 in 1-1
20
1 -against- All
• Or “one-against-rest”, a tree algorithm
• Decomposed to a collection of binary classifications
• k decision functions, one for each class
(wk)T • x+bk, k Y
• The kth classifier constructs a hyperplane between class
n and the k-1 other classes
Class of x = argmaxi{(wi)T • (x)+bi}
21
1 -against- 1
• k(k-1)/2 classifiers where each one is trained on data
from two classes
• For training data from ith and jth classes, run binary
classification
• Voting strategy: If
Sign(wij)T • x+bij)
says x is in class i, then add 1 to class i. Else to class j.
• Assign x to class with largest vote (Max wins)
22
Kernels to Experiment
• Polynomial kernels
K(Xi, Xj)=(XiXj+1)^d
• Gaussian Kernels
K(Xi, Xj)=e^(-|| Xi - Xj ||/σ^2)
23
SVM Tools - Weka
Data Preprocessing
• To ARFF format
• Import file
24
SVM Tools - Weka
Feature Selection using SVM
• Select Attribute
• SVMAttributeEval
25
SVM Tools - Weka
Multi-class classifier
• Classify
• Meta
• MultiClassClassifier
(Handles multi-class
datasets with 2-class
classifiers)
26
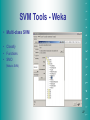
SVM Tools - Weka
• Multi-class SVM
• Classify
• Functions
• SMO
(Weka’s SVM)
27
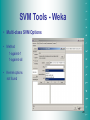
SVM Tools - Weka
• Multi-class SVM Options
• Method
1-against-1
1-against-all
• Kernel options
not found
28
Multi-class SVM Tools
Other Tools include
• SVMTorch (1-against-all)
• LibSVM (1-against-1)
• LightSVM
29
References
• Alizadeh et al. Distinct types of diffuse large B-cell lymphoma
identified by gene expression profiling, 1999
• Cristianini, An Introduction to Support Vector Machines, 2000
• Dor et al, Scoring Genes for Relevance, 2000
• Franc and Hlavac, Multi-class Support Vector Machines
• Furey et al. Support vector machine classification and validation of
cancer tissue samples using microarray expression data, 2000
• Guyon et al. Gene Selection for Cancer Classification using Support
Vector Machines, 2002
• Selikoff, The SVM-Tree Algorithm, A New Method for Handling Multiclass SVM, 2003
• Shipp et al. Diffuse Large B-cell lymphoma outcome prediction by
gene expression profiling and supervised machine learning, 2002
• Weston, Multi-class Support Vector Machines, Technical Report,
1998
30