Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Gene expression wikipedia , lookup
Index of biochemistry articles wikipedia , lookup
List of types of proteins wikipedia , lookup
Immunoprecipitation wikipedia , lookup
Magnesium transporter wikipedia , lookup
Protein moonlighting wikipedia , lookup
Ancestral sequence reconstruction wikipedia , lookup
Protein folding wikipedia , lookup
Homology modeling wikipedia , lookup
Signal transduction wikipedia , lookup
Protein design wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
G protein–coupled receptor wikipedia , lookup
Protein (nutrient) wikipedia , lookup
Protein structure prediction wikipedia , lookup
Western blot wikipedia , lookup
Paracrine signalling wikipedia , lookup
Drug discovery wikipedia , lookup
Interactome wikipedia , lookup
Proteolysis wikipedia , lookup
Nuclear magnetic resonance spectroscopy of proteins wikipedia , lookup
Drug design wikipedia , lookup
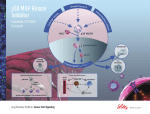
Crystallography, biophysical and cheminformatics studies for next-generation kinase inhibitor design R. A. Engh, Dept of Chemistry, UiT Background: The human genome contains over 500 homologous protein kinases, which control cellular signalling processes. They have become one of the most important target classes for the design of therapeutic inhibitors; many diseases are caused by dysregulation of cellular signalling processes. Most therapeutic inhibitors approved to now are for cancer therapies, but this focus is now broadening. Emerging targets of interest are relevant e.g. for neurodegenerative diseases, inflammation, and other areas. Further protein kinase targets are of non-human origin, with impacts for antibacterial, antifungal, and antiparasitic therapies. Research into protein kinase inhibitor drugs has been progressing for the past 20-30 years with many high-throughput tools, including synthesis, screening, and structural studies. As a result, an enormous body of relevant data exists, including three dimensional structures, binding strengths and selectivity determinants. Further, many and growing numbers of highly active kinase inhibitors are now coming off patent for the first time, including the first approved protein kinase drugs. This creates a drug discovery environment with unparalleled richness. Protein kinase selectivity determinants are known that can retarget such substances to similar but distinct kinases with relatively minor changes. Due to the complexities of drug-target interactions, and especially in connection with the natural flexibility of protein kinases, inhibitor binding properties can be predicted with only limited success. Taken together, these facts highlight the future of protein kinase inhibitor drug discovery: 1) increased interest in new targets, 2) cheminformatics driven and rapid refunctionalization of known binders to these targets, and 3) ongoing discovery of key selectivity determinants. Project goals and activities • (Application writing phase) Evaluation of up-to-date inhibitor and target data to select a protein kinase target area of interest. • Selection of specific constructs for protein production, based on strategies for successful crystallography, variations to identify key binding interactions, and possible related target interactions (to optimize selectivity profiles, avoid toxicities, and forestall drug resistance) • Protein production, crystallography and biophysical binding studies, studying mechanisms of ligand binding strengths and kinetics. • Cheminformatics studies to identify the most promising inhibitor design approaches; modelling and possible synthesis of new compounds The research environment • The project is based at the Norwegian Center for Structural Biology (NORSTRUCT), Department of Chemistry, Tromsø, Norway • NOSTRUCT facilities include state-of-the-art recombinant protein production facilities allowing both prokaryotic and eukaryotic expression with scalable chromatographic purification, and also recently upgraded (2017) systems for automated liquid handling, crystallization, X-ray data collection, and biophysical ligand binding studies (SPR, ITC, MST) • The Department of Chemistry hosts and encourages collaboration with the Center for Computational and Theoretical Chemistry (Norwegian Center of Excellence) and the Norwegian node for the pan-European research infrastructure Elixir • • • • The strategic research focus of the Department of Chemistry prioritizes therapeutic target/ligand interaction studies with a Drug Discovery and Design technology and networking platform, with ongoing projects in antibiotic, anticancer, and other drug discovery areas. Tromsø is a center for marine bioprospecting studies, with potential impact for this projects The project supervisor (https://www.linkedin.com/in/richard-a-engh-77949a88) has extensive experience and a wide ranging publication record beginning with very early protein kinase crystallography efforts (see publications below) Tromsø is a vibrant and unique city north of the Polar Circle with a large international community, often cited in "top ten" lists of places to experience (northern lights, midnight sun, whale watching, unspoiled nature). Selected relevant