Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Metastability in the brain wikipedia , lookup
Psychoneuroimmunology wikipedia , lookup
Activity-dependent plasticity wikipedia , lookup
Aging brain wikipedia , lookup
Stimulus (physiology) wikipedia , lookup
Development of the nervous system wikipedia , lookup
Synaptogenesis wikipedia , lookup
Haemodynamic response wikipedia , lookup
Subventricular zone wikipedia , lookup
Neuroanatomy wikipedia , lookup
Optogenetics wikipedia , lookup
Neuropsychopharmacology wikipedia , lookup
Molecular neuroscience wikipedia , lookup
Alzheimer's disease wikipedia , lookup
Feature detection (nervous system) wikipedia , lookup
Clinical neurochemistry wikipedia , lookup
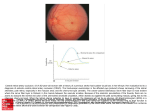
Send Orders for Reprints to [email protected] Current Alzheimer Research, 2017, 14, 1-12 1 REVIEW ARTICLE Seeing Early Signs of Alzheimer’s Disease Through the Lens of the Eye a,b,c,d Brian T. Reed a,b,c,d , Francine Behar-Cohen a,b,c,d,* and Slavica Krantic a Sorbonne Universités, UPMC Univ Paris 06, UMR_S 1138, Centre de Recherche des Cordeliers, F-75006, Paris, France; bINSERM, UMR_S 1138, Centre de Recherche des Cordeliers, F-75006, Paris, France; cUniversité Paris Descartes, Sorbonne Paris Cité, UMR_S 1138, Centre de Recherche des Cordeliers, F-75006, Paris, France; d Centre National de la Recherche Scientifique (ou CNRS) ERL 8228, Centre de Recherche des Cordeliers, Paris, France ARTICLE HISTORY Received: January 10, 2016 Revised: August 01, 2016 Accepted: August 10, 2016 DOI: 10.2174/1567205013666160819 131904 Abstract: Alzheimer's disease (AD) develops undetected for years due to the lack of early diagnostic biomarkers. In advanced AD, visual deficits related to cortical neurodegeneration are well recognized, but recent studies have identified that the retina could be affected prior to vulnerable brain areas such as cortex and hippocampus. In this review, we focus on new evidence suggesting that synaptic dysfunction within the retina may be reminiscent of changes within the brain. The data on the earliest dysfunction of synaptic and neuronal networks in vulnerable brain areas (mostly cortex and hippocampus) are next discussed to point out how they may inspire the analogous research in the retina during the asymptomatic stage of AD. We finally present evidence indicating why putative retinal synaptic dysfunction holds the potential to become the earliest sign of AD, allowing for a non-invasive and easy detection using modern imaging and functional techniques. Translation of these findings to clinical diagnosis could lead to earlier therapeutic interventions and, consequently, better chances to delay or halt AD progression. Keywords: Alzheimer’s Disease, neurodegeneration, amyloid plaques, tau protein, glial activation. 1. INTRODUCTION Alzheimer’s disease (AD), a neurodegenerative disease with poorly understood pathogenesis, is the leading cause of dementia in elderly. The number of patients with AD and related dementias is estimated to 44 million people worldwide (http://www.alz.co.uk/research/world-report-2014). The currently available AD treatments are all palliative. They include rivastigmine, galantamine and donepezil which all target cholinergic neurotransmission. Memantine, an antagonist of N-methyl D-aspartate (NMDA)-type of glutamate receptors, is the last AD drug approved for clinical use. However, the benefit provided by these drugs used to treat mild to moderate stages of AD is only limited. Indeed, the current treatments provide partial relief of cognitive symptoms only in a sub-population of patients, and their efficacy has been recently questioned in some countries, such as France (http://www.has-sante.fr/portail/upload/docs/application/pdf/2011-11/rapport_evaluation_mdc_alzheimer). The lack of curative treatments has recently motivated a “shift in focus” towards earlier diagnosis of AD which should allow for the earlier application of the existing treatments. The rationale is that the earlier application of the available treatments may translate to better efficacy. In addition, due to the long latency between the beginning of AD *Address correspondence to this author at the Centre de Recherche des Cordeliers, UMRS 1138, équipe 17, 15, rue de l’école de médecine, 75 006 Paris, France; Tel: (33) 144 - 278 - 187; Fax: (33) 144 - 279- 036; E-mail: [email protected] 1567-2050/17 $58.00+.00 pathogenesis and the manifestation of the clinical symptoms, earlier diagnosis may allow for application of non-medicinal intervention strategies (cognitive enhancement, healthy diet, exercise, etc.) to prevent the onset of full-blown AD pathology and its progression towards irreversible neurodegeneration. Such a “shift in focus” relies obviously on the discovery of early diagnostic biomarkers. In this review, we will discuss the evidence pointing to the retina as a potential source of the earliest diagnostic tissue for AD. This discussion is preceded by a short presentation of the physiological function of the retina and of the current methods that can be used for non-invasive retinal exploration of neurodegenerative changes. Early diagnosis of AD utilizing changes in the retina remains challenging because of the similarities between the pathogenesis of AD, glaucoma and age-related macular degeneration (AMD). These similarities are confounding in terms of diagnosis because they exhibit a high incidence of comorbidity with AD (e.g. glaucoma and AD). However, these aspects will not be discussed here. Instead, recently published reviews on these topics are recommended [1, 2]. Although all parts of the visual system and, more specifically, the visual cortex, have been reported to be affected in the advanced stages of AD [2], in this review, we focus exclusively on the retina. Finally, we provide arguments in favour of monitoring retinal neuronal activity for the purpose of identification of the earliest AD biomarkers using non-invasive diagnostic exploration. © 2017 Bentham Science Publishers 2 Current Alzheimer Research, 2017, Vol. 14, No. 1 1.1. Background on AD AD comprises a number of progressive and age-related cognitive and behavioral impairments. The main neuropathological hallmark of AD is the accumulation of amyloidbeta (Aβ) peptide produced by successive proteolytic cleavage from its precursor APP (amyloid-beta precursor protein) along the amyloidogenic pathway [3]. Aβ accumulates extracellularly as amyloid plaques (or senile plaques). Intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein are also a prominent pathological feature of AD [4]. The cause of sporadic AD is still unknown and among different theories proposed for AD pathogenesis, the amyloid cascade hypothesis is currently the most generally accepted. This hypothesis postulates that the overproduction of Aβ from its precursor APP triggers a series of events, including synaptic dysfunction, glial activation and hyperphosphorylation of tau, which are associated with widespread neuronal death [4]. The amyloid hypothesis is supported by strong genetic evidence coming from the pathogenesis of rare (25%) familial forms of AD. Patients suffering from the familial forms of AD bear mutations in the APP gene, which yield overproduction of Aβ and is therefore more prone to aggregation. The additional genetic mutations leading to AD were identified in the genes encoding presenilin 1 and 2, PSEN1 and PSEN2, respectively. However, although the familial and the much more frequently occuring (95-98%) sporadic forms of AD share the same hallmark histopathological lesions (notably extracellular plaques and intracellular tangles), the etiology of the sporadic form remains unknown. Remarkably, experimental mutations in APP or presenilins recapitulate many of the pathological features of AD and provide solid support for the amyloid cascade hypothesis. The majority of transgenic mouse models of AD were generated based on the over-expression of mutated human APP, PS1/2 genes, as well as of their combined mutations into the mouse genome. These mice are commonly used to develop and test new treatments. It has however been recently recognized that the transgenic AD mouse models which are based on the amyloid cascade hypothesis, mimic faithfully only the initial stages of AD pathogenesis [5]. 