Survey
* Your assessment is very important for improving the workof artificial intelligence, which forms the content of this project
Gene nomenclature wikipedia , lookup
Point mutation wikipedia , lookup
Gene expression wikipedia , lookup
Promoter (genetics) wikipedia , lookup
Evolution of metal ions in biological systems wikipedia , lookup
Oxidative phosphorylation wikipedia , lookup
Ridge (biology) wikipedia , lookup
Genomic imprinting wikipedia , lookup
Two-hybrid screening wikipedia , lookup
Molecular ecology wikipedia , lookup
Proteolysis wikipedia , lookup
Endogenous retrovirus wikipedia , lookup
Community fingerprinting wikipedia , lookup
Mitogen-activated protein kinase wikipedia , lookup
Silencer (genetics) wikipedia , lookup
Paracrine signalling wikipedia , lookup
Gene regulatory network wikipedia , lookup
Gene expression profiling wikipedia , lookup
Biosynthesis wikipedia , lookup
Biochemical cascade wikipedia , lookup
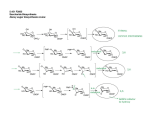
International Journal for Parasitology 34 (2004) 5–13 www.parasitology-online.com Rapid communication A complete shikimate pathway in Toxoplasma gondii: an ancient eukaryotic innovationq S.A. Campbella, T.A. Richardsb, E.J. Muic, B.U. Samuelc, J.R. Cogginsd, R. McLeodc, C.W. Robertsa,* a Department of Immunology, Strathclyde Institute for Biomedical Sciences, University of Strathclyde, 27 Taylor Street, Glasgow, Scotland G4 ONR, UK b Department of Zoology, The Natural History Museum, Cromwell Road, London, UK c Department of Ophthalmology and Visual Sciences, University of Chicago, Chicago, IL 60616, USA d Division of Biochemistry and Molecular Biology, Institute of Biomedical and Life Sciences, University of Glasgow, Glasgow, Scotland, UK Received 19 September 2003; received in revised form 15 October 2003; accepted 16 October 2003 Abstract The shikimate pathway is essential for survival of the apicomplexan parasites Plasmodium falciparum, Toxoplasma gondii and Cryptosporidium parvum. As it is absent in mammals it is a promising therapeutic target. Herein, we describe the genes encoding the shikimate pathway enzymes in T. gondii. The molecular arrangement and phylogeny of the proteins suggests homology with the eukaryotic fungal enzymes, including a pentafunctional AROM. Current rooting of the eukaryotic evolutionary tree infers that the fungi and apicomplexan lineages diverged deeply, suggesting that the arom is an ancient supergene present in early eukaryotes and subsequently lost or replaced in a number of lineages. q 2003 on behalf of Australian Society for Parasitology Inc. Published by Elsevier Ltd. All rights reserved. Keywords: Apicomplexa; Toxoplasma; Plasmodium; Shikimate; AROM; DAHP synthase The shikimate pathway consists of seven enzymes that catalyse the sequential conversion of erythrose-4-phosphate and phosphoenol pyruvate to chorismate, the common precursor of the folates, ubiquinone, the aromatic amino acids and many other aromatic compounds (Herrmann and Weaver, 1999). Previously believed to be confined to bacteria, plants and fungi, the shikimate pathway has recently been shown to function in the apicomplexan parasites, Plasmodium falciparum, Toxoplasma gondii and Cryptosporidium parvum (Roberts et al., 1998). Inhibition of this pathway by the herbicide glyphosate, a specific inhibitor of 5-enolpyruvylshikimate-3-phosphate (EPSP) synthase, restricts the growth of these parasite species in vitro (Roberts et al., 1998). The absence of the pathway in mammals, combined with its essential nature in certain q Nucleotide sequence data reported in this paper are available in the GenBanke EMBL and DDBJ databases under the accession numbers AY341375 and AY314743. * Corresponding author. Tel.: þ44-141-795-4458; fax: þ 44-141-7954406. E-mail address: [email protected] (C.W. Roberts). microorganisms, makes the shikimate pathway enzymes attractive targets for new anti-microbial agents. The molecular organisation and structure of the shikimate pathway enzymes varies considerably between taxonomic groups (Coggins et al., 1987). Bacteria have seven individual polypeptides, each possessing a single enzyme activity, which are encoded by separate genes. Plants have a molecular arrangement similar to bacteria, i.e. separate enzymes encoded by separate genes (Butler et al., 1974), with the exception of dehydroquinase (DHQase) and shikimate dehydrogenase which have been shown to be present as separate domains on a bifunctional polypeptide (Mousdale et al., 1987). Plant enzymes, although nuclear encoded, are largely active in the chloroplast and accordingly possess an N-terminal transit sequence. In contrast, all fungi examined to date have monofunctional 3-deoxy-D arabino-heptulosonate 7-phosphate (DAHP) synthases and chorismate synthases and a pentafunctional polypeptide termed AROM (Duncan et al., 1987). The AROM polypeptide has domains analogous to the bacterial enzymes: dehydroquinate (DHQ) synthase, EPSP synthase, 0020-7519/$30.00 q 2003 on behalf of Australian Society for Parasitology Inc. Published by Elsevier Ltd. All rights reserved. doi:10.1016/j.ijpara.2003.10.006 6 S.A. Campbell et al. / International Journal for Parasitology 34 (2004) 5–13 Fig. 1. Molecular arrangement of the shikimate pathway enzymes are the same in Toxoplasma gondii and fungi, but different from plants and bacteria. (A) The T. gondii arom gene is 19,460 bp and is interrupted by 19 introns (black). Exons are coloured relative to the corresponding domain order in