publications (see also http://www.researchgate.net/profile/Richard_Engh/) • • • • • • • • • • • • • • • • • • • • • • Probing the ATP-binding pocket of protein kinase DYRK1A with benzothiazole fragment molecules; U. Rothweiler, W. Stensen, B.O. Brandsdal, J. Isaksson, F.A. Leeson, R.A. Engh, J.S.M. Svendsen (2016) J. Med.Chem, in press. Addresssing the glycine-rich loop of protein kinases by a multi-dimensional interaction network: inhibition of PKA and a PKB mimic, B.S. Lauber, L.A. Hardegger, A.K. Asraful, B. A. Lund, O.C. Dumele, M. Harder, B. Kuhn, R.A. Engh, F. Diederich (2015) Chemistry doi: 10.1002/chem.201503552. Assessing protein kinase target similarity: examples comparing sequence, structure, and cheminformatics approaches, O.A. Gani, B. Thakkar, D. Narayanan, K.A. Alam, P. Kyomuhendo, U. Rothweiler, V. Tello-Franco, R.A. Engh, (2015) BBA - Proteins and Proteomics, 1854, 1605-1616. The crystal structure of a DYRK1A PKC412 complex reveals disulphide bridge formation with the anomalous catalytic loop HRD(HCD) cysteine, M. Alexeeva, E. Åberg, R.A. Engh, U. Rothweiler, (2015) Acta Cryst D. 71, 1207-1215. Luciferin and derivatives as a DYRK selective scaffold for the design of protein kinase inhibitors, U. Rothweiler, J. Eriksson, W. Stensen, F. Leeson, R.A. Engh, J.S. Svendsen, (2015), Eur. J. Med. Chem. 2015 Feb 25; 94:140-148. doi: 10.1016/j.ejmech.2015.02.035. Evaluating the predictivity of virtual screening for Abl kinase inhibitors to hinder drug resistance, O. Gani, D. Narayanan, R.A. Engh, Chemical Biology and Drug Design (2013), 82, 506-519. Structural origins of AGC protein kinase inhibitor selectivities: PKA as a drug discovery tool. E. Åberg, B. Lund, A. Pflug, O.A.B.S.M. Gani, U. Rothweiler, T.M. de Oliveira, R.A. Engh, Biol. Chem. Hoppe Seyler, (2012), 393, 1121-1129. Anomalous dispersion analysis of inhibitor flexibility-- A case study of the kinase inhibitor H-89. A. Pflug, K.A. Johnson, R.A. Engh, Acta Cryst. F. (2012), 68, 873-7. Differential sensitivity of ErbBB2 kinase domain mutations towards lapatinib, R. K. Kancha, N. von Bubnoff, N. Bartosch, C. Peschel, R. A. Engh, J. Duyster, PLoS One (2011), 6, e26762, doi: 10.1371/journal.pone.0026760. Dynamics of the emergence of dasatinib and nilotinib resistance in imatinib resistant CML patients, F.X. Gruber, T. Ernst, K. Porkka, R.A. Engh, I. Mikkola, J. Maier, T. Lange, A. Hochhaus, Leukemia (2011), doi: 10.1038/leu.2011.187. [Epub ahead of print]. Mutants of protein kinase A that mimic the ATP-binding site of Aurora kinase, A. Pflug, T. M. de Oliveira, D. Bossemeyer, R.A. Engh, Biochem. J. (2011), 440, 85-93. p38 MAP kinase dimers with swapped activation segments and a novel catalytic loop conformation, U. Rothweiler, E. Åberg, K.A. Johnson, T.E. Hansen, J.B. Jørgensen, R.A. Engh, J. Mol. Biol. (2011), 411, 474-85. Epub DOI: 10.1016/j.jmb.2011.06.013. VX680 Binding in Aurora A: π-π Interactions Involving the Conserved Aromatic Amino Acid of the Flexible Glycine-Rich Loop, T.M. Oliveira, R. Ahmad, R. A. Engh, J. Phys. Chem. (2011), 54, 312-319. Epub 2010 Dec 3. Phthalazinone pyrazoles as potent, selective and orally bioavailable inhibitors of Aurora-A kinase, M.E. Prime, S.M. Courtney, F.A. Brookfield, R.W. Marston, V. Walker, J. Warne, A. E. Boyd, N.A. Kairies, W. von der Saal, A. Limberg, G. Georges, R.A. Engh, B. Goller, P. Rueger & M. Rueth, J. Med. Chem. (2011), Epub DOI: 10.1021/jm101346r. Diversity of bisubstrate binding modes of adenosine analogue-oligoarginine conjugates in protein kinase A and implications for protein substrate interactions, A. Pflug, J. Rogozina, D. Lavogina, E. Enkvist, A. Uri, R.A. Engh, D. Bossemeyer, J. Mol. Biol. (2010), 403, 6677. Protein kinase inhibition of clinically important staurosporine analogues, O.A. Gani & R.A.Engh, Nat. Prod. Rep. (2010), 27, 489-498. Crystallography for protein kinase drug design: PKA and SRC case studies; C.B.Breitenlechner, D.Bossemeyer & R.A.Engh; Biochim. Biophys. Acta, Prot. & Proteom. (2005), 1754, 38-49. Protein kinase A in complex with Rho-kinase inhibitors Y-27632, Fasudil (HA-1077) and H-1152P: Structural basis of selectivity; C.B.Breitenlechner, M.Gaßel, H.Hidaka, V.Kinzel, R.Huber, R.A.Engh, & D.Bossemeyer; Structure (2003) 11; 1595-1607. Structural aspects of protein kinase control—Role of conformational flexibility; R.A.Engh & D. Bossemeyer; Pharmacology and Therapeutics (2002) 93; 99-111. Phosphorylation and flexibility of cyclic-AMP dependent protein kinase (PKA) using P-31 NMR Spectroscopy; M.H.J.Seifert, C.B.Breitenlechner, D.Bossemeyer, R.Huber, T.A.Holak & R.A.Engh; Biochemistry (2002) 41; 5968-5977. Crystal Structures of Catalytic Subunit of cAMP-dependent Protein Kinase in Complex with Isoquinolinesulfonyl Protein Kinase Inhibitors H-7, H-8, H-89; R.A.Engh, A.Girod, V.Kinzel, R.Huber & D. Bossemeyer; J. Biol. Chem. (1996) 271; 26157-26164. Phosphotransferase and substrate binding mechanism of the cAMP-dependent protein kinase catalytic subunit from porcine heart as deduced from the 2.0 Å structure of the complex with manganese(2+) adenylyl imidodiphosphate and inhibitor peptide PKI(5-24); D.Bossemeyer, R.A.Engh, V.Kinzel, H.Ponstingl & R.Huber; EMBO Journal (1993) 12; 849-59.