1.2. Current Diagnostic Tools for AD Currently, AD diagnosis is mainly based on the clinical symptoms such as loss of cognitive functions that affect the individuals’ autonomy in executive daily tasks. Cognitive impairments include short-term, which precedes long-term, memory loss as the disease progresses. Cognitive symptoms are often combined with neurological (seizures, parkinsonism-like alterations), behavioral (agitation, sleep disturbances, aggression) and neuropsychiatric (depression, paranoia, hallucinations) symptoms. Some of these symptoms are clinically discernable at the time of diagnosis (anxiety, depression) whilst others (paranoia, hallucinations) appear later in the course of the natural history of AD. However, the clinical panel of symptoms is considerably heterogeneous with important inter-individual differences. Such a heterogeneous panel of symptoms complicates diagnosis. The systematic follow-up of individuals at risk of developing the familial form of AD due to autosomal dominant Reed et al. mutations allowed for the estimation of the age of AD onset based on the age at which the parents began experiencing symptoms. An increased level of Aβ in the cerebrospinal fluid has been detected as early as 25 years before the expected age of AD onset [6]. Relevantly, monitoring Aβ (and hyperphosphorylated tau) levels in the cerebrospinal fluid had initially raised much hope that they could serve as early diagnostic biomarkers for AD [7-9]. However, it turned out that either Aβ or hyperphosphorylated tau alone do not provide sufficient diagnostic accuracy likely because AD is a multifactorial, complex disease. According to the current view, combining Aβ and hyperphosphorylated biomarkers with structural and functional brain changes, as well as with follow-up of memory impairments, may allow more reliable diagnosis (reviewed in [10]). Furthermore, Aβ plaque imaging by Positron Emission Tomography (PET) with radiolabeled (C-11) or F18 counterpart of Pittsburgh compound-B (PiB) was also considered to be a helpful diagnostic tool [7, 11, 12]. Unfortunately, PET screening turned out to detect a high PiB retention in one third of cognitively normal elderly subjects [7, 11, 13]. Limited availability of PET facilities and the high cost of the procedure, combined with a high percentage of the “false-positives” make PiB screening unlikely to become a general tool for early AD diagnosis. 1.3 Alterations of Neuronal Functions in AD Despite the “shift in focus” towards identifying the putative early biomarkers for AD, it has been recognized for at least the last 20 years that synaptic loss represents the best functional correlate for cognitive impairment [14]. More recently, it became clear that the alteration in synaptic activity precedes neuronal loss, and subsequent repercussions at the neuronal circuit and network levels may also be considered as attractive candidates for an early diagnostic tool. 1.3.1. Synaptic Dysfunctions The vast majority of published data on the early synaptic dysfunction investigated brain regions such as hippocampus and cortex that are known to be the most vulnerable to AD. The Aβ-sensitive deviations from normal synaptic function are detectable at both molecular and cellular levels. Gene expression studies have found abnormal patterns of neuronal activity before the onset of overt AD symptoms. For instance, Arc is often over- or under-expressed in the hippocampus of pre-plaque AD model mice that express high soluble Aβ levels [15, 16]. This is an important finding since the fluctuations in expression of this synaptic activitydependent gene indicates network instability. At the cellular level, Aβ-mediated alterations of synaptic transmission involve, among other mechanisms, direct impact on glutamatergic signaling pathways. Ionotropic glutamate receptors (iGluR), α-amino-3-hydroxy-5-methyl-4isoxazolepropionic acid (AMPA) and NMDA receptors are particularly sensitive to Aβ. In the hippocampus of AD mice (hAPPJ20), the expressions of AMPA- and NMDA receptors at the post-synaptic membrane are both reduced [17]. The mechanism of Aβ-mediated decrease in surface AMPA receptor expression involves increased intracellular calcium levels and phosphorylation of glutamate receptor subunit 2 (GluR2) [18]. In the case of NMDA receptors, decreased expression at the post-synaptic membrane likely stems from Seeing Early Signs of Alzheimer’s Disease Aβ-promotion of receptor trafficking from the synaptic- towards the extra-synaptic location [19]. The reported decrease in NMDA and AMPA receptor expression has been further associated with the loss of dendritic spines in the postsynaptic compartment of the excitatory synapses in both AD mice [20-23] and human post-mortem studies [24, 25]. In addition to their direct effect on iGluR, soluble Aβ oligomers have been reported to impact glutamatergic neurotransmission indirectly by modulating the activity of other types of receptors. For example, Aβ oligomers interact with nicotinic acetylcholine receptors (α7-nAChRs) yielding a decreased calcium response [26], which may be related to subsequent alterations in neuronal excitability [17]. Consistently, α7-nAChRs are required for Aβ-mediated increased endocytosis of post-synaptic NMDA receptors subsequent to NR2B subunit dephosphorylation [19]. This results in a rapid and a persistent decrease of NMDA-evoked currents in primary cultures of cortical neurons [19]. All data discussed above that lead to alterations in glutamatergic neurotransmission. However, calcium imaging studies have found that hyper- and hyporeactive neurons coexist in the cortex of APP/PS1 double transgenic mice during the early stages of pathogenesis [27]. More recently, the same group has reported that in the hippocampus, both silent and hyperactive neurons occur in the vicinity of Aβ-plaques [28]. Remarkably, the hyperactive neurons have also been identified in the absence of Aβ-plaques in the hippocampus of young, asymptomatic APP/PS1 mice using bi-photon calcium imaging [28]. Regarding the neuronal hyperactivity, Minkeviciene and colleagues have shown that glutamatergic pyramidal cells in the cortex of APP/PS1 mice have more depolarized resting membrane potentials, lower action potential initiation threshold, and generate more action potentials to a given stimulus at a higher frequency [29]. All of these alterations have been detected well before the onset of the overt AD pathology, and in particular before a significant accumulation of Aβ-plaques [29]. Similar findings were obtained in another strain of AD mice (TgCRND8). In the latter study, the resting calcium levels in CA1 pyramidal neurons were found to be higher in pre-plaque (2 months-old) TgCRND8 mice than in controls [30]. Interestingly, this study has also shown that in young, asymptomatic TgCRND8 mice, there was a narrowing of the action potential waveform, which is likely related to the observed increase in Kv3-type fast delayed rectifier potassium current. Most importantly, these subtle alterations occur weeks and perhaps months before any other synaptic alteration (paired-pulse ratio, AMPA/NMDA receptor ratio, miniature excitatory postsynaptic potentials (ESPS), or expression of other potassium and sodium ionic channels) could be detected [30]. 1.3.2. Neuronal Circuit and Network Impairments Neuronal organization into circuits, and at the higher level, neuronal networks, is superimposed onto neuronal communication via synaptic neurotransmission. This communication has to be coordinated in order to support cognitive function, notably learning and memory. An adaptive value of such organization may be linked to the capacity of the network, at least during the very first stages of AD, to compensate for the aberrant excitatory synaptic activity (dis- Current Alzheimer Research, 2017, Vol. 14, No. 1 3 cussed above) by remodeling of inhibitory synaptic circuits [17]. In addition to its effects on synaptic function, Aβ can induce changes at the higher levels of brain organization complexity. As for neuronal circuits, long term potentiation (LTP) and long term depression (LTD) have been reported as Aβ-sensitive targets [31, 32], in line with the fact that increased and decreased surface expression of AMPA receptors may underlie LTP and LTD, respectively. LTP and LTD are indeed recognized as a cellular basis of learning and memory and deficits are present in early stages of AD [33, 34]. Pioneering work from Palop and colleagues was the first to define the link between Aβ and hippocampal network dysregulation [17]. In particular, excessive network synchronization has been reported and linked to a specific type of inhibitory gamma-amino butyric-acid (GABA) neuron within the hippocampus [35]. More specifically, Verret and colleagues have shown that restoring normal expression of a specific voltage-gated sodium channel (Nav1.1) reduced abnormal network synchronization and memory deficits in hAPPJ20 mice [35]. In addition, the oscillatory activity of neuronal networks is negatively affected by the hyperexcitability and impaired plasticity of synapses. Most importantly, the hippocampal hyperactivation is correlated with cortical thinning (a sensitive and specific marker of AD neurodegeneration) within MCI patients [36]. Increasing evidence suggests that cortical oscillatory activity in the theta [37-39] and gamma [40, 41] ranges are altered in AD patients. While the extent of the changes in hippocampal network activity in asymptomatic AD is still unclear, changes in theta oscillations are viewed as a possible predictor of AD [42]. Furthermore, impairments in thetagamma cross-frequency coupling in hippocampus have been reported in a pre-clinical mouse model of AD [43]. Therefore, at least in the hippocampus (Table 1), subtle disturbances in the communication between groups of neurons and the synchronization of network activities may occur much earlier than any other AD-related pathological lesion, including Aβ accumulation [44]. These recent data suggest that electrophysiological-based approaches may become a sensitive alternative for detection of the early AD-related dysfunction of neuronal activity and networking. 