the T. gondii AROM polypeptide (B), DHQ synthase (pink), EPSP synthase (green), shikimate kinase (red), dehydroquinase (yellow) and shikimate dehydrogenase (blue). The entire polypeptide spans 3332 amino acids. (C) The five central shikimate pathway enzymes are fused in fungi (e.g. Saccharomyces cerevisiae), are monofunctional in plants (e.g. Lycopersicuon escultentum), with the exception of dehydroquinase and shikimate dehydrogenase which are fused to form a bifunctional protein. In general the bacterial enzymes are monofunctional (e.g. Escherichia coli). A gap indicates that the genes are not fused. (D) The DHQ synthase domain of T. gondii has a high degree of sequence conservation with other species, a predicted secondary structure similar to Emericella nidulans and all the residues known to be important for this enzyme. Sequences are: T. gondii (Accession no. AY314743); Pneumocystis carinii (Q12659); S. cerevisiae S.A. Campbell et al. / International Journal for Parasitology 34 (2004) 5–13 shikimate kinase, DHQase and shikimate dehydrogenase (Fig. 1). A number of apicomplexan parasites have a vestigial plastid organelle called an apicoplast, most likely derived from an ancient algal endosymbiont (Kohler et al., 1997; McFadden et al., 1996). This prompted us to seek evidence of plant-like biosynthetic pathways in these parasites and led to the identification of the first apicomplexan shikimate pathway enzyme, chorismate synthase. Chorismate synthase was isolated from T. gondii and a number of Plasmodium species and in all cases the proteins lacked an obvious N-terminal transit sequence, suggesting that they are cytosolically active and unlikely to be located in the apicoplast (Roberts et al., 1998, 2002). P. falciparum chorismate synthase has since been reported to be present in the cytoplasm (Fitzpatrick et al., 2001). Consistent with this, phylogenetic analysis inferred that these apicomplexan chorismate synthases were most closely related to fungal enzymes which also function in the cytoplasm (Keeling et al., 1999). Despite the availability of the P. falciparum genome sequence (Gardner et al., 2002), definitive identification of the genes for the other six shikimate pathway enzymes has proven problematic. Taking advantage of the studies of other apicomplexan genomes we sought to identify the shikimate pathway enzyme genes from T. gondii. A search of the Toxoplasma genome project (ToxoDB 2.1) revealed two contigs (assembled genomic sequences) containing regions that appeared to code for a number of shikimate pathway enzymes. TGG 7014 contained sequences homologous to EPSP synthase, shikimate kinase, DHQase and shikimate dehydrogenase. The order of the elements on this contiguous region of DNA, although spanning some 20 kb, was identical to the genomic arrangement for the same four enzymes of the fungal AROM pentafunctional protein (Duncan et al., 1987). TGG 3535, a fragment of genomic DNA (gDNA) of approximately 5 kb, contained sequences homologous to DHQ synthase, the remaining enzyme present in the fungal AROM. PCR was used to amplify a region spanning the two fragments, the sequence of which confirmed that the fragments were contiguous. (This has since been confirmed in ToxoDB 2.2, TGG 8613.) This established that these five enzymes are clustered in the T. gondii genome. To determine whether the genes were fused to form an AROM-type arrangement the cDNA sequence was determined. Initially, a probe was generated from a region of 7 the putative DHQ synthase to screen a T. gondii (RH strain) tachyzoite cDNA library. This obtained the 50 -region of the putative DHQ synthase gene including the initiation codon. However, as this sequence was truncated, an alternative approach was used. RNA was extracted from T. gondii tachyzoites (RH strain) using Trizol reagent (Invitrogen) and used to generate cDNA using Moloney Murine Leukemia reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. A series of overlapping clones were amplified by PCR and cloned into the pDRIVE vector using the Qiagen PCR Cloning Kit (Qiagen) according to the manufacturer’s instructions. Clones were sequenced commercially (MWG Biotech, Milton Keynes, UK) and assembled using Sequencher (Gene Codes, Ann Arbor, MI). This revealed a 10 kb sequence that had a single open reading frame encoding a polypeptide of 3332 amino acids with a predicted molecular weight of 361.7 kDa. Comparison of the cDNA with the gDNA sequence reveals that the gene consists of 20 exons (Fig. 1A). The predicted T. gondii AROM (TgAROM) polypeptide has all the domains, known to be highly conserved in fungal AROMs with all the enzyme domains arranged in the same order as observed in fungi (Fig. 1B). Nonetheless, TgAROM has a number of obvious differences from the fungal counterparts. Notably the protein is considerably larger than the fungal AROMs, which range in size from 1563 amino acids in Neurospora crassa to 1588 amino acids in Saccharomyces cerevisiae. The T. gondii AROM protein has a number of insertions not present in the fungal counterparts. Analysis of the relative hydrophobicity and charge of these regions, using the ExPASy ProtScale tool (http://us.expasy.org/cgi-bin/protscale.pl), suggests that these areas could form exposed surface loops. The functions of these regions are not obvious although similar hydrophilic insertions have been noted in a number of apicomplexan enzymes including chorismate synthase (Roberts et al., 1998). Early studies established