Survey
* Your assessment is very important for improving the work of artificial intelligence, which forms the content of this project
Enantioselective synthesis wikipedia , lookup
Woodward–Hoffmann rules wikipedia , lookup
2-Norbornyl cation wikipedia , lookup
Aromaticity wikipedia , lookup
Marcus theory wikipedia , lookup
Physical organic chemistry wikipedia , lookup
Ring-closing metathesis wikipedia , lookup
Homoaromaticity wikipedia , lookup
Hofmann–Löffler reaction wikipedia , lookup
Aromatization wikipedia , lookup
1,3-Dipolar cycloaddition wikipedia , lookup
Discodermolide wikipedia , lookup
Ene reaction wikipedia , lookup
Organosulfur compounds wikipedia , lookup
Wolff–Kishner reduction wikipedia , lookup
Vinylcyclopropane rearrangement wikipedia , lookup
Baylis–Hillman reaction wikipedia , lookup
Wolff rearrangement wikipedia , lookup
George S. Hammond wikipedia , lookup
Petasis reaction wikipedia , lookup
Stille reaction wikipedia , lookup
Strychnine total synthesis wikipedia , lookup
Hydroformylation wikipedia , lookup
Tiffeneau–Demjanov rearrangement wikipedia , lookup
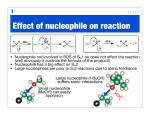
1.3. Substitution of hydroxyl groups of alcohols with halogens Typically: Hlg = Cl, Br thionyl chloride Reagents: inorganic acid chlorides (SOHlg2, SO2Hlg2, POHlg3, PHlg3, PHlg5), ClSO2OH, SF4 sulfuryl chloride BUT! In case of tertiary alcohols even with HHlg can take place the substitution. Reason: high stability of tertiary cation The mechanism of formation of tert-butyl chloride from tert-butyl alcohol and hydrogen chloride. 1.4. Transformation of alkyl halides into each other presipitates 2. SYNTHESIS OF BENZYL HALIDES High regioselectivity (reason: stability of benzyl radical) Finkelstein reaction presipitates Increased reactivity (reason: stability of benzyl cation) cc. HHlg effective reagent, reason: stability of benzyl cation Electron delocalization stabilizes benzyl radical. The unpaired electron is shared by the benzylic carbon and by the ring carbons that are ortho and para to it. The positive charge in benzyl cation is shared by is shared by the carbons ortho and para to the benzylic carbon Chloromethylation of aromatic compounds Similar to the Lucas test of alcohols SE reaction, in presence of activating groups! 3. SYNTHESIS OF ARYL HALIDES 3.1. Halogenation of aromatic compound (SE mechanism) Problems: 1. Hlg = Cl, Br can used 2. Direction, if G is strongly electron withdrawing group the reactivity is decreased 3.2. Substitution of aromatic diazoniumsalts Advantages: preparation of aromatic nitro compounds generally is easy, the position of halogen is given, any halogen atom can build in. HBF4: Tetrafluoroboric acid Chemical properties of C-Hlg compounds substituted compound alkene Basicity: thermodynamic affinity toward H+ Nucleophilicity: affinity toward positively polarized C atom or hetero atom (P, S, etc.) General: Present case: Nucleophile substitution 2. Reaction with O-nucleophiles Williamson’s ether synthesis Note: to prepare an alcohol through an ester within 2 steps is more efficient (no chance for concurrent elimination reaction) Solvolysis: the solvent is itself the nucleophile in the SN process (weaker nucleophile but it is present in high excess!) Reaction with S-nucleophiles Ambident Nu:The negative charge can appeare on both heteroatom Nucleophile substitution 3. Reaction with N-nucleophiles :B can be the excess of the amine Since it can provide the mixture of products, this process is less applied (see later) Gabriel’s synthesis (POTASSIUM PHTALIMIDE) Reaction with C-nucleophiles Another example for ambident nucleophile, the product ratio strongly depends on the reaction conditions Z1,Z2 = electron withdrawing group, e.g. COOR, CN (malonic ester or acetoacetate synthesis) To create a C-C bond !! Importance : prolong a chain, built in a new carbon chain Nucleophile substitution 4. Reaction with Hlg-nucleophiles Equilibrium is a problem since a mixture will be formed. reason: similar reactivity, similar nucleophilicity This reaction can be