2. THE RETINA The vertebrate retina is the layer of light-detecting tissue lining the back of the eye. It contains neurons, glial cells, and vascular tissue, and has a high level of metabolic activity. In addition, the retina has an outermost layer, the retinal pigment epithelium. The retinal pigment epithelium provides regulatory and nutrient factors to the neurons and plays an important role in the homeostasis of the adjacent photoreceptors in the outer nuclear layer. Glial cells of the retina comprise three major types: Müller cells, astrocytes, and microglia [45, 46]. Müller cells are the most common type of retinal glial cells. They span the retina radially and maintain the homeostasis of the extracellular environment. As pigment epithelium cells, Müller cells provide regulatory and nutrient support for the neigh- 4 Current Alzheimer Research, 2017, Vol. 14, No. 1 Table 1. Reed et al. Hippocampal dysfunctions in the brain of mouse AD models. Target Alteration Type of change References Arc Expression +/- [15, 16] Glutamate receptors (R) Post-synaptic membrane - [17] via AMPA R Intracellular Ca2+ level + [18] Glu R2 phosphorylation + [18] Post-synaptic membrane - [17] R trafficking towards extrasynaptic space + [19] Density - [20-23] - [17] + [19] +/- [27, 28, 30] Membrane potential - [29] AP initiation treshold - [29] AP frequency + [29] LTP - [31, 33] LTD + [32, 34] Network communication Synchronization + [35] Network excitability Nav1.1 subunit of Na+ channel expression - [36] Theta-gamma cross-frequency Oscillation coupling - [43] Neuronal level via NMDA R Dendritic spine α7 nAChR Signaling via Ca NR2B Endocytosis Neuronal excitability 2+ 2+ Intracellular Ca level Neuronal circuit and network levels Synaptic plasticity boring neurons [47]. Astrocytes are located primarily in the nerve fiber layer. In addition to fulfilling some of the same roles as Müller cells [48], astrocytes play a critical role in cell communication at the tripartite synapse (the latter involves astrocytes in addition to pre- and post-synaptic terminals which are the constituents of the classical, bipartite synapse) [49]. In contrast to Müller cells and astrocytes, which together with neurons constitute the population of excitable cells of the retina, microglia are classically considered as non-excitable cells, at least in the brain [50]. However, this concept has been challenged [51]. Microglia are located throughout the retina and the main physiological role is to survey the retinal microenvironment. In pathological conditions, microglia can mount a local immune response and undertake a macrophage function after injury, such as cleaning up debris and removing dead neurons [52]. The main physiological function of the retina is to convert a visual scene into a diverse set of electrical signals and relay these signals to the brain along the optic nerve. This function is achieved by retinal neurons through a number of complex signal processing operations to be discussed below [53]. 2.1. Retinal Neurons The vertebrate retina contains complex networks of neuronal circuitry, which can be divided into three layers of neuronal cell bodies and two layers of synapses. The neuronal layers contain the somas of five types of neurons: i) photoreceptor cell bodies are arranged into the outer nuclear layer; ii) horizontal, bipolar and amacrine cell bodies are located in the inner nuclear layer, and iii) ganglion cell bodies constitute the ganglion cell layer (Fig. 1). Photoreceptors, bipolar and ganglion cells are glutamatergic neurons, whereas horizontal and amacrine cells are inhibitory neurons using, GABA as a principal neurotransmitter [54-56]. The neurons extend axonal and dendritic processes into the two synaptic layers (inner and outer plexiform layers) where they connect to each other synaptically (Fig. 1). Such spatial organization is optimized to provide the physiological output of the retina in the form of action potentials. Photoreceptors are the sensory neurons responsible for the light detection and its conversion to an electrophysiological signal, which is transmitted to their post-synaptic partners. There are two types of photoreceptors, rod and cone photoreceptors, in the outer nuclear layer. Rods mediate scotopic (dim) vision, and cones mediate photopic (bright) vi- Seeing Early Signs of Alzheimer’s Disease Current Alzheimer Research, 2017, Vol. 14, No. 1 5 sion. Rods outnumber cones by a ratio of 20:1 [57]. Cones are concentrated in a specific region of the retina known as macula. All photoreceptors contain a light-sensitive photopigment which triggers a phototransduction cascade in which the light stimulus is transduced into an electrical signal [58] resulting in hyperpolarization of the photoreceptor membrane potential. Hyperpolarization reduces the amount of the excitatory neurotransmitter glutamate that the photoreceptors release into the synaptic space. In the dark, photoreceptors are depolarized and steadily release glutamate [59]. Horizontal cells are synaptically connected to photoreceptors and their main function is to measure the intensity of the synaptic output from the photoreceptors and control their excitability [60]. In addition, horizontal cells receive signals from laterally-placed, surrounding photoreceptors and provide inhibitory feedback signals to centrally-located photoreceptors through a process known as lateral inhibition [6164]. Similar to horizontal cells, bipolar cells also receive direct synaptic input from rod and cone photoreceptors along with some modifying input from horizontal cells in the outer plexiform layer [65] (Fig. 1). Bipolar cells further transmit to ganglion cells the visual signal received from each individual photoreceptor after specific processing [66]. Each type of bipolar cell has unique morphological and neurochemical properties as well as unique response properties [67] that are determined principally by the type of neurotransmitter receptors and ion channels expressed. Bipolar cells fall into two general categories, ON and OFF, based on their respective responses to light. ON bipolar cells express mGluR6 glutamate receptors which, upon glutamate binding, trigger closing of TRPM1 channels [68] and inhibit neurotransmission. When the photoreceptor membrane is hyperpolarized by light, less glutamate is released into the synaptic cleft between photoreceptor and ON bipolar cells. ON bipolar cells, which receive less glutamate from the relevant photoreceptor, will therefore have fewer mGluR6 receptors bound by glutamate. Consequently, bipolar cell neurotransmission is less inhibited which results in increased depolarization and subsequent activation of ON bipolar cells. OFF bipolar cells express AMPA and kainite glutamate receptors [69-71]. Engagement of AMPA and kainate receptors (which are both cation channels) by glutamate yields the rapid depolarization of OFF bipolar cells. When the photoreceptor membrane is hyperpolarized by light, less glutamate is released into the synaptic cleft between the photoreceptor and the bipolar OFF cells. Decreased glutamate release results in the opposite response of the OFF bipolar cells (as compared to the ON cells), i.e. an increase in the hyperpolarization (less depolarization) of the OFF bipolar cells. Fig. (1). Schematic diagram of the mammalian retina. In the three neuronal layers are the cell bodies of five major types of neurons: photoreceptors (1), horizontal cells (2), bipolar cells, amacrine cells (3), and ganglion cells. Two subtypes of bipolar cell are depicted: ON (4) and OFF (5) bipolar cells, along with two subtypes of ganglion cell: ON (6) and OFF (7) ganglion cells. The neurons extend their axonal and dendritic processes into the two synaptic layers where they make synaptic connections with each other. In this simplified scheme of ON and OFF pathways, part of the retina is exposed to a light stimulus (area under the white bar) while another part of the retina is dark (area under the gray bar). Depolarized neurons in the ON pathway are red, and hyperpolarized neurons in the OFF pathway are blue. For clarity, only a few bipolar cells of each class are colored and shown connecting to ganglion cells. A horizontal cell provides inhibitory input to a photoreceptor synaptic terminal. Arrows indicate the direction of the flow of the signal, and ‘+’ and ‘-‘ indicate excitatory or inhibitory polarity. Abbreviations: ONL, outer nuclear layer; OPL, outer plexifom layer; INL, inner nuclear layer; IPL, inner plexiform layer; GCL ganglion cell layer. Ganglion cells are distinguished by their unique molecular identities, response properties, and central projections [72-74]. There are about 20 different types of ganglion cells. Each type has a specific preferred feature to which it is optimally responsive [72]. For example, a specific ganglion cell type receives ON and OFF components transmitted by specific ON and OFF bipolar cells. The ON- and OFF- components of the initial signal generated by the photoreceptor is integrated by the ganglion cells which compare the incoming ON- and OFF- inputs from the bipolar cells [63]. The visual signal is integrated by ganglion cells to yield action potentials that are transmitted along the axons, which ultimately come together and form the optic nerve. The axons of ganglion cells form the nerve fiber layer, which exits the back of the eye. Amacrine cells, which have their somas located in the inner nuclear layer, make synaptic contacts with bipolar and ganglion cells in the inner plexiform layer. This layer is