that, although the fungal AROM was highly susceptible to proteolysis, many of the resultant individual domains retained their enzymatic activity. This observation allowed biochemical characterisation of the various enzyme components of the AROM, and encouraged the expression and characterisation of individual or bifunctional domains of the AROM. For example, the DHQ synthase and shikimate dehydrogenase domains from the Emericella nidulans AROM gene can be expressed as individual enzymatically active proteins in Escherichia R (NP010412) and E. nidulans (P07547). Proteins were aligned using MacVector (Oxford Molecular Group). The predicted secondary structure of the T. gondii enzyme domain was obtained by the program PredictProtein (Rost, 1996). The E. nidulans structure was previously determined (Carpenter et al., 1998). Identical amino acids are marked in red, similar amino acids are coloured blue and variable residues are black. Dashes indicate gaps to maximise alignment. The residues identified to be important in the E. nidulans enzyme and conserved in the T. gondii protein are marked by asterisks. The secondary structure prediction of the T. gondii protein is given above the alignment, where arrows represent beta strands and cylinders, a-helical regions. This is compared to the known structure of the E. nidulans DHQ synthase domain given below the alignment. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.) 8 S.A. Campbell et al. / International Journal for Parasitology 34 (2004) 5–13 coli (Moore and Hawkins, 1993). However, the EPSP synthase domain is not active when expressed as a single domain, but only shows activity when expressed as part of a DHQ synthase – EPSP synthase bifunctional protein (Moore and Hawkins, 1993). The DHQ synthase domain from E. nidulans has been expressed in E. coli and the 3D structure determined by X-ray crystallography (Carpenter et al., 1998). As this is the only component of the AROM polypeptide to have been studied in depth, we have compared the DHQ synthase domains from both the T. gondii and E. nidulans AROMs to determine if the key features are conserved between both proteins (Fig. 1D). All the key residues identified by Carpenter et al. (1998) which are known to be involved in the mechanism of the E. nidulans DHQ synthase are conserved within the T. gondii protein. These include the residues corresponding to E. nidulans Glu194, His271 and His287 which interact with the pentacoordinate Zn2þ, and the residues involved in providing a phosphate-binding pocket, Lys152, Asn162, Asn268, His275 and Lys356. In addition, the residues identified as important in the binding of the DAHP substrate analogue, carbaphosphonate (Lys152, Asn268, His275 and Lys356 and Arg130) are conserved within the TgAROM protein. This provides insight into the rational design of other possible inhibitors for the T. gondii DHQ synthase. A secondary structure prediction of the TgAROM generated by the PredictProtein programme (Rost, 1996) (http://cubic. bioc.columbia.edu/predictprotein/) has been aligned with the known secondary structure elements of the E. nidulans enzyme (Fig. 1D). There is a general consensus in the predicted positions of a-helices and b-strand regions between both species. DAHP synthase catalyses the first committed step in the shikimate pathway. Two classes of this enzyme have been described. Class I (AroAI) was originally described as 39 kDa proteins similar to the E. coli enzymes and paralogues, but can now be subdivided into AroAIa and AroAIb exemplified by the E. coli orthologues and the Bacillus subtilis orthologues, respectively (Gosset et al., 2001). Many fungal and one oomycete, Phytophtora infestans, have had Class I (AroAI) genes sequenced, suggesting a wide eukaryote taxonomic distribution. Class II (AroAII) DAHPs were originally described as similar to the 54 kDa higher plant enzymes (Walker et al., 1996), but are now known also to exist in a number of divergent microbes such as Streptomyces and in the fungus N. crassa (Jensen et al., 2002). In plants, AroAII are feedbackinhibited by arogenate, a precursor of phenylalanine and tyrosine. Many bacteria, including E. coli have three paralogous AroAI, DAHPs designated AroF, AroG and AroH that are inhibited by tyrosine, phenylalanine and tryptophan, respectively. Interestingly, the fungi N. crassa and several prokaryotes possess both Class I and II DAHP synthases. Consequently, it has been suggested that the two DAHP classes may have different functions, for example N. crassa and the bacterium Streptomyces hygroscopicus class II enzymes have been linked to secondary metabolism such as the production of antibiotics (Gosset et al., 2001). The tBLASTn alogrithim was used to search ToxoDB 2.1 for evidence of both Class I or/and II DAHP synthases. A portion of Contig TGG_9597 was found to code for a putative protein with similarity to Class II DAHP synthase, but no likely candidates were identified for a Class I DAHP synthase. This region was amplified by PCR and used as a probe to screen a T. gondii cDNA library. This produced a number of overlapping clones that assembled to give the entire T. gondii DAHP synthase, which was confirmed by reverse-transcriptase PCR amplification to produce a full length clone (Genbank accession number AY341375). Initial alignments revealed that the T. gondii DAHP synthase was a member of the AroAII family. The T. gondii DAHP synthase (TgDAHP) is 615 amino acids in length and has a predicted molecular