used only if the solubility of the product is different. soluble! insoluble soluble! insoluble! Finkelstein reaction Mechanisms of the nucleophilic substitution reaction on aliphatic (sp3) carbon 1. SN2 (substitution nucleophilic bimolecular) mechanism sp2-kind C MeI + MeSH Me2S + HI Kinetic is second order: r = d[R-Hlg]/dt = k [R-Hlg] [Nu] Bimolecular process (2) Concertic (synchronous) process (bond cleavage and formation occurs in the same time!): one step A single-step process in which both the alkyl halide and the nucleophile are involved at the transition state. Cleavage of the bond between carbon and the leaving group is assisted by formation of a bond between carbon and the nucleophile. In effect, the nucleophile “pushes off” the leaving group from its point of attachment to carbon. STEREOCHEMISTRY OF SN2 REACTIONS Hybrid orbital description of the bonding changes that take place at carbon during nucleophilic substitution by the SN2 mechanism Only one transtion state! Result: stereochemicaly only one product can form Attack from backside In case of chiral non-racemic substrate the configuration change to the opposite (Walden inversion) stereochemical pathway for substitution of a leaving group (red) by a nucleophile (blue). The nucleophile attacks the substrate from the side opposite the bond to the leaving group. This is called “back-side displacement,” or substitution with inversion of configuration. Substitution by the SN2 mechanism is stereospecific and proceeds with inversion of configuration at the carbon that bears the leaving group. There is a stereoelectronic requirement for the nucleophile to approach carbon from the side opposite the bond to the leaving group. Organic chemists often speak of this as a Walden inversion, after the German chemist Paul Walden, who described the earliest experiments in this area in the 1890s. 1. SN1 (substitution nucleophilic unimolecular) mechanism of nucleophilic substitution slow, rate determining step fast step The first step, a unimolecular dissociation of the alkyl halide to form a carbocation as the key intermediate, is rate-determining. Kinetic is first order: r = d[R-Hlg]/dt = k [R-Hlg] unimolecular process (1) Two steps process The SN1 mechanism is an ionization mechanism. The nucleophile does not participate until after the rate-determining step has taken place. The SN1 mechanism for hydrolysis of tertbutyl bromide. Energy diagram illustrating the SN1 mechanism for hydrolysis of tertbutyl bromide. retention Stereochemistry of SN1 reacion sp2 carbon atom Carbenium ion intermediate inversion Total process: 50% retention + 50% inversion In case of a chiral, enantiopure starting material the product is a racem mixture racemization occurs Two stereochemical possibilities present themselves. The nucleophile simply assumes the position occupied by the leaving group. It attacks the substrate at the same face from which the leaving group departs. This is called “front-side displacement,” or substitution with retention of configuration. In a second possibility the nucleophile attacks the substrate from the side opposite the bond to the leaving group. This is called “back-side displacement,” or substitution with inversion of configuration. stereochemical pathway for substitution of a leaving group (red) by a nucleophile (blue). Factors that can influence the nucleophile substitution 1. Leaving group (quality of halogen ): I > Br > Cl > F 2. Alkyl (R) group 3. Nucleophilicity (nucleophile power) 4. Solvent effect 5. Electrophile catalyst (e. g. Ag ions can modify the mechanism of the reaction) 6. Neighbor groups (groups in position to the leaving group can take part in the reaction) 1. RELATIVE REACTIVITY OF HALIDE LEAVING GROUPS Among alkyl halides, alkyl iodides undergo nucleophilic substitution at the fastest rate, alkyl fluorides the slowest. The order of alkyl halide reactivity in nucleophilic substitutions is the same as their order in eliminations. Iodine has the weakest bond to carbon, and iodide is the best leaving group. Alkyl iodides are several times more reactive than alkyl bromides and from 50 to 100 times more reactive than alkyl chlorides. Fluorine has the strongest bond to carbon, and fluoride is the poorest leaving group. Alkyl fluorides are rarely used as substrates in nucleophilic substitution because they are several thousand times less reactive than alkyl chlorides. 