stratified, and specific types of ganglion cells make selective connections to bipolar and amacrine cells in each strata or sub-layer [75]. Because the axon terminals of specific types 6 Current Alzheimer Research, 2017, Vol. 14, No. 1 of bipolar cells terminate in strata unique to each bipolar cell type, the connections that ganglion cells make with these bipolar cells shape the ganglion cells' response properties [76]. For example, ON ganglion cells receive input from light-depolarizing bipolar cells in the inner portion of the inner plexiform layer, and OFF ganglion cells receive input from hyperpolarizing bipolar cells in the outer portion of the inner plexiform layer. In the inner plexiform layer, amacrine cells provide inhibitory signals received from surrounding bipolar cells to central bipolar cells just as horizontal cells provide inhibitory signals received from surrounding photoreceptors [77, 78]. The major function of the amacrine cells is therefore the “context” detection. This means that, by receiving information from bipolar cells as well as receiving feedback from other amacrine cells, amacrine cells “sense” the global state of activity of the entire set of the neurons that respond to a given light stimulus. Obviously, such a complex processing of visual information requires highly orchestrated communication between the retinal neurons at all levels of organization (synapses, circuits and networks). 2.2. Probing Retinal Neuronal Function Retinal ganglion cells transmit visual information from the retina to the brain by the firing of action potentials. This bioelectrical activity is detectable in vitro by classical methods of electrophysiology such as patch-clamp recording (at the cellular level) and field potential recording (at the level of neuronal populations). In addition, the global output of the electrophysiological activity of the retina can be monitored in vivo by electroretinogram (ERG). 2.2.1. Cellular Electrophysiology The electrophysiological activity in the retina has been extensively studied at the cellular level (more than 2000 citations for “retina cellular electrophysiology” since the first recordings performed in 1950-ies). By contrast, there are only a limited number of studies on the mechanisms individual neurons use to communicate, or in other words, how neuronal activities organize in network wiring patterns [79]. In this context, synchronization of retinal neuronal activity can be considered as an output of communication between distinct neurons or a group of neurons (neuronal circuits). Synchronized firing has been extensively documented in retinal ganglion cells, however the extent and purpose are poorly understood [80]. While little is known about the contribution of synchronized retinal neuronal firing to the coding of visual information, some exploratory studies have shown that synchronized firing can encode information about the presence and direction of certain kinds of motion [80, 81]. Of note, and as a specificity of spatial organization of retinal neurons, ganglion cells can undergo electrical coupling with amacrine cells via gap junctions. Synaptic input from both bipolar and amacrine cells, in combination with coupling of ganglion cells and amacrine cells via gap junction, contributes to the synchronized firing [80, 82] of retinal ganglion cells under certain stimulus conditions [80]. 2.2.2. Electroretinogram (ERG) The ERG is a diagnostic tool used to investigate functional changes in the retina. Several types of ERG have been Reed et al. developed. The full-field flash ERG, for example, is the basic method [83] for measuring the massed bioelectrical response of the retina, and it can isolate contributions of various parts of the retina. Another type of ERG, the pattern ERG, stimulates the retina with patterned stimuli such as reversing checkerboards and can isolate the activity of different components of the retina including the firing of ganglion cells [84]. The stimulus for the flash ERG is usually a full-field flash of light of certain duration that covers the entire visual field. Since the discovery of the flash ERG in the 19th century, a series of four component waves: a, b, c, and d arising from different processes and locations in the retina have been described. The a-wave follows the onset of the stimulus and is generated by the photoreceptors. Immediately following the a-wave is the b-wave, arising from the bipolar and Müller cells of the inner retina. The amplitude of the waves indicates the health of the part of the retina from which they originate [85]. The c wave is generated by the retinal pigment epithelium due to an increase of the trans-epithelial potential by the light-induced bioelectrical activity of photoreceptors. This wave gives insights into the function of epithelial cells and photoreceptors [83]. In 1999, a variant of the full-field flash ERG was developed to provide an objective method to measure the relationship between the function and the integrity of retinal ganglion cells. This variant is designated as the photopic negative response (PhNR). PhNR is a slow corneal negative potential arising after the b-wave of the standard photopic ERG [86, 87]. The scotopic threshold response is another variant of the full-field flash ERG in which dim (scotopic) stimuli are applied to the retina under dark-adapted conditions. Because scotopic vision is mediated by rods, this ERG variant provides insight specifically into rod function. It is a threshold response because it occurs near the threshold of detection for rods [86, 88]. Importantly, certain types of synchronized activity can be observed in the ERG. For example, the synchronous activity of networked amacrine cells is believed to be a source of oscillatory potentials, voltage fluctuations sometimes observed climbing the rising arc of the ERG b-wave [82, 8992]. 2.3. Summary of Retinal Function The vertebrate retina is the sensory tissue that lines the back of the eye. Through a number of complex signal processing operations, the retina converts a visual scene into a diverse set of electrical signals and relays them to the brain along the optic nerve. The retina contains six classes of neurons. In the outer nuclear layer, the photoreceptors transduce the visual stimulus into an electrical signal. Horizontal cells, whose cell bodies are in the inner nuclear layer, mediate lateral interactions between the photoreceptors and bipolar cells in the outer plexiform layer. Bipolar cells transfer the visual signal vertically to the inner plexiform layer. Amacrine cells provide vertical and additional lateral interactions in the inner plexiform layer. Ganglion cells integrate the signal and transfer it Seeing Early Signs of Alzheimer’s Disease along the optic nerve to the brain in the form of action potentials. The output of the retina is in the form of action potentials generated by retinal ganglion cells. The waveforms that make up the ERG can be isolated to show the contributions of various cell types and layers of the retina. 3. RETINA AND ALZHEIMER’S DISEASE According to the classical point of view, AD pathology leads to progressive neurodegeneration, which affects primarily the brain areas involved in cognition such as hippocampus and entorhinal cortex. Visual functions have been considered as relatively spared until late stages of AD. Indeed, routine ophthalmologic examination of AD eyes points to a normal appearance of retina [2]. From the clinical point of view, psychophysical testing has shown that color vision, visual field impairments, contrast sensitivity, depth and motion perception are all altered in advanced stages of AD (for [2]. Consistently, longitudinal studies of normal aging have pointed to a link between decline in visual acuity and cognitive capacities [93]. In an inverse approach, it has been shown that in neurologically normal elderly subjects, poor vision has a predictive value for development of dementia [94]. Taken together, these clinical data indicate that visual deficiencies may be a part of the clinical panel of symptoms in advanced stages of AD. This consensus is further supported by the fact that a number of AD-related alterations in the eye, including retinal changes and optic nerve fiber thinning, have also been identified in advanced AD [95-97]. However, recent research suggests that at the early stages of disease, non-cognitive functions, including visual perception and processing, may also be affected. 3.1. AD-Related Changes in the Retina Both intracellular and extracellular accumulation of the proteins (hyperphosphorylated tau and Aβ) have been reported in the retina. The ectopic accumulation of these proteins is associated with significant structural changes of the retina and optic nerve although these alterations are mainly detectable only when pathology has significantly progressed. 