mass of 67.4 kDa, significantly larger than the previously described Class II enzymes due to the presence of a number of insertions (data not shown) analogous to those observed in the other shikimate pathway enzymes (Roberts et al., 1998). Having identified the genes that encode all seven steps of the shikimate pathway (Roberts et al., 1998 and the current paper) we were intrigued to investigate the evolutionary origins of these genes. Current sampling of shikimate pathway genes is confined primarily to prokaryotes, fungi and plants. Previous work had suggested that the plant shikimate pathway is derived from gene transfer events from prokaryotic genomes, most probably the cyanobacterial endosymbiont that became the plastid (Martin et al., 2002). As such the plants and the fungi do not form a monophyletic eukaryote group on the phylogenetic trees of shikimate pathway genes. Step seven of the apicomplexan shikimate pathway, chorismate synthase, had been demonstrated to cluster with fungal homologues on phylogenetic trees (Keeling et al., 1999). The fungi and apicomplexa lineages are distant relatives within the eukaryotic evolutionary tree (Stechmann and Cavalier-Smith, 2002). As such there are at least two possible explanations for the topology of the chorismate synthase tree, either the shikimate pathway is ancestral to eukaryotes and has evolved through vertical descent, or a horizontal gene transfer (HGT) event has occurred between these two lineages. While investigating the evolution of the six remaining shikimate pathway genes, alternative evolutionary scenarios should also be considered. These include the possibility that the shikimate pathway genes may have been derived from independent HGT events or alternatively, from endosymbiotic gene transfer from either the mitochondrial or the apicoplast endosymbiont. We undertook a full phylogenetic investigation of the remaining six genetic units that encode the T. gondii shikimate pathway. We aimed to test whether the T. gondii shikimate pathway had a single common origin and analyse whether these genes were derived from vertical descent or had been inherited horizontally from either the apicoplast S.A. Campbell et al. / International Journal for Parasitology 34 (2004) 5–13 genome, mitochondrial genome or any other source. Inheritance from the apicoplast would be evident if the T. gondii shikimate pathway genes clustered with the plants or the cyanobacterial taxonomic groups on phylogenetic trees (Kohler et al., 1997). Inheritance from the mitochondria would be evident if the T. gondii shikimate pathway genes clustered with the a-proteobacterial taxonomic groups on phylogenetic trees. All six genes were aligned with the available homologues from GenBank retrieved using tBLASTn. The genes were aligned automatically using the program Clustal_X (Thompson et al., 1997) and refined manually using the program genetic data environment (GDE). The alignments were masked to exclude sequence positions that could not be aligned with confidence such as hyper-variable regions of the protein sequence. The dehydroquinase portion of the AROM is highly variable with few conserved characters identifiable; a reasonable alignment and character sampling could not be achieved, preventing phylogenetic analysis. However, BLAST searches suggest that the T. gondii enzyme is most similar to the type I enzyme normally associated with the AROM protein. The DHQase domain is best aligned by focusing on the lysine residue involved in the formation of the covalent imine intermediate, which is characteristic of the type I family of DHQases (Butler et al., 1974). This residue lies at the centre of an eight-stranded a/b barrel which forms the core of this domain. Secondary structure predictions of the DHQase portion from the T. gondii AROM polypeptide can identify many, but not all the components of this a/b-barrel structure. Further analysis at the structural level maybe required to fully determine if this sequence is capable of forming the correct a/b-barrel structure. The five remaining masked protein alignments were analysed using Bayesian maximum likelihood phylogenetic methods using the program MRBAYES 2.01 (Huelsenbeck and Ronquist, 2001). Gamma distribution and the proportion of invariant site parameters were calculated using the Bayesian Metropolis-coupled Markov chain Monte Carlo (MCMCMC) parameter search. Tree and parameter space was sampled using the MCMCMC method initiated on a random tree, using the JTT matrix. The analysis was run for 1,000,000 generations and sampled every 100 generations. All MCMCMC values reached a plateau within the first 50,000 generations sampled, therefore the trees were sampled from 50,000 to 1,000,000 generations (Bayesian trees represent consensus of 9500 trees). All other trees were excluded as burn in. The level of burn in used was sufficient to guarantee that the parameter searches had stabilised. Bayesian posterior probability values are a product of sampling the MCMCMC plateau and are therefore frequently less informative than bootstrap values, often overestimating support values for phylogenetic tree topologies. Therefore bootstrap support values from distance analysis were calculated with the program PuzzleBoot providing a more rigorous analysis of the level of tree topology support (Schmidt et al., 2002) (Holder, M., and 9 Roger, A.J. PuzzleBoot version 1.03; http://hades.biochem. dal.ca/rogerlab/software/software.html). Gamma correction