2. STERIC EFFECTS IN SN2 REACTIONS There are very large differences in the rates at which the various kinds of alkyl halides—methyl, primary, secondary, or tertiary—undergo nucleophilic substitution. In general, SN2 reactions exhibit the following dependence of rate on substrate structure: The large rate difference between methyl, ethyl, isopropyl, and tert-butyl bromides reflects the steric hindrance each offers to nucleophilic attack. The nucleophile must approach the alkyl halide from the side opposite the bond to the leaving group, and this approach is hindered by alkyl substituents on the carbon that is being attacked. The three hydrogens of methyl bromide offer little resistance to approach of the nucleophile, and a rapid reaction occurs. Replacing one of the hydrogens by a methyl group somewhat shields the carbon from attack by the nucleophile and causes ethyl bromide to be less reactive than methyl bromide. Replacing all three hydrogen substituents by methyl groups almost completely blocks back-side approach to the tertiary carbon of (CH3)3CBr and shuts down bimolecular nucleophilic substitution. Alkyl groups at the carbon atom adjacent to the point of nucleophilic attack also decrease the rate of the SN2 reaction. Compare the rates of nucleophilic substitution in the series of primary alkyl bromides shown in Table. Taking ethyl bromide as the standard and successively replacing its C-2 hydrogens by methyl groups, we see that each additional methyl group decreases the rate of displacement of bromide by iodide. The effect is slightly smaller than for alkyl groups that are attached directly to the carbon that bears the leaving group, but it is still substantial. When C-2 is completely substituted by methyl groups, as it is in neopentyl bromide [(CH3)3CCH2Br], we see the unusual case of a primary alkyl halide that is practically inert to substitution by the SN2 mechanism because of steric hindrance. The relative rate order in SN1 reactions is exactly the opposite of that seen in SN2 reactions: The steric crowding that influences reaction rates in SN2 processes plays no role in SN1 reactions. The order of alkyl halide reactivity in SN1 reactions is the same as the order of carbocation stability: the more stable the carbocation, the more reactive the alkyl halide. An electronic effect, specifically, the stabilization of the carbocation intermediate by alkyl substituents, is the decisive factor. The role of the structure of the alkyl group in the mechanism SN 2 SN 1 Take account: Bond E-s, dissociation E-s SN1 is favored: by the greater stability of the alkyl cation (carbenium ion) Factors that can stabilize the carbocation (e- donation, mesomerism stabilized systems) SN1 mechanism controls the reaction 30 C, benzyl, allyl systems are favored SN2 mechanism is favored: by easier backside attack, smaller steric hindrance (smaller Nu, lower ordered C) Substitution of carbon 3. NUCLEOPHILES AND NUCLEOPHILICITY The Lewis base that acts as the nucleophile often is, but need not always be, an anion. Neutral Lewis bases can also serve as nucleophiles. Common examples of substitutions involving neutral nucleophiles include solvolysis reactions. Solvolysis reactions are substitutions in which the nucleophile is the solvent in which the reaction is carried out. Solvolysis in water converts an alkyl halide to an alcohol. Nucleophilic strength, or nucleophilicity, is a measure of how fast a Lewis base displaces a leaving group from a suitable substrate. As long as the nucleophilic atom is the same, the more basic the nucleophile, the more reactive it is. An alkoxide ion (RO-) is more basic and more nucleophilic than a carboxylate ion (RCO2 - ).