3.1.1. Aβ Plaques and Neurofibriallry Tangles Aβ plaques and neurofibriallry tangles have been identified in the retina in advanced stages of the disease both in AD mouse models [98-100] and in post-mortem human retina from AD patients [101]. At least in mice, neurofibrillary tangles and Aβ plaques spread along all layers of the retina as in Tg2576 mice [102, 103]. By contrast, in APP/PS1 double transgenic mice most of Aβ plaques were preferentially found in the ganglion cell layer and inner plexiform layer [102, 104]. However, in all AD mouse strains studied, the accumulation of retinal Aβ plaques was age-dependent, as detected in the human brain [105]. Some strains of AD mutant mice undergo the accumulation of intracellular Aβ within retinal ganglion cells as well as inner and outer nuclear layers of the retina [106]. Most importantly, the recent study using hyperspectral imaging has shown that they are detectable in the retina of APP/PS1 mice during the asymptomatic stage of AD [107]. The as- Current Alzheimer Research, 2017, Vol. 14, No. 1 7 ymptomatic stage refers to the preclinical phase in human pathology which progresses insidiously from amyloidosis with soluble Aβ oligomers alone (stage 1), amyloidosis combined with neuronal damage (stage 2) up to the subtle cognitive decline, undetectable by standard tests (stage3). The asymptomatic stages precede the clinical stage, characterized by mild, but clinically detectable, cognitive impairment (MCI) and biomarker evidence for AD. The final, more advanced stage is defined in humans as dementia due to AD [108]. Therefore, the detection of soluble Aβ oligomers in mouse retina would correspond to the stage 1 of the preclinical phase which occurs a few months before Aβ plaques are detectable This is a crucial finding since the soluble Aβ oligomers, rather than Aβ plaques, are currently considered the main Aβ toxic species [109, 110]. Soluble oligomers accumulate at synapses yielding impairment in synaptic plasticity and memory before Aβ is deposited in plaques and before neurodegeneration is detectable [111]. The data obtained using the new hyperspectral imaging technology for retina exploration strongly suggest that early Aβ-related synaptic alterations may begin in the retina during the asymptomatic stage. 3.1.2. Structural Changes of the Retina and Optic Nerve The thinning of the retinal neurofibrillary layer, which contains the axons of the ganglion cells which form the optic nerve, has been repeatedly demonstrated in advanced AD [2] and is correlated with a decrease in the density of optic nerve fibers [96, 112]. The neurofibrillary layer thinning is combined with reduction of macula thickness/volume, which is in turn correlated with the degree of cognitive impairment in advanced AD [113]. In the early stages of AD, spectral domain optical coherence tomography (SD-OCT) techniques have detected neurofibrillary layer degeneration in MCI (mild cognitive impairment) [114]. SD-OCT is a non-invasive, non-contact method that allows for a precise and quantitative assessment of retinal morphology with longitudinal, reproducible follow-up. At the cellular level, the thinning of the neurofibrillary layer is accompanied by a loss of ganglion cell neurons [115, 116]. Most importantly, ganglion cell loss is preceded by dendritic atrophy of these neurons, which, in addition, precedes significant dendritic degeneration in hippocampal pyramidal neurons in Tg2576 mice with overt AD pathology [117]. However, photoreceptor neuronal death has not been observed in the advanced stages of AD pathology in a mouse AD model [118]. It has to be stressed that even if the older studies failed to detect early alterations ([119] - for retinal neurofibrillary layer; [120] and [121] - for ganglion cell loss), recent studies using more sophisticated technology, report degenerative changes (thinning of the specific retina regions, loss of optic nerve fibres and ganglion cells), that appear concomitantly with the early cognitive deficits [114]. 3.2. ERG and Electrophysiology of Retinal Changes in Alzheimer’s Disease Alterations in bioelectrical response of the AD eye to light stimulation have been documented by different types of 8 Current Alzheimer Research, 2017, Vol. 14, No. 1 ERG: full-field ERG, pattern ERG (PERG) and multifocal ERG. 3.2.1. ERG Full-field flash ERG has been studied in AD patients under both scotopic and phototopic conditions. The majority of studies did not detect significant differences between patients and age-matched normal subjects [2, 122-124]. By contrast, scotopic full-field flash ERG studies in animal AD models, such as aged APP/PS1, have reported a decrease in the amplitude of a- and b- waves in advanced stages of pathology as compared to non-transgenic littermates [118]. Shimazawa and colleagues provided evidence that increased latency to each peak, after light stimulus, in ERG recordings in Tg2576 and APP/PS1 mice correlate with lower expression levels of the NR2B subunits of NMDA receptors [106]. Based on these finding, the authors postulated that Aβ may impinge on retinal function by impairing NMDA signaling pathways [106], as detected in the vulnerable brain areas [17, 19] (see also the section 1.3.1). Remarkably, it was published this year that subretinal injection of Aβ in C57/BL6 mice yields declined scotopic response [125]. Initial PERG studies of AD retinas failed to detect significant differences between patients and age-matched controls [119,123,126]. However, more recent studies reported significant alterations in amplitude of the positive component peaking at 50 ms (P50) combined with a P50 increase in latency [127-129] or not [130-132]. To sum up, the recent PERG studies using more advanced technology have reported alterations that are suggestive of ganglion cell dysfunction. In animal models, full-field flash ERG a- and b-waves were reduced under scotopic but not photopic conditions (Table 2). However, it should be kept in mind that basic full-field flash ERGs measured in response to a flash of light detect damage only when there is Table 2. Reed et al. a significant loss in the number of retinal neurons. More sensitive methods are needed to detect alterations earlier in the progression of the disease. 3.2.2. Cellular Electrophysiology of Retinal Cells in AD To the best of our knowledge, there is so far not a single study performed in the retina of AD mouse models using cellular electrophysiology. 4. NEW AVENUES FOR REVEALING THE EARLIEST AD BIOMARKERS BY ELECTROPHYSIOLOGICAL EXPLORATION OF THE RETINA? As already discussed in section 3 of this review, visual disturbances are among the complaints reported by AD patients [132]. Furthermore, as in the brain, during the overt stage of AD pathology, Aβ plaques are detectable in the retina ([101]; see section 3.1.1) as well as functional changes in ERG recordings ([130]; see section 3.2.1). Importantly, changes in the retina are also detectable at early stages of AD. For example, imaging the retinal nerve fiber layer using SD-OCT showed that retinal neurofibrillary layer thickness was significantly decreased in patients with MCI compared to controls ([114] and section 3.1.2). In this light, some exciting technological achievements have been accomplished lately. Among them, the methodology such as fluorescence lifetime imaging ophthalmoscopy (FLIO) will certainly become a valuable diagnostic tool. FLIO is based on scanning laser ophtalmoscope using excitation pulse, which yields autofluorescence that is readily detectable in the retina. The comparative pilot study published at the end of the last year with a cohort of 16 patients diagnosed as early AD has convincingly demonstrated that FLIO, but not conventional OCT, could reveal significant correlation with the cognitive status (MMSE score) and phospho-tau levels in the CSF [133]. These findings support the possibility of using the retina as a surrogate for detection of early pathological changes in the brain and point to the exiting possibility of Electrophysiological alterations in AD retina. Methodology Type of alteration Species / Model References Full-field (scotopic and phototopic) No alteration Human [2, 122-124] Full-field (scotopic) Decreased a & b waves Mouse, APP/PS1deltaE9 [118] Full-field (scotopic) Decreased a & b waves Mouse, C57/BL6 [125] Full-field (phototopic) Increased peak latency after light stimulation Mouse, Tg2576 No alteration Human [119, 123, 126] Increased P50 peak & P50 latency Human [127-129] No increase in P50 latency Human [130-132] ERG (retinal Aβ injection) [106] PERG Seeing Early Signs of Alzheimer’s Disease Current Alzheimer Research, 2017, Vol. 14, No. 1 9 identifying these alterations by non-invasive modern imaging such as laser scanning ophtalmoscopy. after a systematic exploration using more advanced technology. In addition to the structural changes detectable by noninvasive imaging methodologies, the functional prodromal changes in neuronal excitability may also be detectable in the retina, which could serve as a biomarker for early diagnostic purposes [1]. The relevance of the search for the earliest electrophysiological AD-associated alterations in the retina is further strengthened by the fact that, at least in the brain, the Aβ-related impairment of synaptic transmission [17,30] precedes destabilization of neuronal network activity and induction of aberrant network synchronization ([35,43]; see also 1.3.2). Indeed, synaptic loss is one of the first functional hallmarks of AD and is the best correlate of cognitive decline [3]. ERG evaluation of synaptic function has been very recently validated for use in the mouse retina [134]. Relevantly, the latter study has also reported that mice carrying the ApoE-ε4 allele of apolipoproteine E4, the most prevalent genetic risk factor for the