and proportion of invariant-site values derived and averaged from the plateau in the MCMCMC parameter space search and the WAG model (Whelan-Goldman, 2000) of aminoacid evolution was used in the bootstrap analysis. The use of two distinct methods, Bayesian and distance analysis with different models of amino acid evolution for tree construction and tree evaluation, respectively, provides increased confidence in tree topological relationships. Where both methods are in agreement and tree topology is consistent between these independent analyses, tree topologies are unlikely to be artefacts of one method of phylogenetic analyses or one model of protein evolution. Unrooted analyses were performed to allow for all possible evolutionary scenarios, some of which may have been falsely excluded if rooted analyses were performed. It has been recognised that ‘the probabilities of obtaining the correct rooted tree are considerably lower than the probabilities of obtaining the correct unrooted tree’, therefore ‘a considerable amount of error in constructing a rooted tree occurs at the time of rooting’ (Sourdis and Krimbas, 1987; Smith, 1994). Following our unrooted analyses that suggest the T. gondii genes group with the fungi, a root was inferred between the division of prokaryotes and eukaryotes and trees were drawn accordingly. Currently it is impossible to reliably identify the evolutionary root of these enzymes. Consequently an ingroup/outgroup approach was used to root these trees. The monophyletic fungi and T. gondii clades supported in the unrooted analyses were used as the ingroup, all bacteria and the plant genes (suggested to be derived from horizontal gene transfer from a bacterial source), were used as an outgroup. This rooting was only possible as the unrooted analysis demonstrated that the fungi and T. gondii eukaryotic clade was monophyletic and separate from all the other groups, an evolutionary relationship additionally supported by the AROM gene arrangement. Interestingly, the plant enzymes were found to group with the prokaryote enzymes, a relationship consistent with the proposal that the plant shikimate pathway is cyanobacterial in origin and has been derived from the bacterial ancestor of the chloroplast. However, the cyanobacteria did not consistently group with the plants in these phylogenetic trees and as such the plant shikimate pathway enzymes may have an alternative evolutionary ancestry. The T. gondii DAHP gene sequence clustered with the Class II homologue from N. crassa with 51% bootstrap support in phylogenetic analysis (Fig. 2). This tree topology is consistent with the phylogenetic relationships seen in the chorismate synthase phylogeny, which show the T. gondii gene clustering with the fungi, and similarly suggests that the T. gondii and the N. crassa DAHP genes have a common origin. These two eukaryotes group strongly (92% bootstrap support) with a DAHP homologue from the d-proteobacteria Stigmatalla aurantiaca. This relationship suggests that the origin of the primitive eukaryote DAHP-synthase enzyme, 10 S.A. Campbell et al. / International Journal for Parasitology 34 (2004) 5–13 Fig. 2. Phylogeny of DAHP (II) shows the Toxoplasma gondii protein clustering with the fungi, Neurospora crassa. The phylogeny was calculated using MRBAYES (Huelsenbeck and Ronquist, 2001) and bootstrap values were calculated using PuzzleBoot (Holder, M. and Roger, A.J. PuzzleBoot version 1.03. http://hades.biochem.dal.ca/rogerlab/ software/software.html). Support values are shown when in excess of 49% in the order of Bayesian posterior probability/distance bootstrap value (see text for further details of the methods used). The phylogeny was calculated from a sampling of 42 taxa with a character sampling of 372 amino acids. present in both T. gondii and N. crassa, is from the d-proteobacteria. Increased sampling of prokaryote genomes may reveal an alternative sister group to the T. gondii/ fungi cluster. The presence of the two distantly related S. auranitiaca homologues suggests that further prokaryote DAHP-synthase genes remain unsampled or alternatively there have been HGT events from a eukaryote to S. auranitiaca. The phylogeny for the DHQ synthase was poorly supported and did not resolve a tree topology with any confidence. Several attempts were made to adjust the alignment and character sampling to improve the resolution of the phylogenetic tree. The phylogeny did not resolve whether the DHQ synthase was more closely related to fungal homologues or clustered within the prokaryote homologues (data not shown). This leaves unsolved the evolutionary origin of the T. gondii DHQ-synthase AROM domain, which plausibly may have evolved from a separate HGT event from a prokaryote source. However, the DHQ-synthase phylogeny confirmed that the T. gondii DHQ-synthase gene did not originate from the apicoplast genome as the T. gondii enzyme did not cluster with either the plants or the cyanobacteria on our phylogenetic tree. The remaining three genetic units of the AROM indicated a monophyletic relationship between the fungi and T. gondii. This relationship was supported with low to moderate bootstrap values of 54, 78 and 64% in the phylogenies of EPSP synthase (Fig. 3A), shikimate kinase (Fig. 3B) and shikimate dehydrogenase (Fig. 3C), respectively. In the case of the EPSP synthase and shikimate dehydrogenase, the T. gondii genes grouped at the base of the fungal cluster consistent with these genetic units being inherited by vertical descent from the common ancestor of fungi and T. gondii. Interestingly, the shikimate kinase Bayesian phylogeny recovered the T. gondii gene within the fungal cluster (Fig. 3B); however, the bootstrap tree topology is consistent with the EPSP synthase and shikimate dehydrogenase phylogenies implying that T. gondii groups at the base of the fungal cluster. This suggests that the position of the T. gondii shikimate kinase gene within the fungal cluster, rather than at the base is an artefact. Overall the phylogenetic analyses are consistent with the proposition that the shikimate pathway genes in fungi and T. gondii are related by vertical descent, from a distant eukaryotic ancestor of both lineages, in spite of the low to moderate bootstrap values, symptomatic of this type of study (Richards et al., 2003). This is not only supported by five shikimate pathway gene phylogenies (this study and Keeling et al., 1999) that show the grouping of T. gondii with the fungi, but also with our demonstration that these organisms have a homologous AROM arrangement. Although HGT between the fungi and T. gondii lineages could explain the tree topologies recovered, this explanation is less parsimonious than the hypothesis of vertical descent as it would require the transfer of three genetic units, the AROM, DAHP synthase and chorismate synthase between these two lineages. There is also an evidence that the shikimate pathway is widespread through out the eukaryote kingdom, for example the oomycete, P. infestans, encodes a DAHP synthase protein (see Genbank accession number AF424663.1). Additionally, there is biochemical evidence of an AROM-like protein in Euglena gracilis (reviewed, Roberts et al., 2002), supporting the hypothesis that the AROM genetic arrangement in particular is both widespread and therefore probably of ancient derivation in the eukaryotic kingdom. Although there are marked differences in intron number and gene length between the T. gondii AROM and the known fungal AROMs, it is highly unlikely that this five-gene fusion would have evolved independently on two separate occasions within the eukaryotic kingdom. It is even more unlikely that the five-gene fusion, if it were to occur independently in the eukaryote kingdom, would produce a fused gene with the same domain order. Thus, the most parsimonious explanation is that the AROM supergene was an ancient eukaryotic innovation and probably occurred by the fusion of the genes encoded on a previously evolved prokaryotic operon (Andersson and Roger, 2002) donated S.A. Campbell et al. / International Journal for Parasitology 34 (2004) 5–13 11 Fig. 3. Phylogenetic trees showing the evolutionary relationships of three of the five functional domains encoded on the AROM polypeptide. All the phylogenies were calculated using MRBAYES (Huelsenbeck and Ronquist, 2001) and bootstrap values were calculated using PuzzleBoot (Holder, M. and Roger, A.J., PuzzleBoot version 1.03. http://hades.biochem.dal.ca/rogerlab/software/software.html). Support values are shown in excess of 49% in the order of Bayesian posterior probability/distance bootstrap value (see text for further details of the methods used). (A) shows the EPSP phylogeny. (B) and (C) show the phylogenies of shikimate kinase and shikimate dehydrogenase, respectively. The taxon and character sampling for these phylogenies is as follows: EPSP 69 taxa and 293 amino acid characters, shikimate kinase 44 taxa and 139 characters, and shikimate dehydrogenase 52 taxa and 166 characters. In all the phylogenies the Toxoplasma gondii AROM domains cluster with the fungal homologues suggesting they are related, given the taxon sampling available. The shikimate kinase phylogeny also revealed a potential cyanobacterial to plant gene transfer, consistent with this plant enzyme originating from the plant chloroplast endosymbiont, although the cyanobacteria were not monophyletic in the Bayesian tree. The bootstrap tree shows a monophyletic cyanobacteria clade sister to the plant clade with a bootstrap support value of 50%. Following preliminary phylogentic analysis nearest neighbour paralogues were excluded unless sequence similarity was low as noted for Listeria moncytogenes (C) and Mesorhizobium loti (B). 12 S.A. Campbell et al. / International Journal for Parasitology 34 (2004) 5–13 from the bacterial progenitor of the eukaryotes (Martin et al., 2001). A survey of some 80 currently available completed prokaryotic genomes found clustering of shikimate pathway genes in a number of taxonomes, some of which are known to be co-transcribed as an operon. The lack of a described clustering in the precise order of the AROM functional domains, may reflect lack of sampling or alternatively that multiple sequential fusion events, coupled with rearrangements in domain order occurred in the evolution of an efficient functional AROM protein. In testing the evolutionary origin of the T. gondii shikimate pathway we had to consider a number of possible evolutionary scenarios that could have arisen during apicomplexan evolution. These include the possibility of direct vertical descent or the acquisition of genes that encode plastid-located enzymes from the algal endosymbiont. In the latter case, these genes would have been derived from the algal plastid genome and may or may not have been transferred to the nuclear genome as proposed for modern plants. We included homologues from the cyanobacteria and the plants, in an attempt to