late-onset AD, which acts in synergy with Aβ, present a significant reduction in mixed rodcone responses, such as decreased a- and b-wave amplitudes [134]. Thus two of the hallmark characteristics of AD lead to changes in retinal function in rodent models. Because retina is a window to the brain, which can be explored non-invasively, and because new data has convincingly shown that AD pathology likely begins in the retina prior to the brain (section 3.1.1), it is sound to search for the earliest AD diagnostic markers in retina. We believe that exploring the retinal neurons by both advanced laser imaging methodologies and future, more sensitive retinal electrophysiology devices, would help identifying the first Aβrelated structural and functional alterations in the retina. During the last years, a tremendous progress has been accomplished in developing laser-based non-invasive imaging technologies. The same efforts should be now made to develop more advanced modalities of ERG. To achieve this goal, researchers, ophthalmologists and physicists should work hand-in-hand. The next-generation tools for monitoring the activity of the cell types, identified by cellular electrophysiology, exhibiting altered synaptic activity, will likely help establish AD diagnosis much earlier than currently possible. For example, if cellular electrophysiology identifies amacrine cells as the specific type of neuron displaying altered synaptic activity during the asymptomatic stage of AD, then development of more sensitive devices recording negative oscillatory potential for use in human, will undoubtedly be a big step towards earlier AD diagnosis. Finally, using electrophysiological exploration of the retina (ERG), instead of brain (EEG) holds a significant advantage. Indeed, it has been reported that modern EEG can be used to detect cross-frequency uncoupling in hippocampus [43, 44]. However, EEG recording of cross-frequency uncoupling in hippocampus is unlikely to become an early diagnostic marker since detection remains challenging because the hippocampus is a relatively deep brain structure. One option would be to transpose the search for diagnostic markers from the brain to the retina, which is a part nervous system and is amenable to non-invasive and uncostly study by using a totally safe ERG exploration [1]. CONCLUSION AND FUTURE DIRECTIONS In spite of the accumulating body of evidence that early disturbances in visual function in AD involve alterations within the retina, some evidence is still elusive or even contradictory. For example, in AD patients, no significant difference was seen in ERG studies (section 3.2.1). By contrast, in animal AD models, scotopic (but not photopic) ERG response appears altered (section 3.2.1), thus suggesting possible rod photoreceptor dysfunction (although contribution of the downstream neurons cannot be excluded). However, structural alterations of rod photoreceptors are not detectable and, as with cones, rods appear to be spared even at the late stages of AD (section 3.1.2). Such inconsistencies may simply be related to the insensitivity of current methods used for detection in humans. This statement is, for example, supported by the data obtained for another type of retinal neuron: the ganglion cell. Early studies (before 2000), using less sophisticated devices, could not resolve the AD-related loss of ganglion cells. However, using more elaborate methods of detection, ganglion cell loss has been systematically found in both AD patients and animal models, with overt pathology (section 3.1.2). These facts strongly suggest that previous views suggesting that the visual system (including retina and optic nerve) is not affected in AD should be reconsidered In conclusion, the major advantages of exploring retina as the source of the earliest AD biomarkers are that it can be studied intact and non-invasively which make the eye suitable for analysis with low-cost and widely available devices such as the more advanced ERG. This goal is among the major challenges for the future, in the light of the everincreasing proportion of the elderly population world-wide and that AD has recently met the World Health Organization’s criteria of “epidemic” disease. At the individual level, achieving this goal will allow all of us to reach old age with the knowledge that moving the borders of the onset and progression of diseases like AD sufficiently far away will improve our quality of life. CONFLICT OF INTEREST The authors confirm that this article content has no conflict of interest. ACKNOWLEDGEMENTS We thank our young colleagues, Aileen Hofer and Matthieu Capelo for constructive discussions. We would also like to acknowledge the support from Fondation pour la Recherche Médicale (operating grant FRM n°DVS 20131228910 to SK) and from Région Ile-de-France (DIM Cerveau et pensée to SK). REFERENCES [1] [2] [3] Krantic S, Torriglia A. Retina: source of the earliest biomarkers for Alzheimer's disease? J Alzheimers Dis 40(2): 237-43 (2014). Tzekov R, Mullan M. Vision function abnormalities in Alzheimer disease. Surv Ophthalmol 59(4): 414-33 (2014). Nelson PT, Braak H, Markesbery WR. Neuropathology and cognitive impairment in Alzheimer disease: a complex but coherent relationship. J Neuropathol Exp Neurol 68(1): 1-14 (2009). 10 Current Alzheimer Research, 2017, Vol. 14, No. 1 [4] [5] [6] [7] [8] [9] [10] [11] [12] [13] [14] [15] [16] [17] [18] [19] [20] [21] [22] [23] [24] [25] Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297(5580): 353-56 (2002). Ashe KH, Zahs KR. Probing the biology of Alzheimer's disease in mice. Neuron 66(5): 631-45 (2010). Bateman RJ, Xiong C, Benzinger TLS, Fagan AM, Goate A, Fox NC, et al. Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med 367(9): 795-804 (2012). Fagan AM, Mintun MA, Mach RH, Lee S-Y, Dence CS, Shah AR, et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol 59(3): 512-19 (2006). Thal LJ. Prevention of Alzheimer disease. Alzheimer Dis Assoc Disord 20(3 Suppl 2): S97-99 (2006). Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 6(3): 131-44 (2010). Hampel H, Prvulovic D, Teipel S, Jessen F, Luckhaus C, Frolich L, et al. The future of Alzheimer's disease: the next 10 years. Prog Neurobiol 95(4): 718-28 (2011). Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, et al. Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiol Aging 31(8): 1275-83 (2010). Morris JC, Roe CM, Grant EA, Head D, Storandt M, Goate AM, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Arch Neurol 66(12): 1469-75 (2009). Rowe CC, Ng S, Ackermann U, Gong SJ, Pike K, Savage G, et al. Imaging beta-amyloid burden in aging and dementia. Neurology 68(20): 1718-25 (2007). Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30(4): 572-80 (1991). Palop JJ, Chin J, Bien-Ly N, Massaro C, Yeung BZ, Yu GQ, et al. Vulnerability of dentate granule cells to disruption of arc expression in human amyloid precursor protein transgenic mice. J Neurosci 25(42): 9686-93 (2005). Palop JJ, Mucke L. Synaptic depression and aberrant excitatory network activity in Alzheimer's disease: two faces of the same coin? Neuromol Med 12(1): 48-55 (2010). Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer's disease. Neuron 55(5): 697-711 (2007). Liu SJ, Gasperini R, Fau L, Small DH, Small DH. Amyloid-beta decreases cell-surface AMPA receptors by increasing intracellular calcium and phosphorylation of GluR2. J Alzheimer’s Dis 21(2): 655-66 (2010). Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, et al. Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8(8): 1051-8 (2005). Knafo S, Venero C, Merino-Serrais P, Fernaud-Espinosa I, Gonzalez-Soriano J, Ferrer I, et al. Morphological alterations to neurons of the amygdala and impaired fear conditioning in a transgenic mouse model of Alzheimer's disease. J Pathol 219(1): 41-51 (2009). Lanz TA, Fici GJ, Merchant KM. Lack of specific amyloid-beta(142) suppression by nonsteroidal anti-inflammatory drugs in young, plaque-free Tg2576 mice and in guinea pig neuronal cultures. J Pharmacol Exp Therap 312(1): 399-406 (2005). Alpar A, Ueberham U, Bruckner MK, Seeger G, Arendt T, Gartner U. Different dendrite and dendritic spine alterations in basal and apical arbors in mutant human amyloid precursor protein transgenic mice. Brain Res 1099(1): 189-98 (2006). Rocher AB, Kinson MS, Luebke JI. Significant structural but not physiological changes in cortical neurons of 12-month-old Tg2576 mice. Neurobiol Dis 32(2): 309-18 (2008). DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol 27(5): 457-64 (1990). Scheff SW, DeKosky ST, Price DA. Quantitative assessment of cortical synaptic density in Alzheimer's disease. Neurobiol Aging 11(1): 29-37.(1990). Reed et al. [26] [27] [28] [29] [30] [31] [32] [33] [34] [35] [36] [37] [38] [39] [40] [41] [42] [43] [44] [45] [46] [47] Ju Y, Asahi T, Sawamura N. Arctic mutant Abeta40 aggregates on alpha7 nicotinic acetylcholine receptors and inhibits their functions. J Neurochem 131(5): 667-74 (2014). Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, et al. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science 321(5896): 1686-9 (2008). Busche MA, Chen X, Henning HA, Reichwald J, Staufenbiel M, Sakmann B, et al. Critical role of soluble amyloid-beta for early hippocampal hyperactivity in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 109(22): 8740-5 (2012). Minkeviciene R, Rheims S, Dobszay MB, Zilberter M, Hartikainen J, Fulop L, et al. Amyloid beta-induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci 29(11): 3453-62 (2009). Wykes R, Kalmbach A, Eliava M, Waters J. Changes in the physiology of CA1 hippocampal pyramidal neurons in preplaque CRND8 mice. Neurobiol Aging 33(8): 1609-23 (2012). Koffie RM, Hyman BT, Spires-Jones TL. Alzheimer's disease: synapses gone cold. Mol Neurodegen 6(1): 63 (2011). Pozueta J, Lefort R, Shelanski ML. Synaptic changes in Alzheimer's disease and its models. Neurosci 251: 51-65 (2013). Hu X, Pickering EH, Hall SK, Naik S, Liu YC, Soares H, et al. Genome-wide association study identifies multiple novel loci associated with disease progression in subjects with mild cognitive impairment. Trans Psychiatry 1: e54 (2011). Jolas T, Zhang XS, Zhang Q, Wong G, Del Vecchio R, Gold L, et al. Long-term potentiation is increased in the CA1 area of the hippocampus of APP(swe/ind) CRND8 mice. Neurobiol Dis 11(3): 394-409 (2002). Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, et al. Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149(3): 708-21 (2012). Putcha D, Brickhouse M, O'Keefe K, Sullivan C, Rentz D, Marshall G, et al. Hippocampal hyperactivation associated with cortical thinning in Alzheimer's disease signature regions in nondemented elderly adults. J Neurosci 31(48): 17680-8 (2011). Moretti DV, Pievani M, Geroldi C, Binetti G, Zanetti O, Rossini PM, et al. EEG markers discriminate among different subgroup of patients with mild cognitive impairment. Am J Alzheimer's Dis Other Dement 25(1): 58-73 (2010). Czigler B, Csikos D, Hidasi Z, Anna Gaal Z, Csibri E, Kiss E, et al. Quantitative EEG in early Alzheimer's disease patients - power spectrum and complexity features. Int J Psychophysiol 68(1): 75-80 (2008). van der Hiele K, Vein AA, Reijntjes RH, Westendorp RG, Bollen EL, van Buchem MA, et al. EEG correlates in the spectrum of cognitive decline. Clin Neurophysiol 118(9): 1931-9.(2007). van Deursen JA, Vuurman EF, Verhey FR, van KranenMastenbroek VH, Riedel WJ. Increased EEG gamma band activity in Alzheimer's disease and mild cognitive impairment. J Neural Transm 115(9): 1301-11 (2008). Herrmann CS, Demiralp T. Human EEG gamma oscillations in neuropsychiatric disorders. Clin Neurophysiol 116(12): 2719-33 (2005). Jelic V, Johansson SE, Almkvist O, Shigeta M, Julin P, Nordberg A, et al. Quantitative electroencephalography in mild cognitive impairment: longitudinal changes and possible prediction of Alzheimer's disease. Neurobiol Aging 21(4): 533-40 (2000). Goutagny R, Gu N, Cavanagh C, Jackson J, Chabot J-G, Quirion R, et al. Alterations in hippocampal network oscillations and thetagamma coupling arise before Abeta overproduction in a mouse model of Alzheimer's disease. Eur J Neurosci 37(12): 1896-902 (2013). Goutagny R, Krantic S. Hippocampal oscillatory activity in Alzheimer's disease: toward the identification of early biomarkers? Aging Dis 4(3): 134-40 (2013). Newman EA. Glial modulation of synaptic transmission in the retina. Glia 47(3): 268-74 (2004). Boycott BB, Hopkins JM. Microglia in the retina of monkey and other mammals: its distinction from other types of glia and horizontal cells. Neuroscience 6(4): 679-88 (1981). Reichenbach A, Bringmann A. New functions of Muller cells. Glia 61(5): 651-78 (2013). Seeing Early Signs of Alzheimer’s Disease [48] [49] [50] [51] [52] [53] [54] [55] [56] [57] [58] [59] [60] [61] [62] [63] [64] [65] [66] [67] [68] [69] [70] [71] [72] [73] [74] Walz W. Controversy surrounding the existence of discrete functional classes of astrocytes in adult gray matter. Glia 31(2): 95103 (2000). Araque A, Parpura V, Sanzgiri RP, Haydon PG. Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22(5): 208-15 (1999). Hung J, Chansard M, Ousman SS, Nguyen MD, Colicos MA. Activation of microglia by neuronal activity: results from a new in vitro paradigm based on neuronal-silicon interfacing technology. Brain Behav Immun 24(1): 31-40 (2010). Jiang X, Newell EW, Schlichter LC. Regulation of a TRPM7-like current in rat brain microglia. J Biol Chem 278(44): 42867-76 (2003). Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20(3): 269-87 (1995). Masland RH. The fundamental plan of the retina. Nat Neurosci 4(9): 877-86 (2001). Lagnado L. The Wellcome Prize Lecture. Visual signals in the retina: from photons to synapses. Exp Physiol 85(1): 1-16 (2000). Marc RE, Liu WL, Kalloniatis M, Raiguel SF, van Haesendonck E. Patterns of glutamate immunoreactivity in the goldfish retina. J Neurosci 10(12): 4006-34 (1990). Marc RE. Structural organization of GABAergic circuitry in ectotherm retinas. Prog Brain Res 90: 61-92 (1992). Curcio CA, Sloan KR, Kalina RE, Hendrickson AE. Human photoreceptor topography. J Comp Neurol 292(4): 497-523 (1990). Lamb TD, Pugh EN, Jr. A quantitative account of the activation steps involved in phototransduction in amphibian photoreceptors. J Physiol 449: 719-58 (1992). Hagins WA, Penn RD, Yoshikami S. Dark current and photocurrent in retinal rods. Biophys J 10(5): 380-412 (1970). Kamermans M, Fahrenfort I, Schultz K, Janssen-Bienhold U, Sjoerdsma T, Weiler R. Hemichannel-mediated inhibition in the outer retina. Science 292(5519): 1178-80 (2001). Kuffler SW. Discharge patterns and functional organization of mammalian retina. J Neurophysiol 16(1): 37-68 (1953). Werblin FS, Dowling JE. Organization of the retina of the mudpuppy, Necturus maculosus. II. Intracellular recording. J Neurophysiol 32(3): 339-55 (1969). Masland RH. The neuronal organization of the retina. Neuron 76(2): 266-80 (2012). Balboa RM, Grzywacz NM. The role of early retinal lateral inhibition: more than maximizing luminance information. Vis Neurosci 17(1): 77-89 (2000). Herrmann R, Heflin SJ, Hammond T, Lee B, Wang J, Gainetdinov RR, et al. Rod vision is controlled by dopamine-dependent sensitization of rod bipolar cells by GABA. Neuron 72(1): 101-10 (2011). Wassle H, Puller C, Muller F, Haverkamp S. Cone contacts, mosaics, and territories of bipolar cells in the mouse retina. J Neurosci 29(1): 106-17 (2009). Wassle H. Parallel processing in the mammalian retina. Nat Rev Neurosci 5(10): 747-57 (2004). Morgans CW, Zhang J, Jeffrey BG, Nelson SM, Burke NS, Duvoisin RM, et al. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc Natl Acad Sci U S A 106(45): 19174-78 (2009). Sasaki T, Kaneko A. L-Glutamate-induced responses in OFF-type bipolar cells of the cat retina. Vision Res 36(6): 787-95 (1996). DeVries SH. Bipolar cells use kainate and AMPA receptors to filter visual information into separate channels. Neuron 28(3): 847-56 (2000). DeVries SH, Schwartz EA. Kainate receptors mediate synaptic transmission between cones and 'Off' bipolar cells in a mammalian retina. Nature 397(6715): 157-60 (1999). Taylor WR, Smith RG. Trigger features and excitation in the retina. Curr Opin Neurobiol 21(5): 672-78 (2011). Fukuda Y, Stone J. Retinal distribution and central projections of Y-, X-, and W-cells of the cat's retina. J Neurophysiol 37(4): 74972 (1974). Kim I-J, Zhang Y, Yamagata M, Meister M, Sanes JR. Molecular identification of a retinal cell type that responds to upward motion. Nature 452(7186): 478-82 (2008). Current Alzheimer Research, 2017, Vol. 14, No. 1 [75] [76] [77] [78] [79] [80] [81] [82] [83] [84] [85] [86] [87] [88] [89] [90] [91] [92] [93] [94] [95] [96] [97] [98] [99] 11 Kolb H, Nelson R, Mariani A. Amacrine cells, bipolar cells and ganglion cells of the cat retina: a Golgi study. Vision Res 21(7): 1081-1114 (1981). Nelson R, Famiglietti EV, Jr., Kolb H. Intracellular staining reveals different levels of stratification for on- and off-center ganglion cells in cat retina. J Neurophysiol 41(2): 472-83 (1978). Euler T, Masland RH. Light-evoked responses of bipolar cells in a mammalian retina. J Neurophysiol 83(4): 1817-29 (2000). Bieda MC, Copenhagen DR. Inhibition is not required for the production of transient spiking responses from retinal ganglion cells. Vis Neurosci 17(2): 243-54 (2000). Dunn FA, Wong ROL. Wiring patterns in the mouse retina: collecting evidence across the connectome, physiology and light microscopy. J Physiol 592(Pt 22): 4809-23 (2014). Shlens J, Rieke F, Chichilnisky E. Synchronized firing in the retina. Curr Opin Neurobiol 18(4): 396-402 (2008). Schwartz G, Taylor S, Fisher C, Harris R, Berry MJ, 2nd. Synchronized firing among retinal ganglion cells signals motion reversal. Neuron 55(6): 958-69 (2007). Trong PK, Rieke F. Origin of correlated activity between parasol retinal ganglion cells. Nat Neurosci 11(11): 1343-51 (2008). Perlman I. The Electroretinogram: ERG. In: Kolb H, Fernandez E, Nelson R, Eds. Webvision: The Organization of the Retina and Visual System. Salt Lake City (UT): University of Utah Health Sciences Center (1995). Shahidi AM, Sampson GP, Pritchard N, Edwards K, Russell A, Malik RA, et al. Exploring retinal and functional markers of diabetic neuropathy. Clin Exp Optom 93(5): 309-23 (2010). Creel DJ. Clinical Electrophysiology. Kolb H, Fernandez E, Nelson R, Eds. In: Webvision: The Organization of the Retina and Visual System. Salt Lake City (UT): University of Utah Health Sciences Center (1995). Viswanathan S, Frishman LJ, Robson JG, Harwerth RS, Smith EL, 3rd. The photopic negative response of the macaque electroretinogram: reduction by experimental glaucoma. Invest Ophthalmol Vis Sci 40(6): 1124-36 (1999). Harwerth RS, Crawford MLJ, Frishman LJ, Viswanathan S, Smith EL, 3rd, Carter-Dawson L. Visual field defects and neural losses from experimental glaucoma. Prog Retin Eye Res 21(1): 91-125 (2002). Sieving PA, Frishman LJ, Steinberg RH. Scotopic threshold response of proximal retina in cat. J Neurophysiol 56(4): 1049-61 (1986). Wachtmeister L, Dowling JE. The oscillatory potentials of the mudpuppy retina. Invest Ophthalmol Vis Sci 17(12): 1176-88 (1978). Wachtmeister L. Oscillatory potentials in the retina: what do they reveal. Prog Retin Eye Res 17(4): 485-521 (1998). Vaney DI. Many diverse types of retinal neurons show tracer coupling when injected with biocytin or Neurobiotin. Neurosci Lett 125(2): 187-90 (1991). Shlens J, Field GD, Gauthier JL, Greschner M, Sher A, Litke AM, et al. The structure of large-scale synchronized firing in primate retina. J Neurosci 29(15): 5022-31 (2009). Anstey KJ, Luszcz MA, Sanchez L. Two-year decline in vision but not hearing is associated with memory decline in very old adults in a population-based sample. Gerontology 47(5): 289-93 (2001). Rogers MA, Langa KM. Untreated poor vision: a contributing factor to late-life dementia. Am J Epidemiol 171(6): 728-35.