exclude an origin from the plastid genome of the progenitor of the apicoplast. We found no evidence to suggest that the T. gondii shikimate pathways genes were inherited from the apicoplast genome. However, as these studies progressed and with the realisation that the shikimate pathway may have been an ancestral trait in eukaryotes another possibility had to be considered. That is, the T. gondii genes may have been derived from the nuclear genome of the algal endosymbiont that became the apicoplast. Our analysis, however, could not exclude this possibility. Given that the shikimate pathway and the arom supergene appear to have a wide eukaryotic distribution, it is plausible that the algal nucleus may have contained the ancient eukaryotic shikimate pathway genes with an AROM-like polypeptide. Eukaryotic gene sampling, currently lacking shikimate pathway homologues from algal groups, the likely progenitors of the apicoplast (Kohler et al., 1997), means that it is impossible to distinguish between vertical descent or alternatively an origin from the nucleus of the algal progenitor of the apicoplast. The phylogenetic investigations fail to show a consistent prokaryotic sister group to the fungi/Toxoplasma eukaryote cluster, thus preventing the identification of a prokaryote donor lineage. However, the phylogenies produced no evidence that the T. gondii genes were inherited from either the mitochondria or the plastid. Our analysis also did not support any incidence of prokaryote to T. gondii horizontal gene transfer as all the phylogenies showed a common ancestor with the fungi. This would suggest any transfer event would have had to occur prior to the division of the fungi and T. gondii lineages. However, the DHQ-synthase phylogeny is currently unresolved and a prokaryote to T. gondii transfer scenario is still possible for this enzyme domain. Re-examination of the completed P. falciparum genome did not provide evidence of an AROM-type protein. However, a potential EPSP synthase/shikimate kinase bifunctional protein is evident (accession no. NP472984) and is likely to be the gene previously reported to have low similarity with S. cerevisiae AROM polypeptide (Gardner et al., 2002). Homologues of this potential EPSP synthase/ shikimate kinase bifunctional protein are present in a number of other Plasmodium species (Plasmodium yoelii accession no. EAA17633 and Plasmodium chrPch002449). This raises the question as to why the remaining enzymes are not readily identifiable. It seems unlikely that these enzymes are absent, as we now have evidence for the final three enzymes of the pathway, providing a route from shikimate to chorismate. We also know that inhibition of one of these enzymes, EPSP synthase, is capable of restricting parasite growth (Roberts et al., 1998). There is no known route to produce shikimate other than by the four missing enzymes and shikimate would not be available within the host. This suggests that there may be enzymes with the same biochemical ability, but vastly different in sequence, thus making them difficult to identify. Alternatively this highlights a potential ongoing challenge for gene prediction and thus complete annotation of the P. falciparum and other Plasmodium genome projects. We have provided the first evidence for the entire set of seven shikimate pathway enzymes in any apicomplexan parasite, their genetic and molecular arrangement and their likely evolutionary origin. The results presented for T. gondii provides the tools for functional studies, structural determination and rational drug design. Phylogenetic comparisons suggest that the AROM-gene fusion was an innovation likely to have been present in the progenitor of modern eukaryotes, as the distantly diverged T. gondii and fungi lineages both posses a homologous arom supergene. Thus, the shikimate pathway, rather than being confined to bacteria, fungi and plants and at least some apicomplexans, is likely to have been an ancient eukaryotic attribute. It has been lost in many taxonomes, including mammals that are now dependent on exogenous aromatic compounds. In plants the ancient gene organisation has not survived and it seems likely that the source of the shikimate pathway genes, which are essentially bacterial like, has been through the acquisition of the chloroplast, although this hypothesis requires further testing. It also seems likely that the list of taxonomes where this ancient pathway has been retained is likely to grow as we see the completion of more eukaryotic genome projects. Acknowledgements Preliminary genomic and/or cDNA sequence data was accessed via http://ToxoDB.org and/or http://www.tigr.org/ tdb/t_gondii/. Genomic data were provided by the Institute for Genomic Research (supported by the NIH grant no. AI05093), and by the Sanger Center (Wellcome Trust). EST sequences were generated by Washington University S.A. Campbell et al. / International Journal for Parasitology 34 (2004) 5–13 (NIH grant no. 1R01AI045806-01A1). The work reported in this manuscript was funded by NIH, USA RO1 AI-43228, the Wellcome Trust, Koshland, Breenan, Blackmon, Langel and Kiewit families. T.A.R. is supported by a BBSRC studentship. References Andersson, J.O., Roger, A.J., 2002. Evolutionary analyses of the small subunit of glutamate synthase: gene order conservation, gene fusions, and prokaryote-to-eukaryote lateral gene transfers. Eukaryot. Cell 1, 304–310. Butler, J.R., Alworth, W.L., Nugent, M.J., 1974. Mechanism of dehydroquinase catalysed dehydration 1. Formation of a shiff base intermediate. J. Am. Chem. Soc. 96, 1617– 1618. Carpenter, E.P., Hawkins, A.R., Frost, J.W., Brown, K.A., 1998. Structure of dehydroquinate synthase reveals an active site capable of multistep catalysis. Nature 394, 299– 302. Coggins, J.R., Duncan, K., Anton, I.A., Boocock, M.R., Chaudhuri, S., Lambert, J.M., Lewendon, A., Millar, G., Mousdale, D.M., Smith, D.D., 1987. The anatomy of a multifunctional enzyme. Biochem. Soc. Trans. 