(2010). Berisha F, Feke GT, Trempe CL, McMeel JW, Schepens CL. Retinal abnormalities in early Alzheimer's disease. Invest Ophthalmol Vis Sci 48(5): 2285-89 (2007). Danesh-Meyer HV, Birch H, Ku JYF, Carroll S, Gamble G. Reduction of optic nerve fibers in patients with Alzheimer disease identified by laser imaging. Neurology 67(10): 1852-54.(2006). Goldstein LE, Muffat JA, Cherny RA, Moir RD, Ericsson MH, Huang X, et al. Cytosolic beta-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer's disease. Lancet 361(9365): 1258-65 (2003). Gasparini L, Crowther RA, Martin KR, Berg N, Coleman M, Goedert M, et al. Tau inclusions in retinal ganglion cells of human P301S tau transgenic mice: effects on axonal viability. Neurobiol Aging 32(3): 419-33 (2011). Koronyo Y, Salumbides BC, Black KL, Koronyo-Hamaoui M. Alzheimer's disease in the retina: imaging retinal abeta plaques for 12 Current Alzheimer Research, 2017, Vol. 14, No. 1 [100] [101] [102] [103] [104] [105] [106] [107] [108] [109] [110] [111] [112] [113] [114] early diagnosis and therapy assessment. Neurodegener Dis 10(1-4): 285-93 (2012). Zhao Y, Bhattacharjee S, Jones BM, Hill JM, Clement C, Sambamurti K, et al. Beta-Amyloid Precursor Protein (betaAPP) Processing in Alzheimer's Disease (AD) and Age-Related Macular Degeneration (AMD). Mol Neurobiol 52(1): 533-44 (2015). Koronyo-Hamaoui M, Koronyo Y, Ljubimov AV, Miller CA, Ko MK, Black KL, et al. Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54(1): S204-17 (2011). Dutescu RM, Li QX, Crowston J, Masters CL, Baird PN, Culvenor JG. Amyloid precursor protein processing and retinal pathology in mouse models of Alzheimer's disease. Graefe's archive for clinical and experimental ophthalmology. Albrecht von Graefes Archiv fur klinische und experimentelle Ophthalmologie 247(9): 1213-21 (2009). Liu B, Rasool S, Yang Z, Glabe CG, Schreiber SS, Ge J, et al. Amyloid-peptide vaccinations reduce {beta}-amyloid plaques but exacerbate vascular deposition and inflammation in the retina of Alzheimer's transgenic mice. Am J Pathol 175(5): 2099-110 (2009). Ning A, Cui J, To E, Ashe KH, Matsubara J. Amyloid-beta deposits lead to retinal degeneration in a mouse model of Alzheimer disease. Invest Ophthalmol Vis Sci 49(11): 5136-43 (2008). Chiu K, Chan TF, Wu A, Leung IY, So KF, Chang RC. Neurodegeneration of the retina in mouse models of Alzheimer's disease: what can we learn from the retina? Age 34(3): 633-49 (2012). Shimazawa M, Inokuchi Y, Okuno T, Nakajima Y, Sakaguchi G, Kato A, et al. Reduced retinal function in amyloid precursor protein-over-expressing transgenic mice via attenuating glutamateN-methyl-d-aspartate receptor signaling. J Neurochem 107(1): 27990 (2008). More SS, Vince R. Hyperspectral imaging signatures detect amyloidopathy in alzheimer's mouse retina well before onset of cognitive decline. ACS Chem Neurosci 6(2): 306-15 (2015). Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on AgingAlzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement 7(3): 280-92 (2011). Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M, et al. High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J 24(8): 271626 (2010). Ono K, Yamada M. Low-n oligomers as therapeutic targets of Alzheimer's disease. J Neurochem 117(1): 19-28 (2011). Ardiles AO, Tapia-Rojas CC, Mandal M, Alexandre F, Kirkwood A, Inestrosa NC, et al. Postsynaptic dysfunction is associated with spatial and object recognition memory loss in a natural model of Alzheimer's disease. Proc Natl Acad Sci U S A 109(34): 13835-40 (2012). Syed AB, Armstrong RA, Smith CU. A quantitative analysis of optic nerve axons in elderly control subjects and patients with Alzheimer's disease. Folia neuropathologica / Association of Polish Neuropathologists and Medical Research Centre, Polish Academy of Sciences 43(1): 1-6 (2005). Iseri PK, Altinas O, Tokay T, Yuksel N. Relationship between cognitive impairment and retinal morphological and visual functional abnormalities in Alzheimer disease. J Neuro-Ophthalmol 26(1): 18-24 (2006). Paquet C, Boissonnot M, Roger F, Dighiero P, Gil R, Hugon J. Abnormal retinal thickness in patients with mild cognitive impairment and Alzheimer's disease. Neuroscience letters 420(2): 97-9 (2007). Reed et al. [115] [116] [117] [118] [119] [120] [121] [122] [123] [124] [125] [126] [127] [128] [129] [130] [131] [132] [133] [134] Blanks JC, Schmidt SY, Torigoe Y, Porrello KV, Hinton DR, Blanks RH. Retinal pathology in Alzheimer's disease. II. Regional neuron loss and glial changes in GCL. Neurobiol Aging 17(3): 38595 (1996). Blanks JC, Torigoe Y, Hinton DR, Blanks RH. Retinal pathology in Alzheimer's disease. I. Ganglion cell loss in foveal/parafoveal retina. Neurobiol Aging 17(3): 377-84 (1996). Williams PA, Thirgood RA, Oliphant H, Frizzati A, Littlewood E, Votruba M, et al. Retinal ganglion cell dendritic degeneration in a mouse model of Alzheimer's disease. Neurobiol Aging 34(7): 1799-806 (2013). Perez SE, Lumayag S, Kovacs B, Mufson EJ, Xu S. Beta-amyloid deposition and functional impairment in the retina of the APPswe/PS1DeltaE9 transgenic mouse model of Alzheimer's disease. Invest Ophthalmol Vis Sci 50(2): 793-800 (2009). Katz B, Rimmer S, Iragui V, Katzman R. Abnormal pattern electroretinogram in Alzheimer's disease: evidence for retinal ganglion cell degeneration? Ann Neurol 26(2): 221-5 (1989). Hinton DR, Sadun AA, Blanks JC, Miller CA. Optic-nerve degeneration in Alzheimer's disease. N Engl J Med 315(8): 485-7 (1986). Curcio CA, Drucker DN. Retinal ganglion cells in Alzheimer's disease and aging. Ann Neurol 33(3): 248-57 (1993). Justino L, Kergoat M, Bergman H, Chertkow H, Robillard A, Kergoat H. Neuroretinal function is normal in early dementia of the Alzheimer type. Neurobiol Aging 22(4): 691-5 (2001). Strenn K, Dal-Bianco P, Weghaupt H, Koch G, Vass C, Gottlob I. Pattern electroretinogram and luminance electroretinogram in Alzheimer's disease. J Neural Transm Supp 33: 73-80 (1991). Rizzo JF, 3rd, Cronin-Golomb A, Growdon JH, Corkin S, Rosen TJ, Sandberg MA, et al. Retinocalcarine function in Alzheimer's disease. A clinical and electrophysiological study. Arch Neurol 49(1): 93-101 (1992). Liu C, Cao L, Yang S, Xu L, Liu P, Wang F, et al. Subretinal injection of amyloid-beta peptide accelerates RPE cell senescence and retinal degeneration. Intern J Mol Med 35(1): 169-76 (2015). Prager TC, Schweitzer FC, Peacock LW, Garcia CA. The effect of optical defocus on the pattern electroretinogram in normal subjects and patients with Alzheimer's disease. Am J Ophthalmol 116(3): 363-9 (1993). Krasodomska K, Lubinski W, Potemkowski A, Honczarenko K. Pattern electroretinogram (PERG) and pattern visual evoked potential (PVEP) in the early stages of Alzheimer's disease. Doc Ophthalmol 121(2): 111-21 (2010). Parisi V, Restuccia R, Fattapposta F, Mina C, Bucci MG, Pierelli F. Morphological and functional retinal impairment in Alzheimer's disease patients. Clin Neurophysiol 112(10): 1860-7 (2001). Sartucci F, Borghetti D, Bocci T, Murri L, Orsini P, Porciatti V, et al. Dysfunction of the magnocellular stream in Alzheimer's disease evaluated by pattern electroretinograms and visual evoked potentials. Brain Res Bull 82(3-4): 169-76 (2010). Trick GL, Barris MC, Bickler-Bluth M. Abnormal pattern electroretinograms in patients with senile dementia of the Alzheimer type. Ann Neurol 26(2): 226-31 (1989). Nesher R, Trick GL. The pattern electroretinogram in retinal and optic nerve disease. A quantitative comparison of the pattern of visual dysfunction. Doc Ophthalmol 77(3): 225-35 (1991). Frost S, Martins RN, Kanagasingam Y. Ocular biomarkers for early detection of Alzheimer's disease. J Alzheimers Dis 22(1): 1-16 (2010). Jentsch S, Schweitzer D, Schmidtke KU, Peters S, Dawczynski J, Bar KJ, et al. Retinal fluorescence lifetime imaging ophthalmoscopy measures depend on the severity of Alzheimer's disease. Acta Ophthalmol 93(4): e241-7 (2015). Antes R, Ezra-Elia R, Weinberger D, Solomon A, Ofri R, Michaelson DM. ApoE4 induces synaptic and ERG impairments in the retina of young targeted replacement apoE4 mice. PLoS One 8(5): e64949 (2013).