15, 754 –759. Duncan, K., Edwards, R.M., Coggins, J.R., 1987. The pentafunctional arom enzyme of Saccharomyces cerevisiae is a mosaic of monofunctional domains. Biochem. J. 246, 375 –386. Fitzpatrick, T., Ricken, S., Lanzer, M., Amrhein, N., Macheroux, P., Kappes, B., 2001. Subcellular localization and characterization of chorismate synthase in the apicomplexan Plasmodium falciparum. Mol. Microbiol. 40, 65–75. Gardner, M.J., Hall, N., Fung, E., White, O., Berriman, M., Hyman, R.W., Carlton, J.M., Pain, A., Nelson, K.E., Bowman, S., Paulsen, I.T., James, K., Eisen, J.A., Rutherford, K., Salzberg, S.L., Craig, A., Kyes, S., Chan, M.S., Nene, V., Shallom, S.J., Suh, B., Peterson, J., Angiuoli, S., Pertea, M., Allen, J., Selengut, J., Haft, D., Mather, M.W., Vaidya, A.B., Martin, D.M., Fairlamb, A.H., Fraunholz, M.J., Roos, D.S., Ralph, S.A., McFadden, G.I., Cummings, L.M., Subramanian, G.M., Mungall, C., Venter, J.C., Carucci, D.J., Hoffman, S.L., Newbold, C., Davis, R.W., Fraser, C.M., Barrell, B., 2002. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511. Gosset, G., Bonner, C.A., Jensen, R.A., 2001. Microbial origin of planttype 2-keto-3-deoxy-D -arabino-heptulosonate 7-phosphate synthases, exemplified by the chorismate- and tryptophan-regulated enzyme from Xanthomonas campestris. J. Bacteriol. 183, 4061–4070. Herrmann, K., Weaver, L., 1999. The shikimate pathway. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50, 473–503. Huelsenbeck, J.P., Ronquist, F., 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. Jensen, R.A., Xie, G., Calhoun, D.H., Bonner, C.A., 2002. The correct phylogenetic relationship of KdsA (3-deoxy-D -manno-octulosonate 8phosphate synthase) with one of two independently evolved classes of AroA (3-deoxy-D -arabino-heptulosonate 7-phosphate synthase). J. Mol. Evol. 54, 416 –423. 13 Keeling, P.J., Palmer, J.D., Donald, R.G., Roos, D.S., Waller, R.F., McFadden, G.I., 1998. Shikimate pathway in apicomplexan parasites. Nature 397, 219 –220. Kohler, S., Delwiche, C.F., Denny, P.W., Tilney, L.G., Webster, P., Wilson, R.J., Palmer, J.D., Roos, D.S., 1997. A plastid of probable green algal origin in apicomplexan parasites. Science 275, 1485– 1489. Martin, W., Hoffmeister, M., Rotte, C., Henze, K., 2001. An overview of endosymbiotic models for the origins of eukaryotes, their ATPproducing organelles (mitochondria and hydrogenosomes), and their heterotrophic lifestyle. Biol. Chem. 382, 1521–1539. Martin, W., Rujan, T., Richly, E., Hansen, A., Cornelsen, S., Lins, T., Leister, D., Stoebe, B., Hasegawa, M., Penny, D., 2002. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl Acad. Sci. USA 99, 12246– 12251. McFadden, G.I., Reith, M.E., Munholland, J., Lang-Unnasch, N., 1996. Plastid in human parasites. Nature 381, 482. Moore, J.D., Hawkins, A.R., 1993. Overproduction of, and interaction within, bifunctional domains from the amino- and carboxy-termini of the pentafunctional AROM protein of Aspergillus nidulans. Mol. Gen. Genet. 240, 92–102. Mousdale, D.M., Campbell, M.S., Coggins, J.R., 1987. Purification and characterisation of a bifunctional dehydroquinase-shikimate: NADP oxidoreductase from peas seedlings. Phytochemistry 26, 2665–2670. Richards, T.A., Hirt, R.P., Williams, B.A., Embley, T.M., 2003. Horizontal gene transfer and the evolution of parasitic protozoa. Protist 154, 17– 32. Roberts, F., Roberts, C.W., Johnson, J.J., Kyle, D.E., Krell, T., Coggins, J.R., Coombs, G.H., Milhous, W.K., Tzipori, S., Ferguson, D.J., Chakrabarti, D., McLeod, R., 1998. Evidence for the shikimate pathway in apicomplexan parasites. Nature 393, 801–805. Roberts, C.W., Roberts, F., Lyons, R.E., Kirisits, M.J., Mui, E.J., Finnerty, J., Johnson, J.J., Ferguson, D.J., Coggins, J.R., Krell, T., Coombs, G.H., Milhous, W.K., Kyle, D.E., Tzipori, S., Barnwell, J., Dame, J.B., Carlton, J., McLeod, R., 2002. The shikimate pathway and its branches in apicomplexan parasites. J. Infect. Dis. 185 (Suppl 1), S25–S36. Rost, B., 1996. PHD: predicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol. 266, 525 –539. Schmidt, H.A., Strimmer, K., Vingron, M., von Haeseler, A., 2002. TREEPUZZLE: maximum likelihood phylogenetic analysis using quartets and parallel computing. Bioinformatics 18, 502 –504. Smith, A.B., 1994. Rooting molecular trees: problems and strategies. Biol. J. Linnean Soc. 51, 279–292. Sourdis, J., Krimbas, C., 1987. Accuracy of phylogenetic trees estimated from DNA sequence data. Mol. Biol. Evol. 4, 159–166. Stechmann, A., Cavalier-Smith, T., 2002. Rooting the eukaryote tree by using a derived gene fusion. Science 297, 89–91. Thompson, J.D., Gibson, T.J., Plewniak, F., Jeanmougin, F., Higgins, D.G., 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882. Walker, G.E., Dunbar, B., Hunter, I.S., Nimmo, H.G., Coggins, J.R., 1996. Evidence for a novel class of microbial 3-deoxy-D -arabino-heptulosonate-7-phosphate synthase in Streptomyces coelicolor A3(2), Streptomyces rimosus and Neurospora crassa. Microbiology 142, 1973–1982.